

Abstract
Spiny neurons in the neostriatum die within 24 hr after transient global ischemia, whereas large aspiny (LA) neurons remain intact. To reveal the mechanisms of such selective cell death after ischemia, excitatory neurotransmission was studied in LA neurons before and after ischemia. The intrastriatally evoked fast EPSCs in LA neurons were depressed ≤24 hr after ischemia. The concentration–response curves generated by application of exogenous glutamate in these neurons were approximately the same before and after ischemia. A train of five stimuli (100 Hz) induced progressively smaller EPSCs, but the proportion of decrease in EPSC amplitude at 4 hr after ischemia was significantly smaller compared with control and at 24 hr after ischemia. Parallel depression of NMDA receptor and AMPA receptor-mediated EPSCs was also observed after ischemia, supporting the involvement of presynaptic mechanisms. The adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine blocked the inhibition of evoked EPSCs at 4 hr after ischemia but not at 24 hr after ischemia. Electron microscopic studies demonstrated that the most presynaptic terminals in the striatum had a normal appearance at 4 hr after ischemia but showed degenerating signs at 24 hr after ischemia. These results indicated that the excitatory neurotransmission in LA neurons was depressed after ischemia via presynaptic mechanisms. The depression of EPSCs shortly after ischemia might be attributable to the enhanced adenosine A1 receptor function on synaptic transmission, and the depression at late time points might result from the degeneration of presynaptic terminals.
Keywords: ischemia, excitotoxicity, AMPA, neuronal death, striatum, interneurons, excitatory synaptic transmission
Large aspiny (LA) neurons only account for <2% of the entire neuronal population in the neostriatum but have great influences on the function of basal ganglia. They have large somata (20–60 μm) with extended smooth dendrites and widespread axonal arborizations (DiFiglia and Carey, 1986; Wilson et al., 1990; Kawaguchi, 1992). LA neurons are the source of acetylcholine in the neostriatum (Butcher and Hodge, 1976; Bolam et al., 1984), and by activation of muscarinic receptors, they affect the output of the neostriatum through the complex modulation of synaptic transmission in spiny projection neurons and interneurons (Kawaguchi et al., 1995;Galarraga et al., 1999; Koos and Tepper, 1999; Calabresi et al., 2000).
The striatum is one of the most vulnerable regions in the brain to cerebral ischemia. Small- to medium-sized neurons (most likely spiny neurons) in the dorsal striatum die 24 hr after 25–30 min of forebrain ischemia (Pulsinelli et al., 1982). However, LA neurons in the same region are resistant to ischemic insult (Francis and Pulsinelli, 1982;Chesselet et al., 1990). The mechanisms underlying this selective vulnerability after ischemia are poorly understood. Excitotoxicity is one of the major causes of neuronal death after ischemia (Rothman and Olney, 1986; Choi and Rothman, 1990). Previous studies have shown that the excitatory synaptic transmission is enhanced in ischemia-vulnerable CA1 pyramidal neurons in the hippocampus after ischemia/hypoxia (Urban et al., 1989; Crepel et al., 1993; Gao et al., 1998a). On the contrary, a depression in excitatory synaptic transmission has been observed in ischemia-resistant CA3 neurons and dentate granule cells after ischemia (Gao et al., 1998b). These results suggest that the enhancement of excitatory neurotransmission might be associated with ischemic cell death, and that the depression of excitation might be involved in neuroprotection after ischemia.
Electrophysiological studies on selective cell death in the striatum after ischemia are less extensive. What are the differences in synaptic transmission between spiny neurons and LA neurons after ischemia? What are the mechanisms underlying these changes and how these changes determine the ischemic outcome of these neurons? In vitrostudies have reported that the membrane potential of spiny neurons was depolarized during hypoxia/hypoglycemia, whereas that of LA neurons was hyperpolarized (Calabresi et al., 1997b; Pisani et al., 1999). Deprivation of oxygen and glucose in brain slices has been shown to induce long-term potentiation (LTP) in spiny neurons but not in LA neurons (Calabresi et al., 2002). Because of the dramatic difference in temperature, microenvironment, and homeostasis, the neuronal responses to hypoxia/hypoglycemia in vitro might differ from those after ischemia in vivo. To reveal the synaptic transmission changes in striatal neurons after ischemia, especially those after reperfusion, studies using an intracellular recording in vivo technique have shown that IPSPs in spiny neurons were abolished, and that the incidence of cortically evoked polysynaptic EPSPs was significantly increased after transient forebrain ischemia (Gajendiran et al., 2001). Little is known about synaptic transmission changes in LA neurons after ischemia in vivo, because recording from LA neurons in vivo is extremely difficult because of their sparse distribution. To circumvent this problem, the present study examined the evoked EPSCs from visually identified LA neurons in brain slices prepared at different intervals after ischemiain vivo.
MATERIALS AND METHODS
Male Wistar rats (100–180 gm; Charles River Laboratories, Wilmington, MA) of ∼5–6 weeks of age were used in the present study. Experimental protocols were institutionally approved in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize both the suffering and number of animals used.
Transient forebrain ischemia. Transient forebrain ischemia was induced using the four vessel occlusion method (Pulsinelli and Brierley, 1979) with modifications (Ren et al., 1997). The animals were fasted overnight to provide uniform blood glucose levels. For surgical preparation, the animals were anesthetized with a mixture of 1–2% halothane, 33% O2, and 66% N2 via a gas mask placed around the nose. A silicon tube loop was placed loosely around each common carotid artery to allow subsequent occlusion of these vessels. The animal was then placed on a stereotaxic frame, and the vertebral arteries were electrocauterized. A very small temperature probe (0.025 inch D; Physitemp, Clifton, NJ) was inserted beneath the skull in the extradural space, and the brain temperature was maintained at 37°C with a heating lamp using a temperature-control system (BAT-10; Physitemp). Transient forebrain ischemia was induced by occluding both common carotid arteries to induce ischemic depolarization for ∼22 min. Cerebral blood flow resumed immediately on release of the carotid artery clasps.
Slice preparation and whole-cell, voltage-clamped recording.Brain slices were prepared from animals before ischemia and at 4–6 and 24 hr after reperfusion as described previously (Chi and Xu, 2000). The animals were anesthetized with ketamine-HCl (80 mg/kg, i.p.) and decapitated. The brains were quickly removed and immersed in ice-cold artificial CSF (ACSF), which was composed of the following (in mm): 130 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. Transverse striatal slices of 280 μm thickness were cut using a vibratome (VT 1000S; Leica, Nussloch, Germany) and incubated in ACSF for ≥1 hr at room temperature before being transferred to the recording chamber. The slice was submerged beneath the fluid surface and superfused continuously with oxygenated ACSF. The flow rate was adjusted to 2–3 ml/min. Recordings were performed at room temperature (∼24°C).
For whole-cell recording, patch electrodes were prepared from borosilicate glass (Warner Instruments, Hamden, CT) using a horizontal electrode puller (P-97; Flaming/Brown; Sutter, Novato, CA) to produce tip openings of 1–2 μm (3–5 MΩ). Electrodes were filled with an intracellular solution containing (in mm): 43 CsCl, 92 CsMeSO4, 5 TEA, 2 EGTA, 1 MgCl2, 10 HEPES, and 4 ATP (Sigma, St. Louis, MO). Neurobiotin (2%) (Vector Laboratories, Burlingame, CA) was included in some experiments to verify the neurons recorded. Neurons were visualized with an infrared–differential interference contrast (DIC) microscope (BX 50 WL; Olympus Optical, Tokyo, Japan) and a CCD camera. Only those with large somata (>20 μm) were selected for recording. Positive pressure was applied to the recording pipette as it was lowered into the medium and approached the cell membrane. Constant negative pressure was applied to form the seal (>1 GΩ) when the recording pipette attached to the membrane. A sharp pulse of negative pressure was applied to open the cell membrane for whole-cell recording. The series resistance of the pipette was ∼10 MΩ. Voltage-clamped recording was performed with an Axopatch 200 B amplifier (Axon Instruments, Foster City, CA). Signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a data-acquisition program (Axograph 4.5; Axon Instruments). Intrastriatal stimulation was delivered every 10–20 sec using a bipolar tungsten electrode (∼5 MΩ; Micro Probe, Potomac, MD), and 0.1 msec current pulses were used to evoke the excitatory responses. Zero to five times of threshold stimulus intensity (0–5T) was used in the present experiments. In some experiments, a train of five stimuli at 100 Hz with the same intensity (4T) and duration (100 μsec) was applied at an interval of 60–90 sec.
In some experiments, neurobiotin was iontophoresed into the cell by passing depolarizing pulses after recording. The slice was then fixed in 4% paraformaldehyde overnight and incubated in 0.1% horseradish peroxidase-conjugated avidin D (Vector Laboratories) in 0.01m potassium PBS, pH 7.4, with 0.5% Triton X-100 for 24 hr at room temperature. After detection of peroxidase activity with 3,3′-diaminobenzidine, slices were examined in potassium PBS. Slices containing labeled neurons were mounted on gelatin-coated slides and processed for light microscopy.
Electron microscopy. At 4–6 hr (n = 2) or 24 hr (n = 2) after transient forebrain ischemia, the animals were anesthetized with ketamine (80 mg/kg, i.p.) and then perfused transcardially with 100 ml of PBS, followed by 400 ml of 2% glutaraldehyde and 2% paraformaldehyde in 0.1 mpotassium PBS. The brain was removed and postfixed in the same fixative for ≥12 hr. The brain blocks, including the striatum between 1.2 and 0.2 mm from the Bregma level, were cut in vibratome. Sections of 50 μm thickness were postfixed with 0.5% osmium tetroxide for 1 hr and incubated in 0.5% uranylacetate for 2 hr. The sections were embedded in Embed 812 and put into an oven at 60°C for polymerization for 48 hr. Ultrathin sections were stained with 2% uranylacetate and 1% lead citrate and examined under a Philips electron microscopic (EM) 400 electron microscope. For quantitative analysis of degenerated terminals at different intervals after ischemia, images at the dorsal striatum were captured randomly at 13,000× magnification (unit area of 42.3 μm2 per image). The pictures were enlarged, and the presynaptic terminals were counted and divided into different groups according to their morphological features. The data were analyzed using StatView 5.0 (Abacus Concepts, Calabasas, CA).
Drug application. (−)-Bicuculline methiodide (BMI), (−)-2-amino-5-phosphonopentanoic acid (d-APV), scopolamine, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), and glybenclamide were obtained from Sigma. Antagonists were applied via bath superfusion. BMI was used to block GABAAreceptors at a concentration of 30 μm, and 50 μmd-APV was used for NMDA receptor (NMDAR) blockade. (±)-4-(4-aminophenyl)-1,2-dihydro-1-methyl-2-propylcarbamoyl-6,7-methylenedioxyphthalazine (SYM 2206), cyclothiazide (CTZ), and (RS)-α-methyl-4-carboxyphenylglycine (MCPG) were purchased from Tocris Cookson (Ellisville, MO). Agonists were applied through a “Y”-tube system (Kiyosawa et al., 2001). The tip of the Y-tube had a diameter between 100 and 150 μm and was placed close to the neuron under study.
Data analysis. A Michaelis–Menten equation using a least-squares fitting was applied for evaluation of the EC50 of glutamate in the concentration–response relationships, as follows: a =bn/(bn + EC50n), where a is relative activation, b is agonist concentration (molar concentration), and n is the Hill coefficient.
The values were presented as mean ± SEM. The unpairedt test (for two groups) or ANOVA (for more than two groups) followed by post hoc Scheffe's test was used for statistical analysis (StatView 5.0; Abacus Concepts). Changes were considered significant if p < 0.05.
RESULTS
LA neurons were easily identified according to their large somata, ranging from 20 to 60 μm in diameter under infrared–DIC optics (Fig.1A). Intracellular staining with neurobiotin revealed that these neurons have large somata with three to five primary dendrites bearing few spines (Fig.1B). Forebrain ischemia that induces an ischemic depolarization of ∼22 min consistently produced >90% cell death in the dorsal striatum (Ren et al., 1997). At 24 hr after ischemia, it was difficult to find small- to medium-sized neurons from brain slices, whereas neurons with large somata remained intact.
Fig. 1.

Photomicrographs of a LA neuron visualized in brain slice with an infrared–DIC microscope before recording (A) and after intracellular staining with neurobiotin (B). The cell body of this neuron is ∼60 μm in diameter, and the dendrites are smooth.
Depression of AMPA receptor-mediated EPSCs after ischemia
Intrastriatal stimulation evoked both EPSCs and IPSCs from LA neurons. To block the IPSCs mediated by GABAAreceptors and muscarinic receptors, BMI (30 μm) and scopolamine (3 μm) were added to the bath solution. Both AMPA receptor (AMPAR)- and NMDAR-mediated components contribute to EPSCs in LA neurons (Kawaguchi, 1992; Calabresi et al., 1998). Inward EPSCs were evoked at a holding potential of −60 mV. Because AMPARs, especially calcium-permeable AMPARs, play an important role in neuronal injury after ischemia (Pellegrini-Giampietro et al., 1994; Gorter et al., 1997), the present study focused on changes in AMPAR-mediated EPSCs in LA neurons after ischemia. Therefore, d-APV (50 μm) was added to the bath solution to block NMDAR-mediated EPSCs. The remaining evoked responses could be reversibly blocked by 30 μm CNQX, indicating non-NMDAR-mediated responses (Fig.2A). To further clarify the receptor subtype that mediated the synaptic response, the AMPAR-specific antagonist SYM 2206 (100 μm) was applied and completely blocked the EPSCs. The current–voltage relationship of AMPAR-mediated EPSCs showed inward rectifications before and after ischemia (Fig. 2B), suggesting that they are mediated by Ca2+-permeable AMPARs (Suzuki et al., 2001).
Fig. 2.

Non-NMDAR-mediated EPSCs in LA neurons.A, Evoked synaptic currents were recorded from a LA neuron at a holding potential of −60 mV. Synaptic currents were elicited by intrastriatal stimulation in the presence of BMI (30 μm) and scopolamine (3 μm).d-APV (50 μm) was added to the bath subsequently. The evoked EPSCs were blocked by 30 μmCNQX, indicating that they are AMPAR-mediated currents.B, Current–voltage relationship of AMPAR-mediated EPSCs in LA neurons. BMI, scopolamine, and d-APV were added to the bath medium. B1, Representative traces showing the EPSCs recorded at different holding potentials with four times the threshold stimulus intensity (4T) before and at 4 hr after ischemia. The amplitude of EPSCs increased with more hyperpolarizing holding potentials, and the inward currents reversed to outward currents at positive holding potentials. B2, Current–voltage (I–V) curves of AMPAR-mediated EPSCs in LA neurons before (n = 9) and after (n = 9) ischemia. Amplitudes of evoked EPSCs at various holding potentials (VH) were normalized to those at various holding potentials of −60 mV. TheI–V curve showed strong inward rectification at positive holding potentials. All traces are the average of 6–10 consecutive recordings. The values of the plotting are mean ± SEM.
To examine the efficacy of synaptic transmission in LA neurons, the threshold of stimulus intensity was compared before and after ischemia. The threshold of stimulus intensity was defined as the stimulating current that evokes the smallest detectable response from LA neurons. There were significant changes in the threshold of stimulus intensity after ischemia (ANOVA; F = 4.07; df 2, 45;p = 0.024). In control animals, the threshold stimulus intensity for inducing EPSCs was 410 ± 61 μA (n= 18) in the present experimental conditions. The threshold stimulus intensity increased to 524 ± 75 μA (n = 17) and 808 ± 16 μA (n = 13; post hocScheffe's test; p < 0.05) at 4–6 and 24 hr after ischemia, respectively. The amplitudes of the evoked EPSCs increased according to the increase in stimulus intensities (Fig.3). In comparison with the control levels, the amplitudes of EPSCs in LA neurons decreased dramatically at most stimulus intensities after ischemia (Fig. 3B). For example, at a stimulus intensity of four times the threshold stimulus intensity (4T), the amplitude of EPSCs was significantly depressed (ANOVA; F = 7.22; df 2, 45; p = 0.002). The amplitude was 129.7 ± 14.4 pA in controls (n= 18) and 68.9 ± 11.9 pA (n = 17; post hoc Scheffe's test; p < 0.01) and 75.1 ± 10.5 pA (n = 13; p < 0.05) at 4–6 and 24 hr after reperfusion, respectively.
Fig. 3.

Changes in EPSCs in LA neurons before and after ischemia. A, Representative traces recorded from LA neurons before and at 4 hr after ischemia. The amplitude of evoked EPSC increased with increasing stimulus intensities (0–5 times threshold intensities, 0–5T), but the amplitude of EPSCs at 4 hr after ischemia was significantly smaller than that of control ones. The traces are the average of six consecutive recordings. B, The input–out relationship of evoked EPSCs before and at different intervals after ischemia. The amplitude of EPSCs was dramatically decreased at 4 and 24 hr after ischemia at all stimulus intensities (1.5–5T). *p < 0.05;†p < 0.01.
No significant change in postsynaptic AMPAR function after ischemia
To reveal whether the depression of evoked EPSCs after ischemia was caused by the attenuation of glutamate receptor functions, postsynaptic responses were examined by focal application of exogenous AMPA or glutamate in the presence of d-APV through a Y-tube system. The application of glutamate induced an inward current at a holding potential of −60 mV. The non-NMDAR antagonist CNQX (data not shown) and the AMPAR-specific antagonist SYM 2206 (Fig.4A1) blocked the glutamate-induced responses in a dose-dependent manner. The AMPAR desensitization was extremely rapid, ranging from 2 to 40 msec in neurons (Kiskin et al., 1986; Trussell et al., 1988; Tang et al., 1989), and the speed with which we could apply agonists to our recorded neurons in slice was limited. The peak current might not be accurately detected in the present configuration. Therefore, the steady-state currents were compared before and after ischemia. The glutamate-induced currents became detectable at a concentration of ∼3 × 10−5m and then increased in a sigmoid manner with increasing glutamate concentrations. The normalized concentration–response curves were approximately the same before and after ischemia (Fig. 4B). The EC50 values were 6 ± 0.31 × 10−4m(n = 13) for control neurons and 6.4 ± 0.29 × 10−4m(n = 11) and 6.8 ± 0.35 × 10−4m(n = 8) for neurons at 4–6 and 24 hr after ischemia, respectively. The Hill coefficients were 1.8 ± 0.2 (n = 13) in the control group and 1.9 ± 0.2 (n = 11) and 1.9 ± 0.2 (n = 8) at 4–6 and 24 hr after ischemia, respectively. To eliminate the impact of cell size, currents were normalized by cell capacitance and expressed as current densities. No significant difference in current density was detected after ischemia (Fig. 4C).
Fig. 4.

Exogenous glutamate induced whole-cell currents before and after ischemia. A, Application of exogenous glutamate induced an inward current at a holding potential of −60 mV in the presence of 50 μmd-APV. The glutamate responses were antagonized by SYM 2206, a specific AMPAR antagonist (A1). The amplitude of glutamate-induced currents was increased accordingly with increasing concentrations of glutamate before (A2) and after (A3) ischemia. B, Dose–response curve of glutamate responses before and after ischemia. The EC50values and Hill coefficients of the dose–response curve after ischemia were approximately the same as those in control neurons.C, Comparison of current densities of glutamate-induced currents in LA neurons before and after ischemia. The current density was slightly decreased at 4 hr after ischemia, but no statistic significance was detected.
AMPARs quickly desensitize, and a decrease in the desensitization of AMPAR-mediated responses has been shown to be involved in cell death after trauma (Goforth et al., 1999). To examine the possible changes in desensitization of AMPARs after ischemia, 100 μm CTZ, a drug that selectively blocks AMPAR desensitization (Bertolino et al., 1993), was used to eliminate the fast desensitization. After coapplication of CTZ, AMPA-induced currents were increased more than twofold, but no significant difference in current densities was detected after ischemia (Fig.5B). To further explore whether the desensitization contributes to the changes in EPSCs after ischemia, the decay time of evoked EPSCs was compared before and after ischemia. The decay phase of EPSCs could be well fitted by a single exponential (Fig. 5C). The decay times were 7.86 ± 0.46 msec (n = 14) in control neurons, and no significant difference was found in neurons after ischemia (8.58 ± 1.05 msec at 4–6 hr, n = 10; 8.08 ± 1.04 msec at 24 hr, n = 11) (Fig. 5D). These data indicated that the desensitization did not contribute to the changes in evoked EPSCs in LA neurons after ischemia.
Fig. 5.

Comparison of desensitization of AMPAR-mediated responses before and after ischemia. A, Examples of whole-cell currents induced by 1 mm AMPA at 4 hr after ischemia before and after application of CTZ. After treatment with 100 μm CTZ, the amplitude of glutamate-induced responses dramatically increased. B, Current density of AMPA-induced responses in LA neurons was approximately the same before and after ischemia. No difference in current density was found between control and ischemic neurons after application of CTZ, despite the current density values of all neurons that were significantly increased. C, Representative traces showing the decay of evoked EPSCs were well fitted by the single exponential method. The traces are the average of six consecutive recordings.D, Histogram showing that the mean decay time of EPSCs in LA neurons did not change after ischemia. The above results indicate that desensitization plays little role in synaptic depression of LA neurons after ischemia.
Alterations of presynaptic mechanisms after ischemia
Another possible cause of the reduction of evoked EPSCs after ischemia is a decrease in presynaptic transmitter release probability. To investigate whether alteration of release probability accounts for the reduction of evoked fast EPSCs, a paired-pulse test (50 msec interstimulus interval) was conducted in LA neurons before and after ischemia. However, no obvious changes were detected in paired-pulse ratio (PPR) at all stimulus intensities after ischemia (data not shown). The changes in release probability were also examined by comparing the EPSCs evoked by a train of five stimuli at 100 Hz (Scheuss et al., 2002). A train of stimuli at high frequency markedly reduces synaptic release by depleting the releasable pool of vesicles (Wang and Kaczmarek, 1998; Bellingham and Walmsley, 1999). A decrease in release probability will decrease the speed of vesicle depletion in the presynaptic terminals, and therefore, the fifth/first EPSC ratio will increase. As shown in Figure6A2, the fifth/first EPSC ratio was significantly increased at 4 hr after ischemia (ANOVA;F = 12.57; df 2, 23; p < 0.001). In control conditions, the amplitude of the fifth EPSC was reduced to 28.7 ± 2.8% of the first one (n = 11). Four hours after reperfusion, the fifth EPSC was only reduced to 53.4 ± 8.2% of the initial EPSC (n = 7; post hoc Scheffe's test; p < 0.01). At 24 hr after reperfusion, the fifth EPSC was reduced to 16.2 ± 4.6% of the initial values, which was close to the control values. The above results suggest that the releasing probability transiently decreased 4–6 hr after ischemia.
Fig. 6.
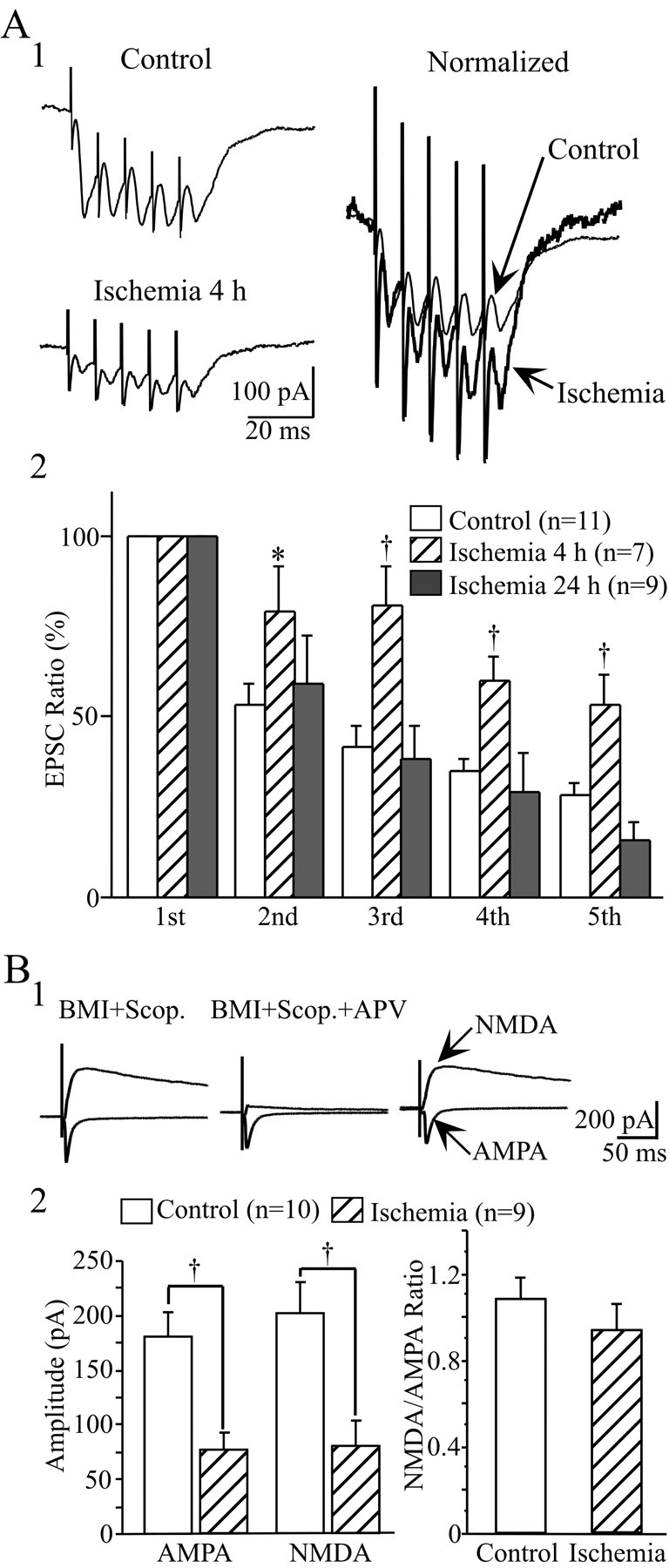
Comparison of EPSCs elicited by a train of high-frequency (100 Hz) stimulation and changes in NMDAR/AMPAR-mediated responses before and after ischemia. A1, Representative traces showing the synaptic responses induced by a train of five stimuli before ischemia and at 4 hr after reperfusion. The two traces were normalized to the same amplitude of the first EPSCs to show the difference in the fifth/first EPSC ratio between these traces (right). All traces are the average of six consecutive recordings. A2, Pooled data showing the changes in EPSCs during train stimuli. All amplitudes were normalized to the first EPSCs at each recording. The fifth/first EPSC ratio was significantly higher at 4 hr after ischemia compared with controls and at 24 hr after ischemia, suggesting that the releasing probability of LA neurons was transiently decreased shortly after ischemia. B, Parallel change in NMDAR-and AMPAR-mediated responses before and at 4 hr after ischemia. B1, left, Evoked EPSC in the presence of 20 μm BMI and 2 μmscopolamine (Scop.), at the holding potentials of −80 mV and +60 mV, respectively. Middle, Evoked AMPAR-mediated responses in the presence of BMI, scopolamine, and 50 nmd-APV. Right, top trace, The NMDAR-mediated response was obtained by subtracting the left two top traces. B2, left histogram, Parallel decrease in NMDAR- and AMPAR-mediated responses after ischemia. Right histogram, Comparison of NMDAR-mediated current with the AMPAR-mediated current ratio before and after ischemia. No significant difference in NMDA/AMPA ratio was detected between these two conditions. *p < 0.05;†p < 0.01.
To further examine the involvement of a presynaptic mechanism in the changes in EPSCs after ischemia, the AMPAR- and NMDAR-mediated responses were compared before and after ischemia, because AMPARs and NMDARs are often colocalized at individual synapses. The changes in presynaptic transmitter release are expected to cause an equal increase or decrease in the synaptic currents mediated by these two receptor subtypes (Perkel and Nicoll, 1993; Liao et al., 1995; Malenka and Siegelbaum, 2001). With BMI- and scopolamine-containing medium, NMDAR-mediated responses were obtained by subtracting the responses at +60 mV with d-APV from those without d-APV (Fig. 6B1). AMPAR-mediated responses were isolated in the presence of BMI, scopolamine, and d-APV at a holding potential of −80 mV. At 4 hr after ischemia, NMDAR- and AMPAR-mediated EPSCs showed parallel decrease (NMDA: control, 201.89 ± 26.87 pA; ischemia, 79.31 ± 23.72 pA; unpairedt test; p < 0.01) (AMPA: control, 181.28 ± 21.31 pA; ischemia, 77.07 ± 15.55 pA;p < 0.01). The ratio of NMDAR-mediated EPSCs to AMPAR-mediated EPSCs was approximately the same before and after ischemia (Fig. 6B2). These data also supported a presynaptic mechanism in the decrease in AMPAR-mediated responses after ischemia.
Adenosine receptors and metabotropic glutamate receptors (mGluRs) have long been recognized to be associated with presynaptic inhibition of transmitter release (Zhang and Schmidt, 1999). To reveal the possible roles of these receptors in synaptic transmission after ischemia, the effects of the nonselective mGluR antagonist MCPG and the adenosine A1 receptor antagonist DPCPX on fast EPSCs were examined before and after ischemia. When MCPG (1 mm) and DPCPX (100 nm) were applied to control slices, no obvious changes were found in the amplitude of EPSCs, indicating that no tonic inhibition was induced by endogenous mGluR and A1 receptor activation before ischemia. Four hours after ischemia, application of MCPG produced no detectable changes, but application of DPCPX significantly increased the amplitude of evoked EPSCs (Fig. 7). The average amplitude increased ≤144 ± 2.1% after DPCPX application (n = 9; unpaired t test; p< 0.01) compared with the control one. However, application of DPCPX did not increase the amplitude of evoked EPSCs at 24 hr after ischemia. These data indicated that activation of the A1 receptor was involved in the reduction of fast EPSCs shortly after ischemia.
Fig. 7.
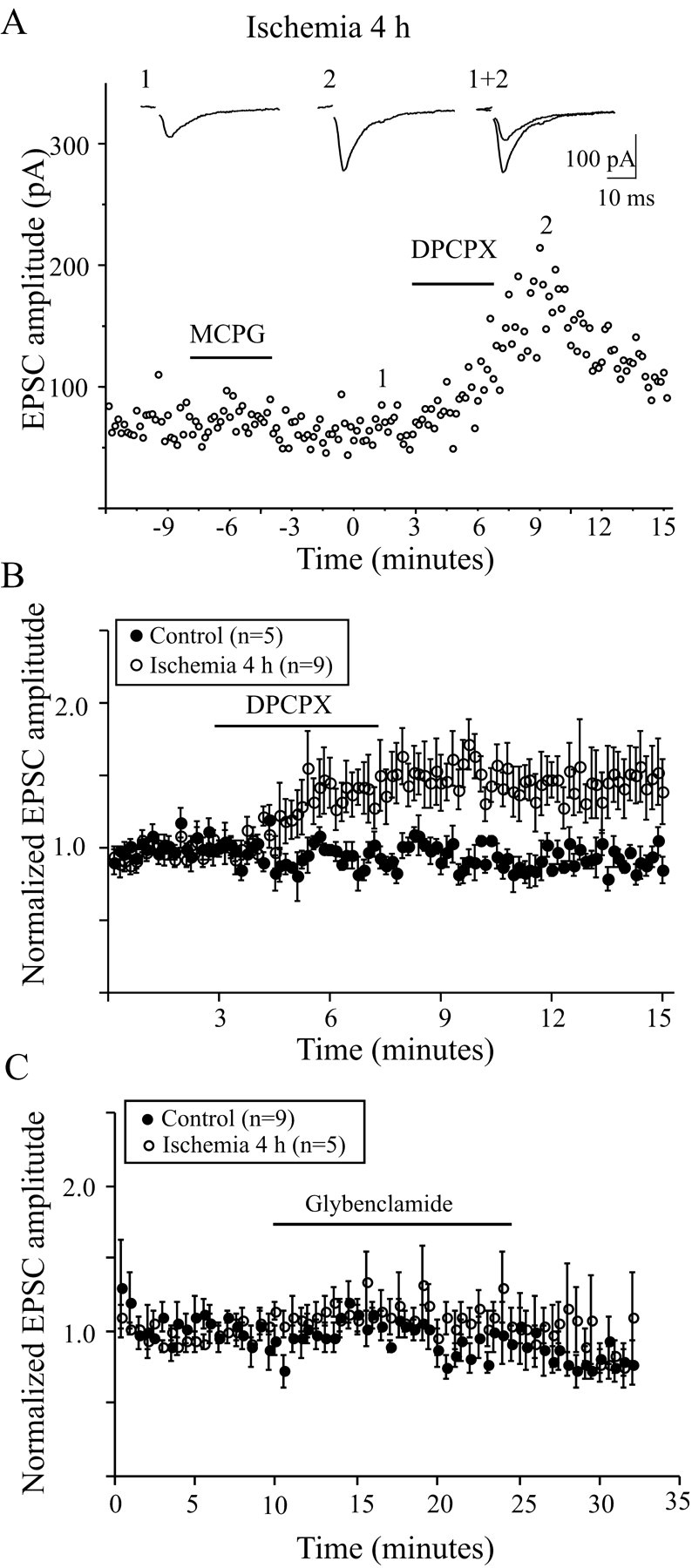
Effects of mGluR and adenosine A1 receptor blockers on EPSCs in LA neurons. A, Effect of bath application of the mGluR blocker MCPG (1 mm) and the A1 receptor DPCPX (100 nm) on the amplitude of EPSCs 4 hr after ischemia. Application of MCPG has no effects on EPSC amplitude, whereas DPCPX significantly enhances the EPSCs. Theinsets are representative traces averaged from six recordings before and after DPCPX application. B, The plotting showing the effects of DPCPX on EPSCs before and after ischemia. DPCPX has no effect on EPSCs in control neurons but enhances the EPSCs of LA neurons 4 hr after ischemia. C, The plotting showing the effects of glybenclamide (10 μm) on the evoked EPSCs from LA neurons. Glybenclamide had no obvious effects on EPSCs before and after ischemia.
The decrease in excitatory transmission during energy depletion has been attributed, at least in part, to ATP-dependent potassium channels (KATP) (Ashcroft, 1988; Calabresi et al., 1997a). To reveal the role of KATP in the changes in fast excitatory synaptic transmission after ischemia, the KATP channel blocker glybenclamide (10 μm) was applied to the medium, and no significant difference in EPSC amplitude was observed before and after ischemia (109.4 ± 0.8%, n = 9, controls; 101.6 ± 1.5%, n = 5, 4 hr after ischemia; 95.9 ± 13.3%,n = 4, 24 hr after ischemia) (Fig. 7C).
Ultrastructural changes in presynaptic terminals of LA neurons
LA neurons receive excitatory inputs from the neocortex and thalamus (Lapper and Bolam, 1992). Certain neurons in the cortex and thalamus are vulnerable to transient cerebral ischemia (Pulsinelli et al., 1982). To investigate whether decreased EPSC amplitude after ischemia is caused by the degeneration of presynaptic terminals, the morphology of presynaptic terminals was examined at different intervals after ischemia. Approximately 6 hr after ∼22 min of ischemia, most of the small- to middle-sized neurons in the dorsal striatum displayed shrunken and electron dense somata with many vacuoles in the cytoplasm. Many swollen processes of astrocytes were observed in the neuropil. The neurons with large somata, most likely LA neurons, remained intact at this time. Asymmetrical synapses were found on the cell bodies or dendrites of large neurons. No degenerating signs were observed in most of these terminals and their postsynaptic targets. A representative axosomatic synapse on a large neuron at 6 hr after ischemia is shown in Figure 8A. The postsynaptic membrane of this synapse had a prominent coating of dense material on its cytoplasmic face, and the presynaptic terminals contained a dense pack of small round vesicles, indicating that this is an asymmetrical synapse and is excitatory in nature. At 24 hr after ischemia, the small- to medium-sized neurons were further degenerated. The plasma membranes were ruptured, and the organelles were collapsed. The only intact structures were those of ischemia-resistant interneurons, including LA neurons. Despite the normal structure of dendrites and somata of these large neurons, some terminals making synaptic contacts on these neurons showed degenerating signs. Some of these terminals were electron dense, and the fine structures within the terminals were obscured. Sometimes the swollen vesicles were visible in these dark terminals (Fig. 8B). Another type of degenerating terminal showed severely swollen structures, but the presynaptic vesicles remained identifiable (Fig. 8C). To quantitatively compare the changes in presynaptic terminals at different intervals after ischemia, a total of 497 terminals at 6 hr after ischemia and 756 terminals at 24 hr after ischemia were analyzed. Within a unit area (42.3 μm2), the terminals with intact structures were 18.6 ± 0.5 per area, and the dark terminals were 7.5 ± 0.5 per area at 6 hr after ischemia (n = 19). No swollen terminals were observed at this time. Compared with those at 6 hr after ischemia, the number of intact terminals at 24 hr after ischemia was decreased to 5.1 ± 0.8 per area (n = 25; unpaired t test;p < 0.01), the dark terminals were increased to 19.5 ± 1.1 per area (p < 0.01), and the swollen terminals were increased to 5.6 ± 0.5 per area (p < 0.01). These changes represented a significant increase in degenerated terminals from 16% at 6 hr after ischemia to 84% (20% swollen, 64% dark) at 24 hr after ischemia (Fig. 8D). The above ultrastructural studies indicate that the neurons with large somata are resistant to transient cerebral ischemia, and most presynaptic terminals in the striatum remain intact at 4–6 hr after ischemia but show degenerating signs at 24 hr after reperfusion.
Fig. 8.
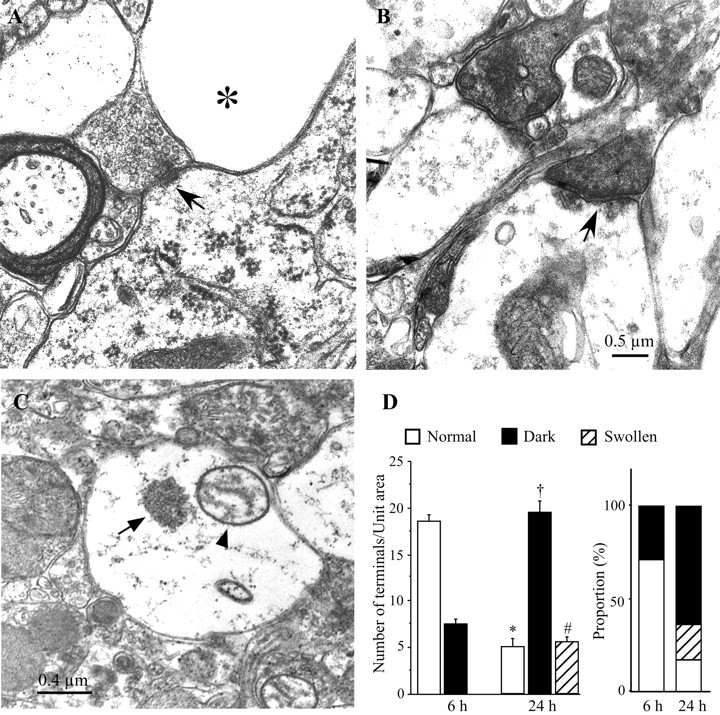
Electron micrographs showing the changes in presynaptic terminals after ischemia. A, An electron micrograph taken from the dorsal striatum at 6 hr after ischemia showing an axosomatic synapse (arrow). The postsynaptic membrane is thicker than the presynaptic one, indicating that this is an asymmetrical synapse. The presynaptic terminal contains many round vesicles. The postsynaptic target is the soma of a large neuron, most likely a LA neuron, with intact cytoplasmic organelles. Next to this synapse are swollen processes of spiny neurons or astrocytes (asterisk). B, Electron micrograph showing the degenerating synapses in the dorsal striatum at 24 hr after ischemia. Two degenerating synapses are filled with electron-dense materials. The dense postsynaptic membrane of one synapse (arrow) is still visible, indicating that this is an asymmetrical synapse. The synaptic vesicles of the other dark terminal are swollen. C, Electron micrograph showing an example of another type of degenerating terminal in the striatum 24 hr after ischemia. The terminal is enlarged dramatically with swollen mitochondria (arrowhead). A cluster of presynaptic vesicles (arrow) is located in the center of the terminal. D, Quantitative comparison of presynaptic terminals in the dorsal striatum at 6 and 24 hr after ischemia. Left, Histogram showing the number of terminals counted in a unit area (42.3 μm2/field). Compared with that at 6 hr after ischemia, the number of terminals with normal appearances in a given area significantly decreases, and the number of degenerated terminals significantly increases at 24 hr after ischemia. No swollen terminals are observed at 6 hr after ischemia. Right, Comparison of percentage change of terminals in the dorsal striatum at different intervals after ischemia. The degenerated terminal significantly increases from 16% at 6 hr after ischemia to 84% at 24 hr after ischemia. *,†,#p < 0.01 compared with its counterpart at 6 hr after ischemia.
DISCUSSION
The present study has shown that transient forebrain ischemia suppresses AMPAR-mediated EPSCs in LA neurons. Such suppression results from the alterations of presynaptic rather than postsynaptic components after ischemia. The decrease in release probability mediated by adenosine A1 receptors contributes, at least in part, to the reduction of EPSCs at 4–6 hr after ischemia, and the degeneration of presynaptic terminals is responsible for the reduction of EPSCs at late time points after reperfusion. The depression of excitatory neurotransmission might be one of the mechanisms protecting LA neurons against transient global ischemia.
Several lines of evidence, based on studies of hippocampal neurons, suggest that enhancement of excitatory synaptic transmission might cause neuronal injury, and that suppression of excitatory synaptic transmission might be neuroprotective after cerebral ischemia. Evoked EPSPs in ischemia-vulnerable CA1 neurons are potentiated after ischemia/hypoxia (Urban et al., 1989; Hori and Carpenter, 1994;Tsubokawa et al., 1994; Gao and Xu, 1996; Gao et al., 1998a). In contrast, the slope and amplitude of EPSPs in ischemic-resistant CA3 neurons and dentate granule cells are reduced 12–36 hr after transient forebrain ischemia (Gao et al., 1998b). Further supporting the notion that synaptic depression is associated with neuronal survival after ischemia, CA1 neurons also exhibit a depression of synaptic transmission after mild ischemia (∼5 min) that does not cause neuronal damage (Xu and Pulsinelli, 1994, 1996). In the neostriatum, small- to medium-sized neurons, most likely spiny neurons, die within 24 hr after transient forebrain ischemia, whereas interneurons, including LA neurons, survive (Francis and Pulsinelli, 1982; Pulsinelli et al., 1982; Chesselet et al., 1990). The incidence of polysynaptic EPSPs was significantly increased in spiny neurons after ∼22 min of ischemia (Gajendiran et al., 2001), indicating the facilitation of excitatory neurotransmission. When the ischemia duration was reduced to 5–8 min, which does not produce cell death in the striatum, the synaptic transmission of spiny neurons was transiently depressed (Xu, 1995). Recent studies have shown that facilitation of synaptic transmission in the form of LTP was induced from spiny neurons but not in LA neurons after deprivation of oxygen and glucose in vitro (Calabresi et al., 2002). Together with the above observations, the reduction of EPSCs in LA neurons after ischemia indicates that the depression of excitatory neurotransmission also participates in neuroprotection in the neostriatum after cerebral ischemia.
Several mechanisms might be involved in the depression of synaptic transmission after ischemia: (1) a decrease in postsynaptic responsiveness after ischemia, which might be caused by the decreased receptor number or reduced receptor sensitivity, and (2) a reduction of presynaptic function, which might result from a decrease in release probability or in the number of active synapses. A differential sensitivity of the presynaptic and postsynaptic component to ischemia/hypoxia has been noticed for sometime. Because presynaptic impulses persisted, whereas postsynaptic potentials were depressed in the hippocampus by hypoxia, it was believed that hypoxia-induced synaptic depression was primarily caused by postsynaptic changes (Kass and Lipton, 1982; Urban et al., 1989; Lee et al., 1991). However, studies using more sensitive techniques suggested that the hypoxia-induced depression was caused by presynaptic alterations. The IPSPs and EPSPs were depressed during hypoxia without obvious change in postsynaptic responses to focal application of GABA or quisqualic acid (Krnjevic et al., 1991; Rosen and Morris, 1993), and the mean amplitude of spontaneous miniature EPSCs was not affected by hypoxia at a time when the evoked synaptic responses were almost completely inhibited (Hershkowitz et al., 1993). In the present study, several lines of evidence indicate that postsynaptic mechanisms are not involved in the reduction of EPSCs after ischemia. First, the amplitude of AMPAR-mediated EPSCs in LA neurons was significantly reduced by ≤24 hr after ischemia, whereas the response to exogenous glutamate remained the same as that before ischemia. Second, the desensitization character of AMPARs does not change after ischemia, because application of CTZ, an agent that selectively blocks AMPAR desensitization (Bertolino et al., 1993), had similar potency on EPSCs of control as well as ischemic neurons. Third, the decay time constant of EPSCs after ischemia was the same as that of control neurons, further suggesting that the desensitization of AMPARs does not contribute to the reduction of EPSCs in LA neurons after ischemia. However, evidence strongly indicates that the presynaptic release probability has been transiently altered and is responsible for the reduction of EPSCs after ischemia. High-frequency stimulation depletes the release pool of synaptic terminals so that the amplitude of consecutive EPSCs progressively decreases (Wang and Kaczmarek, 1998; Bellingham and Walmsley, 1999; Scheuss et al., 2002). The alteration of fifth/first EPSC ratio in different experimental conditions suggests the change in release probability. The high (or low) release probability results in fast (or slow) depletion of the releasing pool and, therefore, the low (or high) fifth/first EPSC ratio. The fifth/first EPSC ratio of LA neurons at 4–6 hr after ischemia was significantly higher than those of controls and at 24 hr after reperfusion, suggesting that the release probability transiently decreases shortly after ischemia. Despite the fact that the paired-pulse experiment with an interstimulus interval of 50 msec failed to show a significant change in PPR after ischemia, the ratio of second/first EPSC in high-frequency train stimulation (interstimulus interval of 10 msec) at 4 hr after ischemia was significantly higher than that seen for controls and at 24 hr after ischemia (Fig.6A2). In addition, a parallel decrease in NMDAR- and AMPAR-mediated responses was observed after ischemia, indicating the decrease in transmitter release from presynaptic terminals 4 hr after ischemia. However, the present study could not exclude other presynaptic or postsynaptic factors that might also contribute to this short-term plasticity (Scheuss et al., 2002).
Adenosine is a modulator that has broad actions on neuronal activities (Song et al., 2000; Dunwiddie and Masino, 2001). It has inhibitory effects on the release of neurotransmitter, especially in glutamatergic systems (Dunwiddie and Hoffer, 1980; Kocsis et al., 1984). Extracellular adenosine significantly increases after ischemia/hypoxia (Winn et al., 1979; Latini et al., 1998), and activation of A1 receptors plays an important role in the depression of excitatory synaptic transmission after ischemia (Fowler, 1989; Katchman and Hershkowitz, 1993; Tanaka et al., 2001), which offers neuroprotection against the insult (Hsu et al., 1994; Mitchell et al., 1995). In the present study, the A1 receptor antagonist DPCPX had no effects on EPSCs in control conditions but strongly facilitated the EPSCs of LA neurons at 4–6 hr after ischemia, suggesting that by activation of A1 receptors, adenosine contributes, at least in part, to the reduction of EPSCs in LA neurons at early time points after ischemia. The inhibitory effects of adenosine on spiny neurons have also been reported in corticostriatal synapses and in GABAergic synapses during anoxia/aglycemia (Calabresi et al., 1997a,b; Centonze et al., 2001). However, different from the present study, these authors have shown that adenosine has endogenous tonic inhibition in control conditions, and paired-pulse facilitation has been observed in these studies. Besides the difference in cell type, one of the possible explanations for these discrepancies is the difference in afferent fiber pathway. LA neurons are predominantly innervated by fibers from the thalamus (Lapper and Bolam, 1992). Thus, the EPSCs in the present study are mediated primarily by thalamostriatal synapses, whereas the synaptic transmission in other studies is mediated by corticostriatal or GABAergic synapses.
Based on the above observations, transient changes in presynaptic components, most likely by activation of adenosine A1 receptors, may account for the reduction of EPSCs in LA neurons at 4–6 hr after ischemia. However, the amplitude of EPSCs remained reduced at 24 hr after ischemia, whereas the release probability returned to control levels, suggesting that other mechanisms must be involved in the synaptic depression at late time points after reperfusion. The excitatory inputs to LA neurons are from the cerebral cortex and thalamus (Lapper and Bolam, 1992). The neurons in the cortex and thalamus die within 24 hr after ∼15 min of forebrain ischemia (Pulsinelli et al., 1982). The forebrain ischemia induced in the present study lasts for ∼22 min, and the ischemia-vulnerable neurons in the cortex and thalamus certainly have died at 24 hr after reperfusion. EM examination has found that most of the presynaptic terminals in the striatum are degenerated at 24 hr after ischemia, suggesting that the reduction in excitatory inputs attributable to degeneration of presynaptic terminals is the major cause of EPSC reduction in LA neurons at 24 hr after ischemia. The ultrastructure of most presynaptic terminals remains intact at 4–6 hr after ischemia, suggesting that the reduction of EPSCs in LA neurons at early time points after ischemia is primarily caused by the transient malfunction of the presynaptic components rather than the structural damages.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS38053. Z.-P.P. and P.D. are recipients of American Heart Association postdoctoral fellowships. We thank Dr. Yuan Fan for helpful discussions.
Correspondence should be addressed to Dr. Zao C. Xu, Department of Anatomy and Cell Biology, Indiana University School of Medicine, 635 Barnhill Drive, MS 507, Indianapolis, IN 46202. E-mail:zxu@anatomy.iupui.edu.
REFERENCES
- 1.Ashcroft FM. Adenosine 5′-triphosphate-sensitive potassium channels. Annu Rev Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- 2.Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 3.Bertolino M, Baraldi M, Parenti C, Braghiroli D, DiBella M, Vicini S, Costa E. Modulation of AMPA/kainate receptors by analogues of diazoxide and cyclothiazide in thin slices of rat hippocampus. Receptors Channels. 1993;1:267–278. [PubMed] [Google Scholar]
- 4.Bolam JP, Ingham CA, Smith AD. The section-Golgi-impregnation procedure. III. Combination of Golgi-impregnation with enzyme histochemistry and electron microscopy to characterize acetylcholinesterase-containing neurons in the rat neostriatum. Neuroscience. 1984;12:687–709. doi: 10.1016/0306-4522(84)90164-7. [DOI] [PubMed] [Google Scholar]
- 5.Butcher LL, Hodge GK. Postnatal development of acetylcholinesterase in the caudate-putamen nucleus and substantia nigra of rats. Brain Res. 1976;106:223–240. doi: 10.1016/0006-8993(76)91022-2. [DOI] [PubMed] [Google Scholar]
- 6.Calabresi P, Centonze D, Pisani A, Bernardi G. A possible mechanism for the aglycemia-induced depression of glutamatergic excitation in the striatum. J Cereb Blood Flow Metab. 1997a;17:1121–1126. doi: 10.1097/00004647-199710000-00014. [DOI] [PubMed] [Google Scholar]
- 7.Calabresi P, Ascone CM, Centonze D, Pisani A, Sancesario G, D'Angelo V, Bernardi G. Opposite membrane potential changes induced by glucose deprivation in striatal spiny neurons and in large aspiny interneurons. J Neurosci. 1997b;17:1940–1949. doi: 10.1523/JNEUROSCI.17-06-01940.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. Muscarinic IPSPs in rat striatal cholinergic interneurones. J Physiol (Lond) 1998;510:421–427. doi: 10.1111/j.1469-7793.1998.421bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calabresi P, Centonze D, Bernardi G. Electrophysiology of dopamine in normal and denervated striatal neurons. Trends Neurosci. 2000;23:S57–S63. doi: 10.1016/s1471-1931(00)00017-3. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi P, Saulle E, Centonze D, Pisani A, Marfia GA, Bernardi G. Post-ischaemic long-term synaptic potentiation in the striatum: a putative mechanism for cell type-specific vulnerability. Brain. 2002;125:844–860. doi: 10.1093/brain/awf073. [DOI] [PubMed] [Google Scholar]
- 11.Centonze D, Saulle E, Pisani A, Bernardi G, Calabresi P. Adenosine-mediated inhibition of striatal GABAergic synaptic transmission during in vitro ischaemia. Brain. 2001;124:1855–1865. doi: 10.1093/brain/124.9.1855. [DOI] [PubMed] [Google Scholar]
- 12.Chesselet MF, Gonzales C, Lin CS, Polsky K, Jin BK. Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol. 1990;110:209–218. doi: 10.1016/0014-4886(90)90032-n. [DOI] [PubMed] [Google Scholar]
- 13.Chi XX, Xu ZC. Differential changes of potassium currents in CA1 pyramidal neurons after transient forebrain ischemia. J Neurophysiol. 2000;84:2834–2843. doi: 10.1152/jn.2000.84.6.2834. [DOI] [PubMed] [Google Scholar]
- 14.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 15.Crepel V, Hammond C, Chinestra P, Diabira D, Ben-Ari Y. A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2045–2055. doi: 10.1152/jn.1993.70.5.2045. [DOI] [PubMed] [Google Scholar]
- 16.DiFiglia M, Carey J. Large neurons in the primate neostriatum examined with the combined Golgi-electron microscopic method. J Comp Neurol. 1986;244:36–52. doi: 10.1002/cne.902440104. [DOI] [PubMed] [Google Scholar]
- 17.Dunwiddie TV, Hoffer BJ. Adenine nucleotides and synaptic transmission in the in vitro rat hippocampus. Br J Pharmacol. 1980;69:59–68. doi: 10.1111/j.1476-5381.1980.tb10883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 19.Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- 20.Francis A, Pulsinelli W. The response of GABAergic and cholinergic neurons to transient cerebral ischemia. Brain Res. 1982;243:271–278. doi: 10.1016/0006-8993(82)90250-5. [DOI] [PubMed] [Google Scholar]
- 21.Gajendiran M, Ling GY, Pang Z, Xu ZC. Differential changes of synaptic transmission in spiny neurons of rat neostriatum following transient forebrain ischemia. Neuroscience. 2001;105:139–152. doi: 10.1016/s0306-4522(01)00163-4. [DOI] [PubMed] [Google Scholar]
- 22.Galarraga E, Hernandez-Lopez S, Reyes A, Miranda I, Bermudez-Rattoni F, Vilchis C, Bargas J. Cholinergic modulation of neostriatal output: a functional antagonism between different types of muscarinic receptors. J Neurosci. 1999;19:3629–3638. doi: 10.1523/JNEUROSCI.19-09-03629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao TM, Xu ZC. In vivo intracellular demonstration of an ischemia-induced postsynaptic potential from CA1 pyramidal neurons in rat hippocampus. Neuroscience. 1996;75:665–669. doi: 10.1016/0306-4522(96)00411-3. [DOI] [PubMed] [Google Scholar]
- 24.Gao TM, Pulsinelli WA, Xu ZC. Prolonged enhancement and depression of synaptic transmission in CA1 pyramidal neurons induced by transient forebrain ischemia in vivo. Neuroscience. 1998a;87:371–383. doi: 10.1016/s0306-4522(98)00150-x. [DOI] [PubMed] [Google Scholar]
- 25.Gao TM, Howard EM, Xu ZC. Transient neurophysiological changes in CA3 neurons and dentate granule cells after severe forebrain ischemia in vivo. J Neurophysiol. 1998b;80:2860–2869. doi: 10.1152/jn.1998.80.6.2860. [DOI] [PubMed] [Google Scholar]
- 26.Goforth PB, Ellis EF, Satin LS. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershkowitz N, Katchman AN, Veregge S. Site of synaptic depression during hypoxia: a patch-clamp analysis. J Neurophysiol. 1993;69:432–441. doi: 10.1152/jn.1993.69.2.432. [DOI] [PubMed] [Google Scholar]
- 29.Hori N, Carpenter DO. Functional and morphological changes induced by transient in vivo ischemia. Exp Neurol. 1994;129:279–289. doi: 10.1006/exnr.1994.1170. [DOI] [PubMed] [Google Scholar]
- 30.Hsu SS, Newell DW, Tucker A, Malouf AT, Winn HR. Adenosinergic modulation of CA1 neuronal tolerance to glucose deprivation in organotypic hippocampal cultures. Neurosci Lett. 1994;178:189–192. doi: 10.1016/0304-3940(94)90756-0. [DOI] [PubMed] [Google Scholar]
- 31.Kass IS, Lipton P. Mechanisms involved in irreversible anoxic damage to the in vitro rat hippocampal slice. J Physiol (Lond) 1982;332:459–472. doi: 10.1113/jphysiol.1982.sp014424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katchman AN, Hershkowitz N. Adenosine antagonists prevent hypoxia-induced depression of excitatory but not inhibitory synaptic currents. Neurosci Lett. 1993;159:123–126. doi: 10.1016/0304-3940(93)90814-2. [DOI] [PubMed] [Google Scholar]
- 33.Kawaguchi Y. Large aspiny cells in the matrix of the rat neostriatum in vitro: physiological identification, relation to the compartments, and excitatory postsynaptic currents. J Neurophysiol. 1992;67:1669–1682. doi: 10.1152/jn.1992.67.6.1669. [DOI] [PubMed] [Google Scholar]
- 34.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 35.Kiskin NI, Krishtal OA, Tsyndrenko A. Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci Lett. 1986;63:225–230. doi: 10.1016/0304-3940(86)90360-5. [DOI] [PubMed] [Google Scholar]
- 36.Kiyosawa A, Katsurabayashi S, Akaike N, Pang ZP, Akaike N. Nicotine facilitates glycine release in the rat spinal dorsal horn. J Physiol (Lond) 2001;536:101–110. doi: 10.1111/j.1469-7793.2001.t01-1-00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kocsis JD, Eng DL, Bhisitkul RB. Adenosine selectively blocks parallel-fiber-mediated synaptic potentials in rat cerebellar cortex. Proc Natl Acad Sci USA. 1984;81:6531–6534. doi: 10.1073/pnas.81.20.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- 39.Krnjevic K, Xu YZ, Zhang L. Anoxic block of GABAergic IPSPs. Neurochem Res. 1991;16:279–284. doi: 10.1007/BF00966091. [DOI] [PubMed] [Google Scholar]
- 40.Lapper SR, Bolam JP. Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. 1992;51:533–545. doi: 10.1016/0306-4522(92)90293-b. [DOI] [PubMed] [Google Scholar]
- 41.Latini S, Bordoni F, Corradetti R, Pepeu G, Pedata F. Temporal correlation between adenosine outflow and synaptic potential inhibition in rat hippocampal slices during ischemia-like conditions. Brain Res. 1998;794:325–328. doi: 10.1016/s0006-8993(98)00304-7. [DOI] [PubMed] [Google Scholar]
- 42.Lee KS, Brooks P, Lowenkopf T. Transient ischemia attenuates neuronal afterdischarges induced in the absence of synaptic transmission. Brain Res. 1991;553:171–174. doi: 10.1016/0006-8993(91)90247-s. [DOI] [PubMed] [Google Scholar]
- 43.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 44.Malenka RC, Siegelbaum SA. Synaptic plasticity: diverse targets and mechanisms for regulating synaptic efficacy. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. The Johns Hopkins University; Baltimore: 2001. pp. 393–453. [Google Scholar]
- 45.Mitchell HL, Frisella WA, Brooker RW, Yoon KW. Attenuation of traumatic cell death by an adenosine A1 agonist in rat hippocampal cells. Neurosurgery. 1995;36:1003–1008. doi: 10.1227/00006123-199505000-00017. [DOI] [PubMed] [Google Scholar]
- 46.Pellegrini-Giampietro DE, Pulsinelli WA, Zukin RS. NMDA and non-NMDA receptor gene expression following global brain ischemia in rats: effect of NMDA and non-NMDA receptor antagonists. J Neurochem. 1994;62:1067–1073. doi: 10.1046/j.1471-4159.1994.62031067.x. [DOI] [PubMed] [Google Scholar]
- 47.Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol (Lond) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pisani A, Calabresi P, Centonze D, Marfia GA, Bernardi G. Electrophysiological recordings and calcium measurements in striatal large aspiny interneurons in response to combined O2/glucose deprivation. J Neurophysiol. 1999;81:2508–2516. doi: 10.1152/jn.1999.81.5.2508. [DOI] [PubMed] [Google Scholar]
- 49.Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- 50.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 51.Ren Y, Li X, Xu ZC. Asymmetrical protection of neostriatal neurons against transient forebrain ischemia by unilateral dopamine depletion. Exp Neurol. 1997;146:250–257. doi: 10.1006/exnr.1997.6525. [DOI] [PubMed] [Google Scholar]
- 52.Rosen AS, Morris ME. Anoxic depression of excitatory and inhibitory postsynaptic potentials in rat neocortical slices. J Neurophysiol. 1993;69:109–117. doi: 10.1152/jn.1993.69.1.109. [DOI] [PubMed] [Google Scholar]
- 53.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 54.Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song WJ, Tkatch T, Surmeier DJ. Adenosine receptor expression and modulation of Ca2+ channels in rat striatal cholinergic interneurons. J Neurophysiol. 2000;83:322–332. doi: 10.1152/jn.2000.83.1.322. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki T, Miura M, Nishimura K, Aosaki T. Dopamine-dependent synaptic plasticity in the striatal cholinergic interneurons. J Neurosci. 2001;21:6492–6501. doi: 10.1523/JNEUROSCI.21-17-06492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka E, Yasumoto S, Hattori G, Niiyama S, Matsuyama S, Higashi H. Mechanisms underlying the depression of evoked fast EPSCs following in vitro ischemia in rat hippocampal CA1 neurons. J Neurophysiol. 2001;86:1095–1103. doi: 10.1152/jn.2001.86.3.1095. [DOI] [PubMed] [Google Scholar]
- 58.Tang CM, Dichter M, Morad M. Quisqualate activates a rapidly inactivating high conductance ionic channel in hippocampal neurons. Science. 1989;243:1474–1477. doi: 10.1126/science.2467378. [DOI] [PubMed] [Google Scholar]
- 59.Trussell LO, Thio LL, Zorumski CF, Fischbach GD. Rapid desensitization of glutamate receptors in vertebrate central neurons. Proc Natl Acad Sci USA. 1988;85:2834–2838. doi: 10.1073/pnas.85.8.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsubokawa H, Oguro K, Masuzawa T, Kawai N. Ca2+-dependent non-NMDA receptor-mediated synaptic currents in ischemic CA1 hippocampal neurons. J Neurophysiol. 1994;71:1190–1196. doi: 10.1152/jn.1994.71.3.1190. [DOI] [PubMed] [Google Scholar]
- 61.Urban L, Neill KH, Crain BJ, Nadler JV, Somjen GG. Postischemic synaptic physiology in area CA1 of the gerbil hippocampus studied in vitro. J Neurosci. 1989;9:3966–3975. doi: 10.1523/JNEUROSCI.09-11-03966.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 63.Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winn HR, Rubio R, Berne RM. Brain adenosine production in the rat during 60 seconds of ischemia. Circ Res. 1979;45:486–492. doi: 10.1161/01.res.45.4.486. [DOI] [PubMed] [Google Scholar]
- 65.Xu ZC. Neurophysiological changes of spiny neurons in rat neostriatum after transient forebrain ischemia: an in vivo intracellular recording and staining study. Neuroscience. 1995;67:823–836. doi: 10.1016/0306-4522(95)00096-2. [DOI] [PubMed] [Google Scholar]
- 66.Xu ZC, Pulsinelli WA. Responses of CA1 pyramidal neurons in rat hippocampus to transient forebrain ischemia: an in vivo intracellular recording study. Neurosci Lett. 1994;171:187–191. doi: 10.1016/0304-3940(94)90636-x. [DOI] [PubMed] [Google Scholar]
- 67.Xu ZC, Pulsinelli WA. Electrophysiological changes of CA1 pyramidal neurons following transient forebrain ischemia: an in vivo intracellular recording and staining study. J Neurophysiol. 1996;76:1689–1697. doi: 10.1152/jn.1996.76.3.1689. [DOI] [PubMed] [Google Scholar]
- 68.Zhang C, Schmidt JT. Adenosine A1 and class II metabotropic glutamate receptors mediate shared presynaptic inhibition of retinotectal transmission. J Neurophysiol. 1999;82:2947–2955. doi: 10.1152/jn.1999.82.6.2947. [DOI] [PubMed] [Google Scholar]