

Abstract
Microglial activation and oxidative stress are significant components of the pathology of Parkinson's disease (PD), but their exact contributions to disease pathogenesis are unclear. We have developed an in vitro model of nigral injury, in which lipopolysaccharide-induced microglial activation leads to injury of a dopaminergic cell line (MES 23.5 cells) and dopaminergic neurons in primary mesencephalic cell cultures. The microglia are also activated by PD IgGs in the presence of low-dose dopa-quinone- or H2O2-modified dopaminergic cell membranes but not cholinergic cell membranes. The activation requires the microglial FcγR receptor as demonstrated by the lack of activation with PD IgG Fab fragments or microglia from FcγR−/− mice. Although microglial activation results in the release of several cytokines and reactive oxygen species, only nitric oxide and H2O2appear to mediate the microglia-induced dopaminergic cell injury. These studies suggest a significant role for microglia in dopaminergic cell injury and provide a mechanism whereby immune/inflammatory reactions in PD could target oxidative injury relatively specifically to dopaminergic cells.
Keywords: inflammatory, microglia, IgG, oxidative stress, DAergic neurons, Parkinson's disease
Parkinson's disease (PD) is a neurodegenerative disorder characterized by loss of dopaminergic neurons of the substantia nigra (SN) and the presence of Lewy body inclusions in residual neurons (Fearnley and Lees, 1994). The most significant pathological features of PD are the presence of oxidative stress (Dexter et al., 1994; Jenner and Olanow, 1998) and immune/inflammatory activity (McGeer et al., 1988a,b; Hirsch et al., 1998). Large numbers of reactive human leukocyte antigen-DR (HLA-DR)-positive microglia have been detected in the SN in PD, particularly in areas of maximal neurodegeneration, namely the ventral and lateral portion of the SN (McGeer et al., 1988b; Hirsch et al., 1998). Activated microglia are also associated with nigral injury in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism (Langston et al., 1999), and in Theiler, canine distemper, and Japanese encephalitis virus-infected animal models of nigral injury (Bencsik et al., 1997; Ogata et al., 1997; Oliver et al., 1997).
Evidence for a pathogenic role for such activated microglia and other immune/inflammatory constituents in dopaminergic cell injury in PD is primarily circumstantial and is based on the presence of elevated levels of cytokines. Interleukin-1β (IL-1β), interferon-γ (INF-γ), and tumor necrosis factor-α (TNF-α) are increased by 7- to 15-fold in the SN of PD patients (Mogi et al., 1996; Hirsch et al., 1998). TNF-α is also increased in PD CSF (Mogi et al., 1994; Le et al., 1999). In addition, in PD there is the induction of major histocompatibility complex class I (MHC-I) and MHC-II, complement-activated oligodendrocytes, increased expression of FcγRII/CD23 in glial cells, and deposition of specific antibodies in the brain (Loeffler et al., 1992; Yamada et al., 1992; Hunot et al., 1999).
Additional evidence for inflammatory/immune mechanisms in dopaminergic cell injury relevant to PD includes the experimental animal models of immune-mediated nigral damage produced in guinea pigs after inoculation with bovine mesencephalic tissues or dopaminergic cell line MES 23.5 (Appel et al., 1992; Le et al., 1995a). The sera from these immunized guinea pigs were cytotoxic for nigral dopaminergic cells after stereotaxic microinjection in rat SN in vivo (Le et al., 1996). Similar relatively specific cytotoxicity was demonstrated with PD IgGs (Chen et al., 1998) and bacterial lipopolysaccharide (LPS) (Castano et al., 1998). In all such models, reactive microglia are extremely prominent (Le et al., 1995a, 1996; Chen et al., 1998).
The key question is whether activated microglia can initiate or amplify injury to the nigral dopaminergic neurons, or is their role merely phagocytic. Furthermore, do immune/inflammatory processes participate in the oxidative stress known to be present in many of these models as well as in PD? To characterize the potential roles of PD IgG and microglia in dopaminergic nigral cell injury, we have developed anin vitro system in which PD IgG, in the presence of DA-quinone (DA-Q) and H2O2-modified dopaminergic cell membranes, can activate microglia and target a free radical-mediated injury to dopaminergic cells.
MATERIALS AND METHODS
Cultures of microglia, MES 23.5 cells, and primary mesencephalic cells. Microglia were isolated and purified from brains of 3- to 4-d-old Spraque Dawley rats (Harlan, Houston, TX). Briefly, after brains were dissected and the meninges removed, the tissues were minced and digested with trypsin (0.2%; Sigma, St. Louis, MO) and DNase I (0.01%; Sigma). After mechanical dissociation, the cells were resuspended in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal calf serum (FCS; Life Technologies) and seeded in 75 cm2 flasks at a density of 107 cells per flask. One week after the seeding, the flasks were shaken at 180 rpm for 15 hr, and floating cells were collected and allowed to adhere to a flask for 3 hr before being gently shaken. The cells attached on flasks were collected and plated to 24-well plates for further experimental treatment. To study the role of Fc receptors (FcRs) in microglia activation and MES 23.5 cell injury, mouse microglia were purified as above from the brains of 3- to 4-d-old homozygous FcγR knock-out (FcγR−/−) mice (Taconic Co., New York, NY). The FcγR−/− mice are deficient in the γ subunit of the FcRIII (a low-affinity receptor for IgG) and FcRI receptors (a high-affinity receptor for IgG). The FcγR−/− mice are maintained on a mixed stock (C57BL/6 × 129) resulting from the mating of the original chimera that encodes the targeted inactivation of the FcγR gene and the C57BL/6 strain (Takai et al., 1994). The genetic background of control wild-type (FcγR+/+) mice was the same as FcγR−/− mice (Taconic Co.).
The dopaminergic cell line MES 23.5 was generated in our own laboratory derived from somatic cell fusion of rat embryonic mesencephalic cells with murine N18TG2 neuroblastoma cells (Crawford et al., 1992). MES 23.5 cells display many properties of developing neurons of the SN zona compacta (Crawford et al., 1992) and offer several advantages for such initial studies, including greater homogeneity than primary cultures and susceptibility to both free-radical-mediated cytotoxicity and calcium-dependent cell death (Le et al., 1993, 1995b). MES 23.5 cells were seeded on polyornithine-precoated 24-well plates (Corning, Corning, NY) at a density of 104cells/cm2 and maintained in DMEM with Sato's components (Sigma) at 37°C in a 95% air/5% CO2 humidified atmosphere incubator. Some of the cultured MES 23.5 cells were cocultured with microglia.
Primary culture of neurons from embryonic rat mesencephalon was performed according to the method described previously (Crawford et al., 1992) with some modification. Briefly, the regions of the mesencephalon were dissected out from embryonic 14 d rat brain and then minced and treated with trypsin (0.02%) and DNase I (0.01%). After mechanical dissociation by pipetting, the cells were seeded at a density of 105 on 13 mm coverslips in 24-well plates previously coated with laminin (2 μg/ml) and grown in defined DMEM medium (Crawford et al., 1992). Some of the primary mesencephalic cell cultures were incubated with purified rat microglia 7 d after plating at a ratio of 8:1 (primary mesencephalic cells to microglia).
To study the interaction of reactive microglia with MES 23.5 cells, microglia and MES 23.5 cells were cocultured in 24-well culture plates. Briefly, the purified microglia were plated at a density of 104 1 d before addition of MES 23.5 cells at a ratio of 2:1 (MES 23.5 to microglia). The cocultures were maintained in Sato's conditioned medium containing 2% heat-inactivated fetal bovine serum. The cultures of microglia or MES 23.5 cells alone or together were treated for 2–3 d with LPS (0.1–4 μg/ml; Sigma) as a positive control, human IgG (20–400 μg/ml), or MES 23.5 cell membrane constituents (15–150 μg/ml).
Preparation of MES 23.5 cell membrane fraction. After exposure to DA-Q (50 μm) or H2O2 (10 μm) for 24 hr, the MES 23.5 cells were harvested in a buffer containing 0.25 m sucrose, 100 mm PBS, 1 mmMgCl2, 1 mm EGTA, and 2 μm protease inhibitorp-amidinophenyl methanesulfonyl fluoride hydrochoride, and homogenized with a Teflon homogenizer. Then the homogenate was centrifuged at 8000 × g for 10 min at 4°C to remove the crude nuclear fractions. The supernatants were again centrifuged at 100,000 × g for 60 min at 4°C. The precipitates were homogenized and suspended in culture medium and used as the neuronal membrane fractions.
Preparation of DA-Q. As described previously (Rowe et al., 1998), DA was dissolved in sterilized PBS and added to a final concentration of 1 mm with freshly made copper (II) sulfate (final concentration 0.1 mm). After 20 hr at 37°C, the incubations were dispensed in 250 μl volumes, and the reaction was stopped at −80°C. DA-Q (10–200 μm) was incubated in MES 23.5 cell cultures for 12–24 hr. The cells were harvested for the purification of cell membrane proteins.
Preparation of human IgG. Seven PD patients and eight disease controls (two amyotrophic lateral sclerosis, three Alzheimer's disease, two peripheral neuropathy, and one stroke) were enrolled in this study. The mean age of the PD patients was 67 ± 11 yr (mean ± SD) ranging from 45 to 79 yr, the duration of the disease was 4.2 ± 3.8 yr, and all patients had been medicated with levodopa/carbidopa. The mean age of the disease controls was 62 ± 14 yr, ranging from 41 to 76 yr. The IgG was purified from sera using ferric ammonium sulfate precipitation, ion exchange chromatography, and filtration dialysis, as described previously (Smith et al., 1992). The IgG was stored at −80°C until used. All of the patient's diagnoses were established by clinical history, examination, and laboratory investigation.
Microglial activation assay. Microglial activation was determined by measuring the levels of TNF-α and IL-1β in the culture media of microglia using a sandwich ELISA (R & D Systems, Minneapolis, MN). Briefly, 50 μl of media from microglial cultures was incubated in the 96-well TNF-α or IL-1β assay plates for 2 hr at room temperature. After any unbound substances were washed away, enzyme-linked polyclonal antibodies specific for rat TNF-α or IL-1β were added to the wells. After the addition of peroxidase substrate solution, the enzyme reactive color product was detected by an ELISA reader with the absorbency wavelength set at 450 nm. Each sample of medium was measured in duplicate, and two experiments were performed in a separate manner.
O2−, H2O2, and NO measurements. O2−, H2O2, and NO were measured in microglial incubations after phorbol 12-myristate 13-acetate (PMA) (1 μm; Sigma) stimulation for 2 hr at 37°C. Production of O2−is estimated by spectrophotometric measurement of superoxide dismutase (SOD)-inhibitable reduction of ferricychrone C as described previously (Mayer, 1998). The production of NO was determined by the measurement of nitrite (NO2) levels in the harvested media using the Greiss reaction (Mayer et al., 1998). H2O2 in the microglia medium was measured according to the description by Bianca et al. (1999).
Inducible nitric oxide synthase and nicotinamide adenine dinucleotide phosphate oxidase immunoblot. Rat microglia were incubated at 37°C under stirring with and without the agonists. At the indicated time, samples containing 1.5 × 107 cells were withdrawn and disrupted by sonication (6 sec at 60 W at 4°C). The homogenates were loaded to SDS-PAGE on 12% gel and incubated overnight with anti-inducible nitric oxide synthase (iNOS) rabbit antibody (1:500; Chemicon, Temecula, CA) or anti-nicotinamide adenine dinucleotide phosphate (NADPH) oxidase rabbit antibodies [p67phox, p47phox, andp40phox at 1:1000 dilution (a generous gift of Dr. F. Wientjes, University College, London, UK)]. All of the subsequent steps for ECL Western blotting detection were performed as described in detail elsewhere.
Scanning electron microcopy. Cultured cells were rinsed in 0.1 m PBS and fixed with 2% glutaraldehyde (Electron Microscopy Sciences, Ft. Washington, PA) in PBS containing 0.1 m sucrose, pH 7.4, at 4°C overnight. The cells were then post-fixed with 0.1% osmium tetroxide (Electron Microscopy Sciences) and dehydrated in a series of dilution of ethanol starting from 10 to 100%, then 50% ethanol/50% acetone, and finally in 100% acetone. Samples were critical point dried (Denton Vacuum, Norristown, NJ) and sputter coated using a vacuum Desk II cold sputter etch unit (Denton Vacuum). Photographs were taken with the secondary scanning attachment (ASID-4S) to the JEOL, JEM-100CX electron microscope (Peabody, MA) at magnifications from 500 to 300× using Polaroid type 55 film.
Cell injury assay. Conventional cytotoxicity assays measuring LDH release or MTT reduction were not possible because microglia and MES 23.5 have a similar morphological appearance and response. Instead, we determined tyrosine hydroxylase (TH) activity, which is present in MES 23.5 cells but not in microglia, to monitor the injury effects of activated microglia on MES 23.5 cells (Le et al., 1999). Briefly MES 23.5 cells were incubated with 25 μl aliquots of homogenate buffer containing [14C]-tyrosine (Dupont NEN, Boston, MA; specific activity 48.6 mCi/mmol) and cofactors at 37°C for 20 min. The [14C]-dopa formed was decarboxylated by adding 30 mm potassium ferricyanide and heating at 55°C for 30 min. The14CO2 released was absorbed on filter paper impregnated with hyamine hydroxide and quantified by counting the radioactivity on the paper covering each well.
The dopaminergic neuron injury in primary mesencephalic cell cultures was determined by quantitatively counting the TH-positive neurons. Briefly, the primary mesencephalic cell cultures with or without microglial addition were fixed in 4% paraformaldehyde for 20 min, then washed and treated with 1% H2O2. After 5% normal goat serum blocking for 2 hr, the polyclonal anti-TH antibody (1:1000 dilution; Protos Biotech, New York, NY) was incubated with the cells for 16 hr at 4°C, followed by secondary anti-rabbit biotinylated antibody with peroxidase labeling (Vectastain ABC kit; Vector Laboratories, Burlingame, CA). Some of the cultures were immunostained with parvalbumin antibody to detect GABAergic neurons. TH-positive cells in primary mesencephalic cell cultures with microglia were counted by an unrelated investigator. To quantitatively analyze the TH-positive or parvalbumin-positive neurons, we counted the cultures in a blind manner, and each experiment was performed in triplicate. Ten fields per well (113 mm2 surface area) were counted using a premarked frame lens. The size of field was 1 mm2, and the 10 fields consisted of ∼10% of the whole surface of the well. In control cultures, the percentage of TH-positive cells in the total cell population was ∼2.5%. Some of the cocultures of primary mesencephalic cells and microglia were double stained with antibodies to TH and OX-42 (1:200; Serotec, Oxford, UK) followed by second antibodies coupled with Alexa 546 and Alexa 488 (Molecular Probe, Eugene, OR) to label dopaminergic neurons and microglia, and visualized with fluorescent microscopy.
RESULTS
Microglial activation induced by LPS, high-dose PD IgG, and high-dose DA-Q-modified dopaminergic cell membranes
Microglia, isolated from the cerebral tissue of 3- to 4-d-old SD rats by selective adhesion to plastic, possessed highly homogeneous morphology and 1,1′-dioctadecyl-3,3,3′,3′,-tetramethyllindocarbocyanine perchlorate-acetylated low-density lipoprotein (DIL-ac-LDL) labeling. The highly purified microglia, grown alone or cocultured with intact MES 23.5 cells in 2% FBS DMEM for 2 d or longer, displayed either a ramified shape or bipolar or multipolar processes (Fig.1). Exposure to LPS or high-dose PD IgG, or coculture with MES 23.5 cells pretreated with 50 μmDA-Q, activated the microglia, resulting in enlarged flat cell bodies with vacuoles (Fig. 1). Under scanning electron microscopy examination, activated microglia frequently formed contacts with DA-Q-treated MES 23.5 cells; in control cocultures, cell contact was far less common (Fig. 1).
Fig. 1.

A, The morphology of rat microglial cells labeled with DIL-ac-LDL. Rat microglia were incubated for 2 d with vehicle (Aa–c), LPS (4 μg/ml) (Ad), high-dose PD IgG (200 μg/ml) (Ae), and high-dose DA-Q-M MES 23.5 cell membranes (150 μg/ml) (Af). Note that microglia after being treated with LPS, PD IgG, or DA-Q-M MES 23.5 cell membranes became larger and round. B, Scanning electron microscopy of microglia and MES 23.5 cells. Ba, Individual activated microglia showed spikes and cell-surface features. Bb,Bc, Activated microglia (left) in contact (arrow) with MES 23.5 cell (right).
We first characterized the LPS-induced microglial activation by measuring the levels of TNF-α and IL-1β, two well documented cytokines reflecting microglial activation, and the levels of several reactive oxygen species (ROS) released from activated microglia. We documented that after exposed to LPS (4 μg/ml), the levels of TNF-α and IL-1β were increased by 22- to 25-fold, and the levels of PMA-induced release of ROS (O2−, H2O2, and NO) were elevated up to 5- to 15-fold in the microglia culture media (Fig.2). To investigate the activation effects of PD IgG and DA-Q-treated MES 23.5 cells on microglia, we incubated microglia with high-dose PD IgG (200 μg/ml) from seven patients. The profile of PD IgG-induced microglial activation was similar to that seen with LPS. PD IgG at high dose (200 μg/ml) increased TNF-α and IL-1β by 17- to 21-fold (Fig. 2) and enhanced the PMA-induced release of H2O2 and NO by 14- to 17-fold and O2−by fivefold (Fig. 2). Because MES 23.5 cells activated microglia only after DA-Q treatment, we examined the membrane fractions and supernatants of MES 23.5 cells treated with DA-Q. Incubation with high-dose DA-Q-modified (DA-Q-M) P2 membrane fraction (150 μg/ml) in microglia significantly increased the levels of TNF-α and IL-1β by 3.6- to 4.2-fold (Fig. 2). However, this increase was far less than noted with LPS or high-dose PD IgG. The supernatant fraction from DA-Q-treated MES 23.5 cells had minimal activating effects. We also measured the levels of O2−, H2O2, and NO from the DA-Q-M membrane fraction-treated microglia cultures and found that they were increased 4- to 14-fold (Fig. 2).
Fig. 2.

Activating effects of LPS, PD IgG, and DA-Q-M membranes on microglia. Microglial activation was determined by measuring the levels of TNF-α, IL-1β, O2−, H2O2, and NO in the culture media. Microglia were incubated with LPS (4 μg/ml), high-dose PD IgG (200 μg/ml; n = 7), and high-dose DA-Q-M membranes (150 μg/ml) for 2 d, and medium was collected for measurement of TNF-α and IL-1β levels. Some of the microglial cultures were stimulated with 1 μg/ml PMA for 2 hr before assaying the released levels of O2−, H2O2, and NO.
Incubation with LPS, PD IgG, and DA-Q-M membranes also elevated microglial-immunoreactive iNOS and NADPH oxidase (Fig.3), with corresponding increases of NO and O2−(Fig. 2).
Fig. 3.

iNOS (A, B) and NADPH oxidase (C) induction in activated microglia. A, B, iNOS was detected (A, B) by immunoblot with iNOS antibodies. A, Microglia were incubated with (1) vehicle, (2) high-dose (150 μg/ml) DA-Q-M MES 23.5 cell membranes, (3) high-dose (200 μg) PD IgG, and (4) LPS (4 μg/ml) for 2 d.B, Microglia were incubated with (1) vehicle, (2) low-dose (15 μg/ml) DA-Q-M membranes, (3) low-dose (20 μg/ml) PD IgG, and (4) low-dose DA-Q-M membranes plus IgG. Note that increased iNOS in microglia after treatment with DA-Q-M membranes, IgG and LPS, and a synergetic effect of DA-Q-M membranes + IgG on microglia iNOS.C, NADPH oxidase was detected by three antibodies (p67phox, p47phox, and p40phox) in microglia treated with (1) vehicle, (2) high-dose PD IgG, (3) high-dose DA-Q-M membranes, (4) trypsin-treated DA-Q-M membranes, (5) low-dose PD IgG, (6) low-dose DA-Q-M membranes, and (7) low-dose DA-Q-M membranes + PD IgG. Arrows indicate three isoforms of NADPH oxidase reacting with antibodies ofp67phox, p47phox, andp40phox, respectively.
The specific microglial activation by low-dose PD IgG and DA-Q-M membranes
At high doses of IgG (200 μg/ml), microglia could be equivalently activated by either PD IgG or DC IgG (data not shown). Incubation with low-dose (20 μg/ml) PD IgG or disease control (DC) IgG had minimal microglial activating effects (Fig.4). Activation with low-dose (15 μg/ml) DA-Q-M membranes also had minimal microglial activating effects. However, incubation with low-dose PD IgG plus low-dose DA-Q-M membranes significantly increased TNF-α levels (Fig. 4) and elevated O2−, H2O2, and NO production (Fig. 4). Incubation of microglia with low-dose DC IgG in combination with DA-Q-M membranes had significantly fewer microglial activating effects (Fig. 4).
Fig. 4.

Microglial activation by IgG from PD and disease control (DC). The levels of TNF-α, O2−, H2O2, and NO in the microglial cultures treated with low-dose PD IgG (20 μg/ml), low-dose DC IgG (20 μg/ml), low-dose PD IgG + low-dose DA-Q-M MES 23.5 cell membranes (15 μg/ml) +, low-dose DC IgG + low dose DA-Q-M MES 23.5 cell membranes, and low-dose DA-Q-M MES 23.5 cell membranes alone. *p < 0.05 and **p < 0.01 compared with DC IgG +DA-Q-M.
The specificity of membrane modification by DA-Q versus H2O2 and the specificity of modified MES 23.5 cell membranes versus modified non-DAergic cell membranes in microglial activation
To determine whether microglial activation was specific for the method of cell membrane modification and the dopaminergic or cholinergic phenotype of the cell, we contrasted modification by DA-Q or H2O2 in membranes isolated from dopaminergic or cholinergic cell lines. Both the dopaminergic cell line (MES 23.5) and the cholinergic cell line (SN56) (Wainer et al., 1991) are derived from the same parental neuroblastoma line. Both MES 23.5 cells and SN56 cells were separately incubated with 10 μm H2O2 or 50 μm DA-Q, and the isolated membrane fractions were incubated with primary rat microglia in the presence and absence of low-dose PD IgG. Both high-dose (150 μg/ml) SN56 and MES 23.5 cell membranes had significant microglial activating effects, whether modified by DA-Q or by H2O2. At low doses (15 μg/ml), neither activated microglia. Low-dose DA-Q- or H2O2-modified MES 23.5 cell membranes incubated with low-dose PD IgG were able to activate microglia to a similar extent. However, low-dose DA-Q- or H2O2-modified SN56 cell membranes incubated with low-dose PD IgG had minimal activating potential (Fig. 5).
Fig. 5.

The specificity of microglial activation induced by DA-Q-M- or H2O2-M membranes from MES 23.5 cells as compared with SN 56 cells. The cells were treated with DA-Q (50 μm) or H2O2 (10 μm) for 24 hr, and the cell membranes alone or in combination with PD IgG were incubated with rat microglia for 2 d. TNF-α levels in the culture medium were determined by ELISA. **p < 0.01 versus SN 56 cell membrane addition.
Role of Fc receptors in microglial activation
FcRs are highly expressed on the microglial surface and are involved in microglial activation (Janeway and Travers, 1995; van Vugt et al., 1996). To determine whether the microglial FcγRs were involved in activation by LPS, high-dose PD IgG, high-dose DA-Q-M membranes, or low-dose PD IgG plus low-dose DA-Q-M membranes, we used FcγR-deficient microglia isolated from the brains of FcR γ chain knock-out mice (FcγR−/−). The microglia isolated from FcγR−/− mouse brain were morphologically indistinguishable from control mice. Furthermore, microglia from FcγR−/− mice were as readily activated by LPS (4 μg/ml) as microglia from FcγR+/+ mice as assayed by the release of TNF-α (Fig. 6). However, high-dose PD IgG (200 μg/ml) had fewer activating effects, whereas low-dose PD IgG (20 μg/ml) plus low-dose DA-Q-M membranes (15 μg/ml) had practically no activating potential when incubated with microglia isolated from FcγR−/− mouse brain (Fig. 6). As a further test of the importance of the microglial FcR in the PD IgG immune complex activation, we incubated microglia with Fab fragments (lacking Fc) from PD IgG (n = 3). Incubation of Fab fragments of IgG from all three PD patients failed to increase release of TNF-α from either FcγR−/− or FcγR+/+ microglia, suggesting the importance of the microglial FcR for PD IgG-induced activation (Fig.6).
Fig. 6.

Role of FcR in microglial activation. Mouse microglia were purified from the brains of 4- to 5-d-old mice with intact FcR γ chain (FcγR+/+) or with deleted FcR γ chain (FcγR−/−). The microglia were incubated with LPS (4 μg/ml), high-dose PD IgG (200 μg/ml), low-dose PD IgG (20 μg/ml; n = 3), high-dose DA-Q-M membranes (150 μg/ml), low dose DA-Q-M membranes (15 μg/ml), low-dose PD IgG + dose DA-Q-M membranes, and high-dose Fab fragment of PD IgG (200 μg/ml; n = 3) for 2 d. Microglial activation was determined by TNF-α release in the culture medium. *p < 0.005 and **p< 0.001 versus control FcγR+/+ microglia.
Activated microglia can induce MES 23.5 cell injury
MES 23.5 cell injury in cocultures was determined by measuring the activity of the rate-limiting enzyme in DA synthesis, TH, which is present in MES 23.5 cells but not in microglia. Because of the similar sizes of activated microglia and MES 23.5 cells, we could not readily quantify injury by monitoring changes in MES 23.5 cell morphology. Incubation of MES 23.5 cells directly with LPS, IgG from PD or DC, or DA-Q-M membranes did not alter the morphology, TH activity, or viability of the dopaminergic cells. In MES 23.5 cells cocultured with resting microglia, TH activity was not altered. When MES 23.5 cells were incubated with microglia activated with LPS (4 μg/ml), TH activity was significantly decreased (Fig.7A). Incubation with high-dose IgG (200 μg/ml) from PD (n = 7) or DC (n = 8) or high dose DA-Q-M MES 23.5 cell membranes (150 μm) also had remarkable effects on TH activity (Fig. 7A), and the effects of high-dose PD IgG alone did not differ significantly from the effects of high-dose DC IgG alone (Fig. 7A).
Fig. 7.

Reactive microglia induced MES 23.5 cell injury. MES 23.5 cell injury was determined by measuring TH activity in the cocultures with microglia. A, Cocultures were treated with vehicle, LPS (4 μg/ml), high-dose PD IgG (200 μg/ml), high-dose DC IgG (200 μg/ml), and high-dose DA-Q-M MES 23.5 cell membranes (150 μg/ml). B, Specificity of low-dose PD IgG (20 μg/ml) + DA-Q-M membranes (15 μg/ml) induced MES 23.5 cell injury. Column 1, Low-dose PD IgG (20 μg/ml;n = 7) + low-dose DA-Q-M MES 23.5 cell membranes (15 μg/ml). Column 2, Low-dose DC IgG (n = 8) + low-dose DA-Q-M MES 23.5 cell membranes (15 μg/ml). Dashed lines represent the mean value of TH activity in the cocultures treated with PD IgG (n = 7) or DC IgG + DA-Q-M MES 23.5 cell membranes.p < 0.05; PD IgG + DA-Q-M membranes versus DC IgG + DA-Q-M membranes. *p < 0.01 and **p < 0.005 versus control cocultures.
Neither low-dose PD IgG (20 μg/ml) nor low-dose DA-Q-M membranes (15 μg/ml) had any effects on microglia/MES 23.5 cocultures when used alone. However, combination of low-dose DA-Q-M membranes and PD IgG significantly decreased TH activity of microglia/MES 23.5 cocultures (Fig. 7B). Combinations of DC IgG and DA-Q-M membranes were far less effective (Fig. 7B). Statistical analysis demonstrated a significant difference between PD IgG + DA-modified MES 23.5 cell membranes and DC IgG + DA-Q-M MES 23.5 cell membranes at low doses (p < 0.05).
MES 23.5 cell injury is mediated by NO and H2O2
Because significant levels of TNF-α, IL-1β, and several ROS (O2−, H2O2, and NO) were detected in the activated microglia cultures, we next determined which constituents contributed to microglia-induced MES 23.5 cell injury. To microglia/MES 23.5 cell incubations we added neutralizing antibodies specific for rat TNF-α (monoclonal anti rat TNF-α at 50 μg/ml; PharMingen, San Diego) or for IL-1β (monoclonal anti-rat IL-1β at 10 μg/ml; R & D Systems) 1 hr before the addition of LPS-, PD IgG-, or DA-Q-modified MES 23.5 cell membranes. In some cases, the combination of these two neutralizing antibodies was used in microglia/MES 23.5 cell cocultures. Table1 shows that pretreatment with either neutralizing antibody did not reduce microglia-induced MES 23.5 cell injury. We then added selective ROS inhibitors: catalase (50–200 U; Sigma), reduced glutathione (GSH; 10–100 μm; Sigma), SOD (100–400 U; Sigma), iNOS inhibitorl-N6-1-iminoethyl-lysine hydrochloride (L-NIL; 10–100 μm; RBI, Natick, MA), or nNOS inhibitor 7-nitroindazole (10–100 μm; RBI). These inhibitors were incubated in the microglia/MES 23.5 cell cocultures 1 hr before the addition of DA-Q-M MES 23.5 cell membranes, PD IgG, or LPS. Pretreatment with catalase, GSH, and iNOS inhibitor L-NIL significantly attenuated MES 23.5 cell injury, suggesting that the reactive species NO and H2O2 were responsible for dopaminergic cell injury (Table 1).
Table 1.
Protective effects of catalase, SOD, GSH, NO inhibitors, FcγR−/−, TNF-α, and IL-1β neutralized antibodies on microglia-induced MES 23.5 cell injury
| Treatment | % Protection | |||
|---|---|---|---|---|
| LPS | PD IgG | DA-Q-M | DA-Q-M + PD IgG | |
| Catalase | 90 ± 15.11-165 | 85 ± 18.41-165 | 88 ± 12.21-165 | 81 ± 7.51-165 |
| GSH | 62 ± 9.11-160 | 51 ± 4.21-160 | 65 ± 111-160 | 59 ± 6.41-160 |
| SOD | 10 ± 3.0 | 7 ± 0.8 | 4 ± 1.2 | −2 ± 0.3 |
| L-NIL | 75 ± 6.41-160 | 69 ± 11.01-160 | 58 ± 9.2* | 70 ± 12.51-160 |
| 7-NIO | 8 ± 1.8 | 9 ± 2.2 | 0 ± 0.1 | 11 ± 1.8 |
| GSH + L-NIL | 91 ± 12.21-165 | 89 ± 5.51-165 | 93 ± 121-165 | 84 ± 7.91-165 |
| FcγR−/− | 10 ± 2.9 | 78 ± 18.21-160 | 24 ± 9 | 70 ± 8.21-160 |
| TNF-α antibody | 3 ± 0.6 | −5 ± 0.8 | −2 ± 0.4 | 3 ± 0.4 |
| IL-1β antibody | −4 ± 1.7 | 2 ± 0.5 | −8 ± 1.0 | 4 ± 0.5 |
| TNF-α + IL-1β antibodies | 5 ± 1.2 | 8 ± 1.4 | 6 ± 2.2 | 2 ± 0.5 |
Catalase 100 U/ml, GSH 50 μm, SOD 200 U/ml, iNOS inhibitor L-NIL 100 μm, nNOS inhibitor 7-NIO 100 μm, and neutralized antibodies to TNF-α (50 μg/ml) and to IL-1β (10 μg/ml) were incubated in the cocultures of MES 23.5 cells and microglia 20 min before LPS, PD IgG, and DA-Q-modified MES 23.5 cell membranes. Cell injury was determined by measuring TH activity in MES 23.5 cells cocultured with reactive microglia. Protection was estimated as percentage of inhibition of cell injury in cocultures pretreated with different protective agents versus cocultures treated with 4 μg/ml LPS, high-dose PD IgG (200 μg/ml), high-dose DA-Q-M membranes (150 μg/ml), and low-dose DA-Q-M membranes (15 μg/ml) + low-dose PD IgG (20 μg/ml). Values represent mean ± SEM of triplicate determinations made in three separate experiments.
p < 0.05,
F1-160: p < 0.01, and
F1-165: p < 0.005 versus cultures treated with LPS, PD IgG, DA-Q-M, or DA-Q-M + PD IgG alone.
Cytotoxic effects of activated microglia on primary mesencephalic neurons
To test whether reactive microglia can cause cell injury in primary cultures of mesencephalon, the cocultures of microglia and mesencephalic cells were incubated with LPS, PD IgG, and DA-Q-M membranes, and low-dose PD IgG plus DA-Q-M membranes. Two days after incubation, we found a significant loss (58–82%) of TH-positive neurons in the cocultured cells treated with 4 μg/ml LPS, 200 μg/ml PD IgG, 150 μg/ml DA-Q-M membranes, or 20 μg/ml PD IgG plus 15 μg/ml DA-Q-M membranes (Fig. 8). Incubation with these stimuli in primary mesencephalic cells in the absence of added microglia did not cause any significant TH-positive cell loss. Double staining with anti-TH and OX-42 in the cocultures revealed microglial activation as well as the injury and phagocytosis of primary dopaminergic neurons (Fig. 8). The neurites of TH-positive neurons were attenuated and shortened, and phagocytosis by activated microglia could be demonstrated (Fig. 8h). Incubation with low-dose PD IgG or DA-Q-M membranes had no injury effects on dopaminergic neurons. These effects of activated microglia on primary dopaminergic neurons were identical to the effects on the MES 23.5 cells, except that the primary dopaminergic neurons were more sensitive to microglia-mediated injury.
Fig. 8.
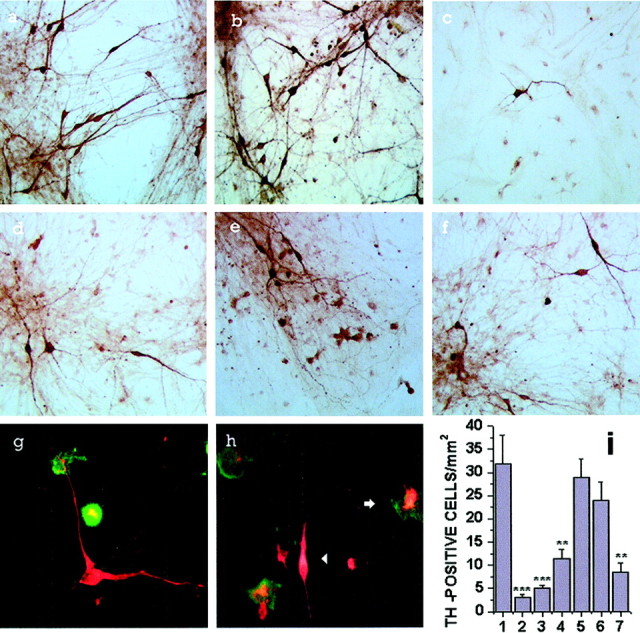
Reactive microglia-induced primary mesencephalic cell injury. Representative photographs of TH immunostaining in primary mesencephalic cell cultures (a) or cocultures with microglia (b–f) in addition of vehicle (b), LPS (4 μg/ml) (c), high-dose PD IgG (200 μg/ml) (d), high-dose DA-Q-M membranes (150 μg/ml) (e), and low-dose PD IgG (20 μg/ml) + low-dose DA-Q-M membranes (15 μg/ml) (f) for 2 d. Double staining of TH-positive neurons (red) and OX-42-positive microglia (green) in the vehicle-treated cocultures (g) and PD IgG-activated cocultures (h). Inh, note a reactive microglia surrounding and phagocytosing an injured dopaminergic neuron (arrow), and a TH-positive neuron with attenuated neurite and shortened cell body (arrowhead). I, Quantitative counting of TH-positive cells in cocultures. Control cocultures of primary mesencephalic cells with microglia treated with vehicle (column 1), 4 μg/ml LPS (column 2), high-dose PD IgG (200 μg/ml) (column 3), high-dose DA-Q-M membranes (150 μg/ml) (column 4), low-dose DA-Q-M membranes (15 μg/ml) (column 5), low-dose PD IgG (20 μg/ml) (column 6), and low-dose PD IgG + low-dose DA-Q-M membranes (column 7). *p < 0.05, **p < 0.01, and ***p < 0.001 versus control cocultures (column 1).
To determine whether other populations of neurons were affected by activated microglia, we examined GABAergic neurons (i.e., parvalbumin-positive neurons) in cocultures of microglia and primary mesencephalic cells. After a 2 d incubation with LPS (4 μg/ml) or PD IgG (200 μg/ml), the 39 and 35%, respectively, of parvalbumin-positive neurons were lost, which was significantly less than the 85–89% loss of TH-positive cells in the LPS- or PD IgG-treated cocultures.
DISCUSSION
Activated microglia are significant components of the pathology in the brain of PD and are often associated with injured pigmented SN cells and the presence of immune/inflammatory factors. However, the specific interactions of microglia with SN cells in PD have not been defined, nor has their potential role in DAergic cell injury been clarified. Microglia comprise up to 20% of the total glial cell population in the brain. The SN has an extremely high density of resting microglia (Lawson et al., 1990), which can be readily transformed to an activated state in response to a wide range of stimuli (Kreutzberg, 1996; Mayer, 1998). In PD brain, activated microglia are present in proximity to damaged nigral cells, suggesting their possible role in initiating or amplifying neuronal injury as well as in removing the debris of injured cells (McGeer et al., 1988b;Hirsch et al., 1998).
To investigate potential mechanisms of immune/inflammatory injury of SN relevant to PD, we first demonstrated that LPS can activate microglia in vitro to release TNF-α and IL-1β as well as O2−, H2O2, and NO. The secreted ROS caused injury of dopaminergic MES 23.5 cells and of primary cultures of mesencephalic dopaminergic cells. We then demonstrated that low levels of PD IgG combined with low levels of DA-Q-M dopaminergic cell membranes could also activate microglia relatively specifically through the FcγR. These activated microglia, in turn, caused injury of dopaminergic MES 23.5 cells as well as primary cultures of mesencephalic dopaminergic cells through the release of NO and H2O2 either in direct contact or in close proximity. The dopaminergic cell line, MES 23.5, has been fully characterized as possessing high levels of TH, DA, DA transporter, and SN neuronal antigens (Crawford et al., 1992; Le et al., 1995a; Zhang et al., 1999). Of significance is the fact that dopaminergic cells were even more sensitive than GABAergic cells to injury by activated microglia in primary cultures, which may reflect a different vulnerability of the mixed populations of neurons to these immune-mediated insults.
Several FcRs are expressed in microglia including FcγRI, the high-affinity receptor for IgG, FcγRIII, the low-affinity receptor for IgG, and FcγRII, a receptor for phagocytosis (Janeway and Travers, 1995). Two experiments support the involvement of microglial FcγR in the activation produced by low-dose PD IgG plus low dose DA-Q-M MES 23.5 cell membranes. (1) Microglia isolated from FcγR-deficient mice were not activated by low-dose PD IgG in the presence of low-dose DA-Q-M MES 23.5 cell membranes (although such microglia were fully activated by LPS, suggesting the integrity of other receptors in FcγR-deficient microglia), and (2) microglia were not activated by the Fab fragment of PD IgG alone. Activation of microglial FcγR requires not only Fab occupancy with its specific antigens but also the presence of the Fc component of IgG (Janeway and Travers, 1995). The demonstration that immune complexes can activate microglia has precedence in the study of canine distemper encephalitis, in which antibody can induce brain macrophages to generate ROS and demyelination (Griot et al., 1989).
In the presence of DA-Q-M membrane proteins, microglial activation and microglia-mediated injury to MES 23.5 cells were relatively specific for PD IgG compared with disease control IgG. A possible explanation for the relative specificity of PD IgG is that the Fab portion of IgG from PD may bind modified dopaminergic cell membrane constituents more effectively than disease control IgG. The resulting immune complexes would then interact more effectively with the microglial FcγR. In accord is our recent demonstration that ∼33% of PD sera versus 7% of disease controls had antibodies to DA-Q-M-soluble ovalbumin (Rowe et al., 1998). DA-Q or H2O2 could modify dopaminergic MES 23.5 cellular constituents to generate neoantigens, possibly recognized by antibodies from the sera of PD. DA-Q has been reported to oxidize as well as cross-link proteins, glycoproteins, and lipoproteins of dopaminergic cells (Montine et al., 1995). H2O2 can also modify cellular constituents. Aldehydes such as 4-hydroxynonenal are increased in SN neurons in PD and could form protein adducts by covalent binding to cysteine, lysine, and histidine residues (Yoritaka et al., 1996). In turn, altered proteins have been documented to be potent microglial activators (Newcombe et al., 1994; Keller et al., 1999) and could release toxic compounds, injuring neurons and altering DA homeostasis (McMillian et al., 1997). H2O2 was just as effective in altering MES 23.5 cells and inducing microglial activation. In ourin vitro studies, the specific membrane constituents responsible for microglial activation are probably proteins because tryptic digestion removed the ability to activate microglia (W. Le, unpublished results).
Activation of microglia resulted in the release of increased levels of TNF-α, IL-1β, and several ROS (O2−, H2O2, and NO). At high concentrations, these cytokines and ROS are generally cytotoxic, but the susceptibility of different neurons is quite variable (Chao et al., 1992; Merrill and Benveniste, 1996; Mayer, 1998; Neumann and Wekerle, 1998). TNF-α and IL-1β are among the most well studied cytokines released from microglia, but their significance in mediating cell injury in PD is unclear (Neumann and Wekerle, 1998). In the presence of TNF-α and IL-1β neutralizing antibodies, MES 23.5 cell injury was not reduced, suggesting that neither TNF-α nor IL-1β was directly responsible for the in vitro effects. We cannot preclude involvement of these cytokines in vivo because it is possible that MES 23.5 cells do not express the receptors required to induce neuronal injury (Merrill and Benveniste, 1996; Neumann and Wekerle, 1998).
The profile of increasing O2−, H2O2, and NO from microglia was similar regardless of the activating stimuli. Activated microglia released three- to fourfold more H2O2 and NO than O2−. However, one must be cautious in interpreting superoxide measurements because of limitations in most methodologies, including those used in the present studies (Mayer, 1998). The protective effects of catalase, GSH, and the iNOS inhibitor L-NIL in our experiments suggested that hydroxyl radicals and NO may contribute to microglia-induced MES 23.5 cell injury. The protective effects of catalase implicated H2O2 in dopaminergic cell injury.
The ability of PD IgG to activate microglia and induce dopaminergic cell injury in vitro provides a potential explanation for the SN cell injury noted after stereotaxic injections of PD IgG in vivo (Chen et al., 1998). The presence of activated microglia 4 weeks after injection in vivo in that study suggested a role for such microglia in initiating or, at the very least, amplifying neuronal injury. The present demonstration of microglial activation in vitro after incubation with immune complexes of PD IgG with modified dopaminergic cell membrane proteins suggests a potential mechanism of dopaminergic cell injury by cytotoxic ROS released from the activated microglia. In the in vivomodel the needle tract may injure SN constituents and the injected PD IgG may amplify neuronal damage by targeting microglial-mediated injury to dopaminergic neurons. These models provide potential mechanisms whereby the presence of modified dopaminergic cell membrane constituents in PD in combination with PD IgG could activate microglia and in turn amplify dopaminergic cell injury.
In PD, nigral cell degeneration is associated with or even preceded by oxidative stress that is possibly initiated by environmental or endogenous toxic reactions (Beal, 1998; Olanow et al., 1998). The neurotransmitter DA itself or its metabolites can generate ROS by chemical or enzymatic means and can damage dopaminergic neuronsin vitro and possibly in vivo (Jenner and Olanow, 1998). Elevated levels of iron, decreased complex I activity, decreased levels of GSH, and increased lipid peroxidation and DNA damage have been demonstrated in the SN of patients with PD (Schapira et al., 1990;Dexter et al., 1994; Yoritaka et al., 1996; Zhang et al., 1999). What initiates or propagates the oxidative stress is presently unknown. MPTP can induce parkinsonism in human and in animal models, possibly by upregulating inducible nitric oxide synthase (Liberatore et al., 1999) and impairing neuronal mitochondrial complex I activity. Activated microglia are an important source of the inducible nitric oxide synthase in the MPTP model and were significantly upregulated in ourin vitro model. In fact, the release of H2O2 and NO from activated microglia were the major reactive species mediating dopaminergic cell injury. Thus our experiments support the hypothesis that the immune/inflammatory pathology and oxidative stress may be tightly linked in PD, with activated microglia playing an important role in propagating and amplifying oxidative neuronal injury and possibly even initiating such injury (McNaught and Jenner, 1999). The presence of activated microglia inducing cytotoxicity would raise a warning regarding the long-term viability of transplanted embryonic neurons in human PD (Brundin et al., 2000). Suppression of the inflammatory response, particularly the microglial activation, could improve the survival of transplanted neurons in patients with PD, reduce the need for human embryonic donor tissue, and increase the likelihood of a successful outcome.
Footnotes
This study was supported by Research Grant NS40370-01 from the National Institute of Neurological Disorders and Stroke and grants from the Claude and Marie Hammill Foundation and the Parkinson Disease Foundation.
W.L. and D.R. contributed equally to this work.
Correspondence should be addressed to Dr. Stanley H. Appel, Professor and Chairman, Department of Neurology, Baylor College of Medicine, 6501 Fannin Street, Houston, TX 77025. E-mail:Sappel@bcm.tmc.edu.
REFERENCES
- 1.Appel SH, Le WD, Tajti J, Haverkamp LJ, Engelhardt JI. Nigral damage and dopaminergic hypofunction in mesencephalon-immunized guinea pigs. Ann Neurol. 1992;32:494–501. doi: 10.1002/ana.410320403. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Excitotoxicity and nitric oxide in Parkinson's disease pathogenesis. Ann Neurol. 1998;44[Suppl 1]:S110–S114. doi: 10.1002/ana.410440716. [DOI] [PubMed] [Google Scholar]
- 3.Bencsik A, Akaoka H, Giraudon P, Belin MF, Bernard A. Inhibition of tyrosine hydroxylase expression within the substantia nigra of mice infected with canine distemper virus. J Neuropathol Exp Neurol. 1997;56:673–685. [PubMed] [Google Scholar]
- 4.Bianca VD, Dusi S, Bianchin E, Pra ID, Rossi F. β-Amyloid activates the O2- forming NADPH oxidase in microglia, monocytes, and neurotrophis. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 5.Brundin P, Karlsson J, Emgard M, Schierle GS, Hansson O, Petersen A, Castilho RH. Improving the survival of grafted dopaminergic neurons: a review over current approaches. Cell Transplant. 2000;9:179–195. doi: 10.1177/096368970000900205. [DOI] [PubMed] [Google Scholar]
- 6.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 7.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 8.Chen S, Le WD, Xie WJ, Alexianu ME, Engelhardt JI, Siklos L, Appel SH. Experimental destruction of substantia nigra initiated by Parkinson disease immunoglobulins. Arch Neurol. 1998;55:1075–1080. doi: 10.1001/archneur.55.8.1075. [DOI] [PubMed] [Google Scholar]
- 9.Crawford GD, Le WD, Smith RG, Xie WJ, Appel SH. A novel N18TG2 mesencephalon cell hybrid expresses properties which suggest a dopaminergic cell line of substantia nigra origin. J Neurosci. 1992;12:3392–3396. doi: 10.1523/JNEUROSCI.12-09-03392.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dexter DT, Holley AE, Flitter WD. Increase levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 11.Fearnley J, Lees A. Pathology of Parkinson's disease. In: Calne DB, editor. Neurodegenerative diseases. W. B. Saunders; Philadelphia: 1994. pp. 545–554. [Google Scholar]
- 12.Griot C, Burge T, Vandevelde M, Peterhans E. Antibody-induced generation of reactive oxygen radicals by brain macrophages in canine distemper encephalitis: a mechanism for bystander demyelination. Acta Neuropathol. 1989;78:396–403. doi: 10.1007/BF00688176. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch EC, Hunot S, Damier P, Brugg B, Faucheux BA, Michel PP, Ruberg M, Muriel MP, Mouatt-Prigent A, Agid Y. Glia cells and inflammation in Parkinson's disease: a role in neurodegeneration. Ann Neurol. 1998;44[Suppl 1]:S115–S120. [Google Scholar]
- 14.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FceRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janeway CA, Travers P. Fc receptor-bearing accessory cells in humoral immunity. In: Janeway CA, Travers P, editors. Immunobiology, Ed 2. Current Biology Ltd; Philadelphia: 1995. pp. 8:23–8:31. [Google Scholar]
- 16.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44[Suppl 1]:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 17.Keller JN, Hanni KB, Gabbita SP, Friebe V, Mattson MP, Kindy MS. Oxidized lipoproteins increase reactive oxygen species formation in microglia and astrocyte cell lines. Brain Res. 1999;830:10–15. doi: 10.1016/s0006-8993(99)01272-x. [DOI] [PubMed] [Google Scholar]
- 18.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 19.Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karlak D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 20.Lawson LJ, Perry VH, Dri P, Gorden S. Heterogeneity in the distribution and morphology of microglia in the normal mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 21.Le WD, Xie WJ, Smith RG, Appel SH. Protective effects of IGF-1, b-FGF, EGF, NGF, and cAMP on oxidation and hypoglycemia-induced damage in hybrid dopaminergic cells. Neurodegeneration. 1993;2:227–236. [Google Scholar]
- 22.Le WD, Engelhardt J, Xie WJ, Schneider L, Smith RG, Appel SH. Experimental autoimmune nigral damage in guinea pigs. J Neuroimmunol. 1995a;57:45–53. doi: 10.1016/0165-5728(94)00160-p. [DOI] [PubMed] [Google Scholar]
- 23.Le WD, Colom L, Xie WJ, Smith RG, Alexianu M, Appel SH. Cell death induced by β-amyloid 1–40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995b;686:49–60. doi: 10.1016/0006-8993(95)00450-5. [DOI] [PubMed] [Google Scholar]
- 24.Le WD, Engehardt JI, Xie WJ, Appel SH. Immune invasion with serum against DA neurons induces a Parkinsonian model in rats. Mov Disord. 1996;8:409. [Google Scholar]
- 25.Le WD, Rowe DB, Jankovic J, Xie WJ, Appel SH. Effects of cerebrospinal fluid from patients with Parkinson disease on dopaminergic cells. Arch Neurol. 1999;56:194–200. doi: 10.1001/archneur.56.2.194. [DOI] [PubMed] [Google Scholar]
- 26.Liberatore GT, Jackson-Lewis W, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 27.Loeffler DA, Brickman CM, Kapatos G, Smithson IL, Peter JB, LeWitt PA. Antineuronal antibodies and markers of immune system activation in Parkinson's disease. Neurodegeneration. 1992;1:145–153. [Google Scholar]
- 28.Mayer AM. Therapeutic implications of microglia activation by lipopolysaccharide and reactive oxygen species generation in septic shock and central nervous system pathologies: a review. Medicina (B Aires) 1998;58:377–385. [PubMed] [Google Scholar]
- 29.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988a;24:574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 30.McGeer PL, Itagaki S, Boyes BE, McGreer EG. Reactive microglia are positive for HLA-DA in the substantia nigra of Parkinson's and Alzheimer's disease brain. Neurol. 1988b;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 31.McMillian MK, Vainio PJ, Tuominen RK. Role of protein kinase C in microglia-induced neurotoxicity in mesencephalic cultures. J Neuropathol Exp Neurol. 1997;56:301–307. doi: 10.1097/00005072-199703000-00009. [DOI] [PubMed] [Google Scholar]
- 32.McNaught KS, Jenner P. Altered glia function causes neuronal death and increases neuronal susceptibility to 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced toxicity in astrocytic/ventral mesencephalic co-cultures. J Neurochem. 1999;73:2496–2476. doi: 10.1046/j.1471-4159.1999.0732469.x. [DOI] [PubMed] [Google Scholar]
- 33.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 34.Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 35.Mogi M, Harada M, Narabayashi H, Inogaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease. Neurosci Lett. 1996;211:13–16. doi: 10.1016/0304-3940(96)12706-3. [DOI] [PubMed] [Google Scholar]
- 36.Montine TJ, Farris DB, Graham DG. Covalent cross-linking of neurofilament proteins by oxidized catechols as a potential mechanism of Lewy body formation. J Neuropathol Exp Neurol. 1995;54:311–319. doi: 10.1097/00005072-199505000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Neumann H, Wekerle H. Neuronal control of immune response in the central nervous system: linking brain immunity to neurodegeneration. J Neuropathol Exp Neurol. 1998;57:1–9. doi: 10.1097/00005072-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Newcombe J, Li H, Cuzner ML. Low density lipoprotein uptake by macrophages in multiple sclerosis plaques: implications for pathogenesis. Neuropathol Appl Neurobiol. 1994;20:152–162. doi: 10.1111/j.1365-2990.1994.tb01174.x. [DOI] [PubMed] [Google Scholar]
- 39.Ogata A, Tashiro K, Nukuzuma S, Nagashima K, Hall WW. A rat model of Parkinson's disease induced by Japanese encephalitis virus. J Neurovirol. 1997;3:141–147. doi: 10.3109/13550289709015803. [DOI] [PubMed] [Google Scholar]
- 40.Olanow CW, Jenner P, Tatton NA, Tatton WG. Neurodegeneration and Parkinson's disease. In: Jankovic J, Tolosa E, editors. Parkinson's disease and movement disorders, Ed 3. Williams & Wilkins; Baltimore: 1998. pp. 67–104. [Google Scholar]
- 41.Oliver KR, Brennan P, Fazakerley JK. Specific infection and destruction of dopaminergic neurons in the substantia nigra by Theiler's virus. J Virol. 1997;71:6179–6182. doi: 10.1128/jvi.71.8.6179-6182.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowe DB, Le WD, Smith RG, Appel SH. Antibodies from patients with Parkinson's disease react with protein modified by dopamine oxidation. J Neurosci Res. 1998;53:551–557. doi: 10.1002/(SICI)1097-4547(19980901)53:5<551::AID-JNR5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 43.Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:499–510. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 44.Smith RG, Hamilton S, Hofmann F, Schneider T, Nastainczyk W, Birnbaumer L, Stefani E, Appel SH. Serum antibodies to skeletal muscle-derived L-type calcium channels in patients with amyotrophic lateral sclerosis. N Engl J Med. 1992;327:1721–1728. doi: 10.1056/NEJM199212103272405. [DOI] [PubMed] [Google Scholar]
- 45.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcRγ chain deletion results in pleiotropic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 46.van Vugt MJ, Heijnen AF, Capel PJ, Parki SY, Ra C, Saito T, Verbeek JS, vande Winketl JG, Smith LC, Gotto AM, Jr, Dresel HA. FcR γ-chain is essential for both surface expression and function of human FcγRI (CD64) in vivo. Blood. 1996;87:3593–3599. [PubMed] [Google Scholar]
- 47.Wainer BH, Lee HT, Roback JD, Hammond DN. In vitro cell cultures as a model of the basal forebrain. In: Napier TC, Kalivas PW, Hanin I, editors. The basal forebrain. Plenum; New York: 1991. pp. 415–434. [DOI] [PubMed] [Google Scholar]
- 48.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 49.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]