

Abstract
Epilepsy is a common outcome of traumatic brain injury (TBI), but the mechanisms of posttraumatic epileptogenesis are poorly understood. One clue is the occurrence of selective hippocampal cell death after fluid-percussion TBI in rats, consistent with the reported reduction of hippocampal volume bilaterally in humans after TBI and resembling hippocampal sclerosis, a hallmark of temporal-lobe epilepsy. Other features of temporal-lobe epilepsy, such as long-term seizure susceptibility, persistent hyperexcitability in the dentate gyrus (DG), and mossy fiber synaptic reorganization, however, have not been examined after TBI. To determine whether TBI induces these changes, we used a well studied model of TBI by weight drop on somatosensory cortex in adult rats. First, we confirmed an early and selective cell loss in the hilus of the DG and area CA3 of hippocampus, ipsilateral to the impact. Second, we found persistently enhanced susceptibility to pentylenetetrazole-induced convulsions 15 weeks after TBI. Third, by applying GABAA antagonists during field-potential and optical recordings in hippocampal slices 3 and 15 weeks after TBI, we unmasked a persistent, abnormal APV-sensitive hyperexcitability that was bilateral and localized to the granule cell and molecular layers of the DG. Finally, using Timm histochemistry, we detected progressive sprouting of mossy fibers into the inner molecular layers of the DG bilaterally 2–27 weeks after TBI. These findings are consistent with the development of posttraumatic epilepsy in an animal model of impact head injury, showing a striking similarity to the enduring behavioral, functional, and structural alterations associated with temporal-lobe epilepsy.
Keywords: traumatic brain injury, hippocampal sclerosis, mossy fiber synaptic reorganization, sprouting, seizures, neuron death, epilepsy, optical recording, voltage-sensitive dye di-2-ANEPEQ
Traumatic brain injury (TBI) is a major risk factor for epilepsy (Feeney and Walker, 1979; Salazar et al., 1985; Annegers et al., 1996, 1998). Human studies showed a strong correlation between posttraumatic epilepsy (PTE) and loss of brain volume after head injury (Salazar et al., 1985). Others have reported reduced hippocampal volume bilaterally in humans after TBI (Bigler et al., 1997; Tate and Bigler, 2000). Likewise, in rats, extensive cortical and subcortical neuronal loss was reported after cortical weight-drop injury (Weisend and Feeney, 1994) or fluid percussion (Cortez et al., 1989; Lowenstein et al., 1992), where damaged neurons were found in hippocampal CA3 and hilus as early as minutes after cortical impact (Hicks et al., 1996; Toth et al., 1997). This hilar cell loss resembled “hippocampal sclerosis,” a hallmark of temporal-lobe epilepsy, suggesting hippocampal involvement in PTE (Lowenstein et al., 1992). This idea, based on an impact model of TBI, differs from findings of regionally restricted seizure susceptibility around focal cortical injury by cold, undercut, needle insertion, heat, or excitotoxins (Grafstein, 1957; Prince and Tseng, 1993; Jacobs et al., 1996; Eysel, 1997; Hagemann et al., 2000).
The connection between PTE and impact-induced hippocampal sclerosis has remained elusive for two reasons. First, a persistent susceptibility to seizures has not been reported after experimental TBI. Others have reported acute posttraumatic seizures during the first hours (Krobert et al., 1992; Nilsson et al., 1994) and enhanced seizure susceptibility during the first week after TBI (Coulter et al., 1996; Reeves et al., 1997). However, PTE, defined as a persistent seizure susceptibility beyond 2 weeks after TBI (Feeney and Walker, 1979; Salazar et al., 1985), has not been demonstrated experimentally. Second, temporal-lobe epilepsy and hippocampal sclerosis are associated with forms of neuronal plasticity that are implicated in epileptogenesis but not previously examined after TBI. This plasticity includes an early and persistent, NMDA receptor-dependent hyperexcitability among granule cells (Mody et al., 1988; Lynch et al., 2000) and abnormal sprouting of granule cell axons (mossy fibers) synapsing in the molecular layer (Frotscher and Zimmer, 1983; Sutula et al., 1988). This mossy fiber synaptic reorganization (MFSR) is found in human temporal-lobe epilepsy (Sutula et al., 1989; Babb, 1997) and is well characterized in experimental models (Cotman, 1979; Sutula et al., 1988, 1998;Stanfield, 1989; Golarai et al., 1992; Okazaki et al., 1995; Ribak et al., 1998; Mitchell et al., 1999). There is evidence that NMDA receptor-mediated hyperexcitability among granule cells promotes MFSR, which in turn may enhance recurrent excitation, leading to persistent hyperexcitability in the dentate gyrus (DG) (Sutula et al., 1996). Such a positive feedback loop between functional and structural changes in the DG could facilitate epileptogenesis (Tauck and Nadler, 1985; Cronin et al., 1992; Golarai and Sutula, 1996).
In the present study, we addressed three hitherto unanswered questions pertaining to epileptogenesis after TBI. First, is impact-induced TBI associated with an enduring susceptibility to behavioral seizures? Second, is the circuitry of the DG persistently susceptible to seizure activity after TBI? Third, is the TBI-induced functional plasticity in the DG associated with persistent structural changes such as MFSR?
MATERIALS AND METHODS
Surgical methods: weight-drop contusion. Subjects were male Sprague Dawley rats (250–400 gm) maintained with equal daily periods of light and dark and free access to food and water. All protocols adhered to the guidelines of the National Institutes of Health. TBI was induced in anesthetized (2% halothane in O2, 2 l/min) rats as described previously (Feeney et al., 1981). Briefly, rats underwent a craniotomy, in which a circular region of skull (3.0 mm diameter, centered 2.3 mm caudal and 2.3 mm lateral to bregma) was removed over the right somatosensory cortex. A weight-drop device was placed stereotaxically over the dura and adjusted to stop an impact transducer (foot plate) at a depth of 2.5 mm below the dura. Then, a 20 gm weight was dropped from 20 cm above the dura, through a guide tube onto the foot plate. These methods result in epileptiform activity in region CA3 and mild to moderate acute convulsions during the first hours after TBI (i.e., class II–V) (Krobert et al., 1992), reproducible contusions, selective subcortical neuronal death, and specific behavioral deficits (Feeney et al., 1981,1982; Feeney, 1997). Similar epileptiform activity was recorded in the hours after compression TBI (Nilsson et al., 1994). In the present study, in addition to similar acute post-TBI convulsions, late convulsions were incidentally observed during routine handling of the animals over weeks.
Controls were age matched and included three groups. First, nine controls received no treatment at all (normal controls). Second, 11 control rats underwent craniotomy without weight drop (craniotomy controls). Third, six un-operated and three craniotomy rats each received a single subconvulsant dose of pentylenetetrazole, as described below (PTZ controls).
Histological methods for assessing cell damage and death. To assess cell damage and loss after TBI, rats were anesthetized (Nembutal, 100 mg/kg) and transcardially perfused with physiological saline, followed by a 4% paraformaldehyde solution. Brains were then post-fixed and cryoprotected in a 4% paraformaldehyde–20% sucrose solution, and 20 μm frozen sections were cut in the coronal plane.
To visualize damaged cells 1–5 d and 2 and 8 weeks after TBI, brain sections were stained with Fluoro-Jade (Histo-Chem Inc., Jefferson, AR) (Schmued et al., 1997). Briefly, hemispheric 20 μm brain sections were pretreated with a 0.06% solution of potassium permanganate and then stained with a 0.01% Fluoro-Jade–0.1% acetic acid solution. A subset of sections was also stained with cresyl violet.
To verify gross neuron loss, we applied cresyl violet stain to alternate horizontal sections from another group of rats 2–27 weeks after TBI. The intervening sections were used for Timm histochemistry as described below. Areas of Fluoro-Jade-labeled neurons and gross loss of cresyl violet-stained neurons were plotted on templates of hippocampus at three anteroposterior levels: 2.5, 3.8, and 5.6 mm posterior to bregma.
Administration of pentylenetetrazole and behavioral seizure classification. To detect any enhanced seizure susceptibility in post-TBI rats, we used a single, normally subconvulsant dose of PTZ (30 mg/kg; Sigma, St. Louis, MO) to challenge rats 15 weeks after TBI and compared these with age-matched controls that received the same dose of PTZ. The dose of 30 mg/kg PTZ was based on earlier work showing that 20–30 mg/kg of PTZ (intraperitoneal) is subconvulsant for adult male Sprague Dawley rats, whereas 50–60 mg/kg is a convulsant dose (Sacks and Glaser, 1941).
After PTZ injection, each rat was placed in a clear plastic cage and observed for 1 hr. Convulsions were scored into five classes by a standard method (Racine, 1972): class I, arrest of motion; class II, myoclonic jerks of the head and neck, accompanied by brief twitching movements of forelimbs and hindlimbs; class III, unilateral clonic activity; class IV, bilateral tonic–clinic forelimb activity; and class V, generalized tonic–clonic activity with rearing and loss of postural tone. For each rat, the highest seizure class during the first hour after PTZ injection was reported. We compared scores between control and TBI rats, using a two-tailed Mann–Whitney Utest.
Rats were prepared for Timm histology (as described below) 18–24 hr after PTZ injection to minimize the contribution of PTZ-evoked seizures to MFSR.
In vitro field potential and optical recording. Excitability of the DG was assessed in coronal hemispheric brain slices from rats 2–3 and 14–15 weeks after TBI and from normal controls. Brain slices were prepared by standard procedures. Rats were anesthetized (85 mg/kg ketamine plus 15 mg/kg xylazine, i.m.). Brains were carefully removed, chilled, and sliced in a partly frozen cutting solution, containing (in mm): 252 sucrose, 3 KCl, 0.2 CaCl2, 6 MgSO4, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, and saturating 95% O2 and 5% CO2, pH 7.4. Slices (400 μm thick) were cut using a Vibratome (Ted Pella) and transferred to artificial CSF (ACSF) containing (in mm): 124 NaCl, 2 KCl, 2 CaCl2, 1 MgSO4, 1.3 NaH2PO4, 26 NaHCO3, 10 dextrose, saturated with 95% O2 and 5% CO2, pH 7.4, at 10–15°C, warmed for 1 h, and maintained at 25–27°C.
For electrophysiological recording, brain slices were placed in an interface chamber at 33–35°C, perfused with ACSF, and aerated with a 95:5% O2/CO2 mixture. Perforant-path axons were activated with single shocks (100 μsec duration at 0.1–0.25 Hz) by a bipolar stimulating electrode placed in the DG molecular layer. In some experiments, pairs of shocks were delivered at these frequencies, with an interpulse interval of 50–60 msec. Field-potential responses were recorded with a glass pipette filled with 3 m NaCl (tip resistance 3–7 MΩ), in the granule cell layer, 1–2 mm away from the tip of the stimulating electrode. Duration of each evoked population EPSP (pEPSP) was estimated as the time interval between the first rise of the positive-going pEPSP and the intercept of the return to baseline of the pEPSP (or its tangent).
To examine granule cell propensity for repetitive firing, GABAA receptors were blocked with (−) bicuculline methiodide and/or picrotoxin. In some experiments, NMDA-type glutamate receptors were blocked withdl-2-amino-5-phosphonovaleric acid (APV). All drugs were obtained from Sigma.
Simultaneous optical recording of voltage-sensitive fluorescent signals. For optical recording, brain slices were stained with the voltage-sensitive fluorescent dye di-2-ANEPEQ (Molecular Probes, Eugene, OR) by incubation in a 0.1% di-2-ANEPEQ–ACSF solution at 20–25°C while being aerated with a 95:5% O2/CO2 mixture. Excess dye was removed by washing in dye-free oxygenated ACSF. Stained slices were placed in a recording interface chamber, and perforant-path-evoked population responses were recorded by field and optical methods simultaneously. To create an inactive reference region for data processing (see below), we coated a small piece of filter paper with fluorescent latex beads (FluoSphere, Molecular Probes) and placed it on or near the slice.
For optical recording of perforant-path-evoked depolarizations in the DG, excitation light (Xenon lamp XBO75, filtered at 530 ± 5.0 nm) was delivered to the surface of the slice via a fluid-filled light guide (Oriel, Stratford, CT). Emission light was collected by a long working distance 5× objective (Mitotoyu, Tokyo, Japan), filtered at 550 nm (glass long-pass filter), and captured by a cooled CCD camera (312 × 586 pixels; Roper Scientific Inc., Tucson, AZ). A pair of 4 msec images were recorded, one before a single shock to the perforant path (“pre-image”) and one at a variable delay (0–40 msec) after the shock (“post-image”). Temporal resolution of 4 msec was obtained by illuminating the slice with a 4 msec flash of excitation light using a Uniblitz shutter (12.5 mm diameter) during each of the two (100 msec long) camera shutter openings. Optical data were recorded, processed, and stored using IPLab software (Signal Analytic, Vienna, VA). Field potentials were monitored simultaneously during optical recordings.
In these experiments, we tested the relative amplitude of depolarization in the hilus compared with the molecular and granule cell layers after single, perforant-path shocks. We first verified that robust voltage-sensitive optical signals were detected in the hilus, when the hilus was directly stimulated with single shocks in a set of pilot experiments (data not shown).
Processing of optical data. For each pre-image and post-image pair, a difference ratio [(post-pre)/pre] was calculated. Twenty ratio images (from consecutively recorded image pairs with the same timing relative to the shock) were averaged and filtered. Data were normalized and color coded according to a standard method, as follows. For each averaged ratio image, the average intensity value of pixels from an inactive reference region (see Fig.7A1,B1, filled green box) was designated baseline and assigned pseudocolor indigo/purple (see Fig.7A1,B1, arrowhead on color bar). Baseline plus 1.5 SD or greater was assigned the saturation color of pink (see Fig. 7A1,B1, abovered on the color bar). No image pixels reached this saturation intensity level in the data shown in this paper. When no fibers were stimulated in normal ACSF, the pixel intensity values in the entire image were similar to the baseline, as appropriate, and corresponded to pseudocolor indigo to purple (see Fig. 7A4,No. Stim.). To demonstrate the anatomical location of optical signals evoked by fiber activation, optical signals were superimposed on a single image of the slice preparation. For this purpose, all ratio-image pixels near baseline (previously assigned pseudocolors indigo to purple) (see Fig. 7A1,B1, below arrowhead on color bar) were made transparent.
Fig. 7.
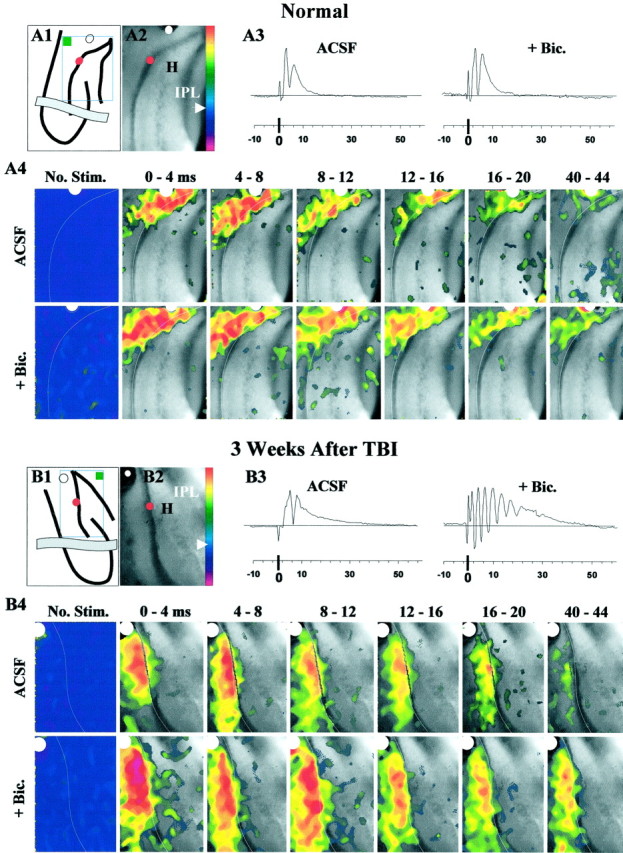
During simultaneous field and voltage-sensitive optical recordings, perforant-path stimulation evoked burst responses in post-TBI slices in the DG even after isolation from CA3a and CA3b. This activity peaked in the molecular and granule cell layers as shown by the optical data. A1, This line drawing shows the orientation of the hippocampus in a normal slice, an isolating knife cut (gray band), the positions of the stimulating (white dot) and recording (red dot) electrodes, the image frame (open rectangle), and the inactive reference region (filled green box). Color bar was used to display optical signals in pseudocolor as described in Materials and Methods. A2, This representative image of normal DG was acquired during a 4 msec exposure to excitation light. Positions of stimulating (white dot) and recording (red dot) electrodes are marked. A3, Field potentials evoked by single shocks (100 μsec) consisted of a pEPSP and a single population spike in standard ACSF. Addition of bicuculline (+ Bic., 50 μm) did not lead to epileptiform responses. Each trace is an average from 24 consecutive responses evoked every 10–20 sec during collection of the optical data shown in A4 (see Materials and Methods). A4, Panels show color-coded, averaged fluorescence-ratio maps of voltage-sensitive dye signals collected over the specified 4 msec periods after single shocks (at 0 msec). The No Stim. panels show the ratio map with no stimulus, and near zero relative change in optical signals corresponding to pseudocolors indigo topurple. White lines trace the hilar margin of the granule cell layer. Pixels corresponding to pseudocolors below indigo (arrowhead) on thecolor bar in A1 were turned transparent in all panels of A4 except No Stim.ACSF, In standard ACSF in this normal DG slice, perforant-path-evoked depolarization peaked in the molecular and granule cell layers, with minimal spread into the hilus, CA3c, or the opposite blade. + Bic., Bath application of bicuculline (50 μm) did not change the spatial pattern of depolarization. B1, For a rat 3 weeks after TBI,line drawing shows slice orientation as inA1. B2, This representative image of the post-TBI DG is analogous to A2. B3, As inA3, however, addition of bicuculline (50 μm; + Bic) led to burst responses recorded in the granule cell layer. B4, These optical responses correspond to the field potentials in B3.ACSF, In standard ACSF, 3 weeks after TBI, perforant-path stimulation evoked depolarization that peaked in the molecular and granule cell layers of the DG, with minimal spread into the hilus, CA3c, or the opposite blade. + Bic, Bath application of bicuculline (50 μm) increased the amplitude of optical signals at longer latencies (compare corresponding frames at 8–12, 16–20, and40–44 msec with ACSF). During disinhibited burst responses, depolarization peaked in the molecular and granule cell layers, spreading into a limited area in the adjacent infragranular region of the hilus.
Timm histochemistry and histological procedures. The distribution of mossy fiber terminals was examined in the following eight groups: (1) normal controls; (2–3) 2 and 16 weeks after craniotomy alone; (4) 18–24 hr after a single PTZ injection (i.e., PTZ controls); (5–7) 2–3, 15–16, and 24–27 weeks after TBI; (8) 18–24 hr after a single PTZ injection 15 weeks after TBI. The distribution of Zn2+-containing mossy fiber terminals was visualized by Timm histology using standard methods (Danscher, 1981;Sutula et al., 1988). Briefly, rats were anesthetized (85 mg/kg ketamine plus 15 mg/kg xylazine, i.m.) and intracardially perfused with 400 ml of 0.2% (w/v) Na2S, followed by 400 ml of a 1.0% (w/v) paraformaldehyde/1.25% (w/v) glutaraldehyde solution. Brains were removed and left in the fixative solution saturated with 30% (w/v) sucrose. Horizontal 40 μm sections were cut and developed in the dark for 30–60 min in a 12:6:2 mixture of gum arabic (2.0% w/v), hydroquinone (5.6% w/v), citric acid-sodium citrate buffer, to which 1.5 ml of silver nitrate solution (17% w/v) was added. Adjacent sections were stained with cresyl violet to assess gross cell loss.
Timm scoring methods. The distribution of Timm granules in the supragranular layer (SGL) of the DG was rated independently by three observers (who were blinded to the identity of each section) on a scale of 0–5, using a published scoring method (Cavazos and Sutula, 1990; Cavazos et al., 1991; Golarai et al., 1992; Sutula et al., 1992a,1996) adapted as follows: (0) no Timm granules between the tips and crest of the DG; (1) sparse Timm granules in the SGL with occasional clusters; (2) multiple clusters of Timm granules connected by sparse granules in the SGL; (3) more numerous and continuously distributed Timm granules in the SGL with occasional regions of confluence; (4) a confluent dense laminar band of granules in the SGL; and (5) a band as in 4, but extending into the inner molecular layer.
To quantify the regional distribution of MFSR after TBI, Timm-stained sections from both brain hemispheres in each rat were evaluated at two standard locations along the septotemporal axis: (1) a “septal” section at ∼3.6 mm deep with respect to bregma and (2) a “temporal” section at the level of the posterior commissure at ∼4.8 mm deep with respect to bregma. Septal and temporal scores were processed separately. Data were analyzed using two independent methods. First, following previous published methods (Cavazos et al., 1991,1992), we averaged individual Timm scores across the three independent observers for each rat for the septal and temporal locations. Thus levels of MFSR intermediate between scoring levels were often scored differently by different observers, and the average reflected that intermediacy. Using these average scores, we then tested between-group differences for statistical significance in a one-way ANOVA, using two-tailed t tests for paired comparisons (Snedecor and Cochran, 1980). In a second method, we treated MFSR scores as strictly ordinal data, used the median score across the three observers in each case (see Fig. 11), and then tested between-group differences using Kruskal–Wallis nonparametric test statistics, corrected for multiple comparisons (Dunn's test) (Snedecor and Cochran, 1980; Glantz, 1997). These two methods led to similar results with slight differences in α levels. Here we report statistical significance according to the more conservative nonparametric analysis.
Fig. 11.

Summary of time course and regional distribution of MFSR during the weeks after TBI, based on Timm-stain rating on a five-point scale in standard locations. Posttraumatic MFSR was found in septal and temporal regions of DG in both hemispheres and intensified over time. Across all the time points tested, MFSR was more prominent ipsilateral to TBI in a septal location than contralateral to TBI in a temporal location. A, Ipsilateral septal MFSR was statistically significant 2–3 weeks after TBI versus right hemisphere in normal controls (2–3 W TBI vsNC; *p < 0.05). MFSR intensified from 2 to 15 and 24 weeks after TBI (2–3 W TBI vs pooled data from 15–16 W TBI and 16 W TBI+ PTZ; *p< 0.05). B, Contralateral septal MFSR reached statistical significance 15–16 weeks after TBI (pooled data vs left hemisphere NC; †p < 0.01) and persisted beyond 24 weeks after TBI. C, Ipsilateral temporal MFSR reached statistical significance 15–16 weeks after TBI (pooled data vs right hemisphere NC; *p < 0.05) and persisted beyond 24 weeks after TBI. D, Contralateral temporal MFSR was detected at 15–16 weeks after TBI and persisted beyond 24 weeks after TBI.NC, Normal control, n = 9;PTZ, PTZ controls (1 d after 1 injection of PTZ, 30 mg/kg, i.p.), n = 9; 3W Cr, 3 weeks after craniotomy alone (no weight drop), n = 5;16W Cr, 16 weeks after craniotomy alone,n = 3; 2–3 W TBI, 2–3 weeks after TBI,n = 9; 15–16 W TBI: 15–16 weeks after TBI,n = 6; 16 W TBI + PTZ, 16 weeks after TBI and 1 d after one injection of PTZ (30 mg/kg, i.p),n = 7; 24–27 W TBI, 24–27 weeks after TBI, n = 3.
RESULTS
Cortical lesion induced by TBI
TBI by weight drop induced a reproducible lesion in the ipsilateral somatosensory cortex. Figure1A shows a typical cresyl violet-stained coronal section of the somatosensory cortex 1 d after TBI; arrowheads delineate the site of impact by weight drop. As reported earlier (Feeney et al., 1981), this cortical site of impact undergoes massive cell loss during a 2 week period.
Fig. 1.
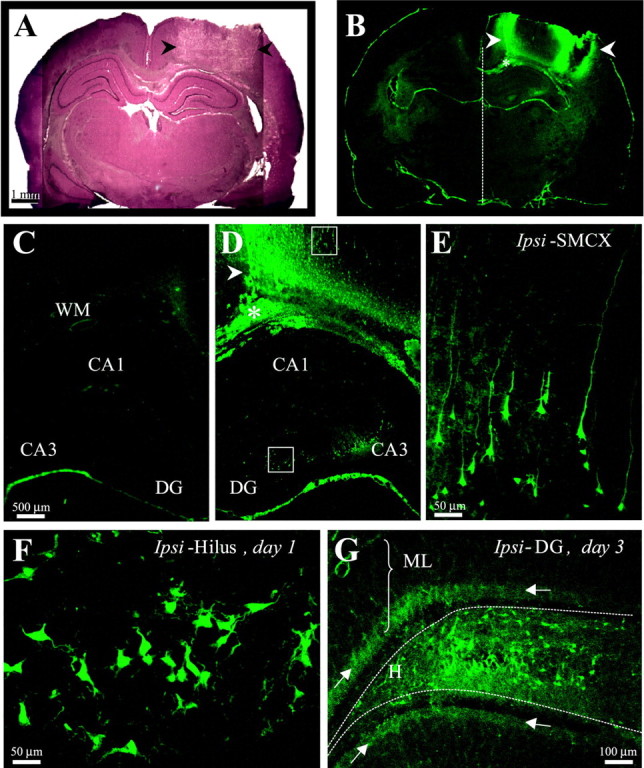
Weight-drop TBI induced reproducible damage in the ipsilateral somatosensory cortex and selective cell loss in the ipsilateral hippocampus. A, A cresyl violet-stained coronal section 1 d after TBI shows the impact site in the right somatosensory cortex (arrowheads). This region underwent massive cell loss and by 2 weeks after TBI turned into a fluid-filled cavity (∼30 mm3) spanning cortical layers external to the white matter. B, A Fluoro-Jade-stained coronal section from the same brain asA shows the pattern of cell damage as detected by specific (bright green) Fluoro-Jade labeling of cells 1 d after TBI. Cortical white matter is marked with anasterisk. Arrowheads same asA. One day after TBI, cortical cell damage appeared mainly at the edges of the impact site, with sparse labeling in between. Specific labeling in the CA3 and hilus of the ipsilateral hippocampus is detectable at this low magnification. C, A higher magnification from B shows the cortex and hippocampus contralateral to TBI. Note scarcity of specific labeling contralateral to TBI, compared with D. D, A higher magnification from B shows the cortex and hippocampus ipsilateral to TBI. Arrowhead andasterisk are same as in B. Note the peak of labeling at the medial edge of the impact site (arrowhead) and sparser labeling laterally. Specific labeling is found in ipsilateral CA3 but not CA1. E, A higher magnification of the boxed somatosensory region from D shows labeled cortical neurons with typical pyramidal shapes, some with extended apical and basal processes.F, A higher magnification of the boxedhilar region of the DG from D shows varied shapes of labeled cells, some with dendrite-like processes. G, A Fluoro-Jade-stained coronal section of the ipsilateral DG 3 d after TBI shows labeled cells across the hilus. Arrowspoint to the punctate staining in the dorsal and ventral inner molecular layer (arrows). DG, Dentate gyrus; Ipsi-SMCX, ipsilateral somatosensory cortex; WM, white matter.
To verify consistency with earlier observations, we examined the time course and regional distribution of TBI-induced cell death using the fluorescence marker Fluoro-Jade, which selectively labels injured neurons (Schmued et al., 1997; Schmued and Hopkins, 2000). One day after TBI, labeled cells were most dense at the edges of the cortical impact site, extending into the interior regions of this site (Fig.1B,D). Labeled cortical cells were pyramidal shaped, with basal and apical processes, some extending for several hundred micrometers (Fig. 1E). During days 2–10 after TBI, the number of labeled cells increased in the interior regions of the impact site (data not shown), whereas labeled cortical cells remained confined to the ipsilateral region around the site of impact at all time points tested (1–5, 7, 10 d, and 2–3 weeks after TBI). A similar distribution of damaged cells was detected by the Fink-Heimer method (data not shown). By 2 weeks after TBI, massive cell loss left a cavity in this region that approached a maximum size of ∼30 mm3, spanning all cortical layers external to the white-matter (Fig.2B). In this study, we examined rats at various latencies after TBI (1–7 d, 2–3, 15–16, and 24–27 weeks), excluding subjects with injuries deviating from the above (11 of 98 rats excluded).
Fig. 2.
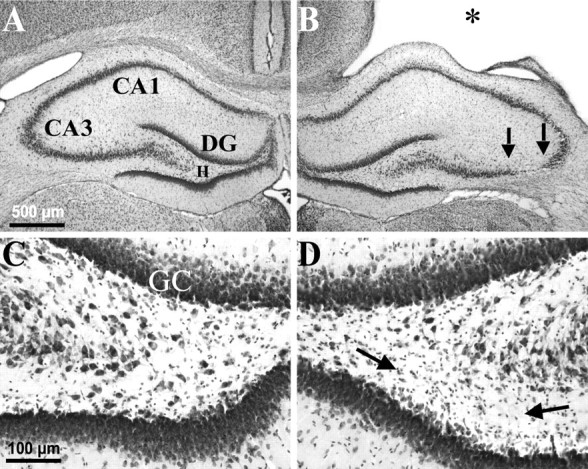
Weight-drop TBI-induced selective, gross cell loss in the ipsilateral hippocampus. A, A cresyl violet-stained coronal section 3 weeks after TBI shows the hippocampus contralateral to TBI with no evidence of macroscopic cell loss.B, From the same brain as A, showing the hippocampus ipsilateral to TBI. Note the TBI-induced cortical cavity (∗). Gross CA3 cell loss is delineated by arrows. There was no obvious cell loss in the CA1 or dentate granule cell layers. C, A higher magnification from Ashows the contralateral hilus with no evidence of gross cell loss after TBI. D, A higher magnification from Bshows the ipsilateral hilus where a subtle loss of large cells can be detected, especially ventrally (between arrows) in this example, as compared with the contralateral side in C.DG, Dentate gyrus; GC, granule cell layer; H, hilus of the dentate gyrus.
Selective cell death in hippocampus after TBI
TBI induced selective neuronal death in the ipsilateral (right hemisphere) subcortical structures, including selective damage in the right hippocampus (Figs. 1-3). In coronal sections 1–7 d after TBI, Fluoro-Jade-labeled damaged cells were numerous in ipsilateral CA3 (Fig. 1D) and hilus (Fig. 1F,G) and occasionally also found in CA1 or the granule cell layer (data not shown) but never observed in any region in the contralateral hippocampus (Fig.1C). One day after TBI, labeled cells were found in the ipsilateral hilus (Fig. 1F), displaying various pyramidal and multipolar shapes with dendrite-like processes, sometimes extending into the granule cell layer. On days 3 and 4 after TBI, labeled cells were more numerous across the hilus (Fig. 1G). We also observed punctate labeling in the ipsilateral inner molecular layer of the DG, consistent with terminal degeneration of the associational pathway arising from the ipsilateral hilus (Fig.1G, arrows). At latencies >7 d after TBI, Fluoro-Jade labeling in the hippocampus was sparse, although labeled cells were readily apparent in subcortical structures such as the caudate nucleus (data not shown).
Fig. 3.
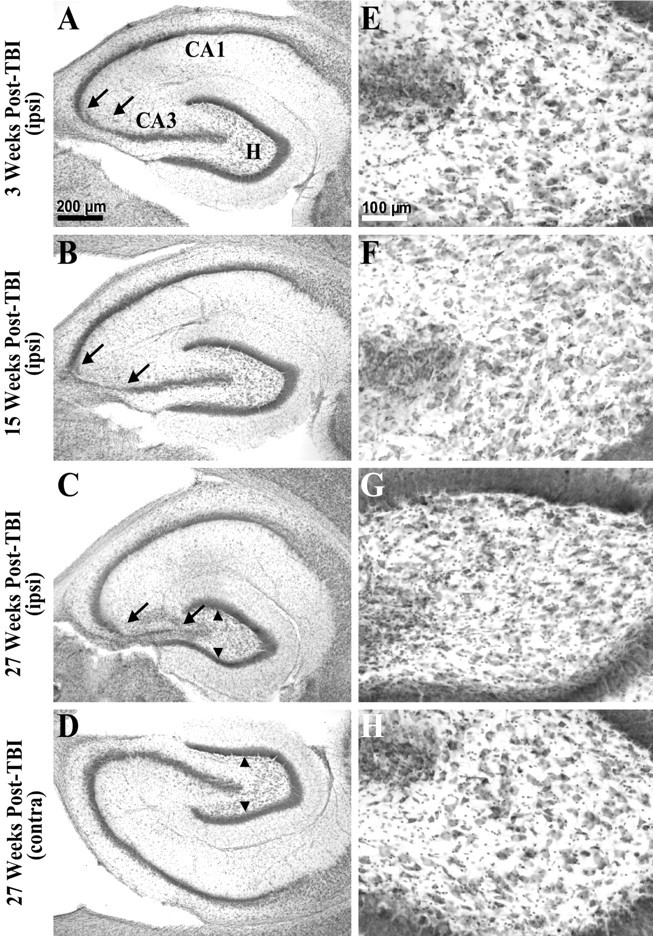
Gross cell loss in the ipsilateral hippocampal CA3 and hilus progressed in temporal regions during the weeks after TBI. A, A cresyl violet-stained horizontal section of the temporal hippocampus from ∼4.5 mm deep with respect to bregma prepared 3 weeks after TBI shows gross cell loss in the ipsilateral CA3 (between arrows). Otherpanels on the left show similarly prepared sections. B, The ipsilateral hippocampus 15 weeks after TBI showed a wider region of gross cell loss in CA3 (between arrows) compared with A.C, The ipsilateral hippocampus 27 weeks after TBI showed a progression of cell loss across the entire CA3. Note atrophy of the ipsilateral hilus, indicated by reduced distance between supragranule and infragranule cell layers (arrowheads) compared withD. D, The contralateral hippocampus 27 weeks after TBI (from the same brain section as C) showed no evidence of gross cell loss.E–H show higher magnifications of hilus from A–D. E, At 3 weeks after TBI, cell loss was subtle in the ipsilateral hilus in this temporal location. F, At 15 weeks after TBI, cell loss remained subtle in the temporal ipsilateral hilus. G, At 27 weeks after TBI, cell loss and atrophy of the temporal ipsilateral hilus was clearly detectable, although some large hilar neurons remain.H, There was no evidence of gross cell loss in the contralateral hilus 27 weeks after TBI at this temporal location. Scale bars in A and E apply toA–D and E–H, respectively.
We further examined the regional specificity of gross neuron loss 2–27 weeks after TBI in cresyl violet-stained sections. In coronal sections stained with cresyl violet, gross cell loss in the hippocampus remained confined to the ipsilateral hilus and CA3. Remaining relatively intact at all time points tested were the corresponding regions in the contralateral hemisphere and the dentate granule cell and CA1 pyramidal cell layers, bilaterally. In the typical example of Figure 2, 3 weeks after TBI, gross cell loss was only apparent ipsilaterally in CA3 and in the hilus between 2.5 and 4.0 mm posterior to bregma, but not among ipsilateral CA1 or dentate granule cells (Fig. 2B) or in any region of the contralateral hippocampus (Fig.2A).
Gross CA3 cell loss along the septotemporal axis of hippocampus was limited to the region between 2 and 6 mm posterior to bregma, 2–3, 15, and 27 weeks after TBI (n = 8, 13, and 3, respectively). Within this septotemporal region, gross cell loss was initially confined to CA3a, but increased over time to include most of CA3 (Fig. 3A–C, between arrows). Note however, that in the ipsilateral hilus large neurons could still be found at all time points tested (Figs. 2D,3E–G), whereas there was also an ipsilateral abundance of small cells consistent with gliosis (Fig.2D, between arrows). In more posterior sections stained with cresyl violet (Fig. 3), hilar cell loss was too subtle to detect without quantitative techniques at 3 and 15 weeks after TBI (n = 8 and 13, respectively) (Fig.3E,F), but gross cell loss and gliosis are apparent by 27 weeks in the ipsilateral hilus compared with the contralateral hilus (n = 3) (Fig. 3, compareC, G with D, H). These findings are consistent with a selective loss of neurons in the ipsilateral hilus after TBI.
Figure 4 summarizes the Fluoro-Jade and cresyl violet data to show the distribution of gross cell loss in the ipsilateral hilar and CA3 regions at various points on the hippocampal septotemporal axis during the weeks after TBI. This distribution resembled the selective cell loss caused by excitotoxicity during seizures or kainic acid treatment (Olney et al., 1986) and showed a progression of gross cell loss from septal to temporal hippocampus during the weeks after TBI.
Fig. 4.

Selective hippocampal cell loss progressed for weeks after TBI. This summary of the time course and regional distribution of selective cell loss in the hippocampus is based on Fluoro-Jade and cresyl violet staining 1 d (1 Day) to 27 weeks (24–27 W) after TBI, as indicated. Regions of visually discernable cell loss 2.5, 3.8, and 5.6 mm posterior to bregma are marked by filled diamonds. Gross cell loss in the CA3 progressed to temporal regions (i.e., posteriorly) and also from CA3a to CA3c over weeks after TBI. In the hilus, gross cell loss was detected in Fluoro-Jade-stained septal sections by 1 d, in cresyl violet-stained septal sections by 3 weeks, and in temporal hippocampus by 27 weeks after TBI.
Persistent susceptibility to PTZ-evoked convulsions after TBI
Selective loss of neurons in the hilus of the DG in temporal-lobe epilepsy is associated with a long-lasting susceptibility to seizures, which can be detected as a lowering of the threshold for seizures evoked by various convulsant agents. Therefore, we compared the PTZ-evoked behavioral responses in rats 15 weeks after TBI with controls. To determine whether seizure threshold was reduced after TBI, we selected a dose of PTZ (30 mg/kg, i.p) that typically does not induce generalized tonic–clonic seizures (class IV or V) in normal rats (Sacks and Glaser, 1941). Each rat was injected once with PTZ (30 mg/kg, i.p.), and its behavior was observed for 1 hr. Figure5 summarizes the behavioral seizure responses of each control (open circles) and TBI rat (filled circles) during the first hour after PTZ injection. Fifteen weeks after TBI, PTZ-evoked convulsions ranged from class I to V. Class V seizures were observed in five of eight subjects and developed into repeated episodes in three of these five rats, ending in fatal status epilepticus in one case. The median convulsion in the post-TBI group was generalized tonic–clonic (i.e., class IV;n = 8). In contrast, PTZ-evoked convulsions in controls ranged from class 0 to III, and the median convulsion was transient arrests of motion (i.e., class I; n = 10). Although the variability within each group was consistent with previously published work with PTZ in normal rats (Mason and Cooper, 1972; Golarai et al., 1992), our observation of dramatically increased susceptibility to PTZ-induced seizures 15 weeks after TBI was highly significant (p < 0.01; two-tailed Mann–WhitneyU test).
Fig. 5.

TBI induced a persistent susceptibility to pentylenetetrazole-evoked seizures in vivo (30 mg/kg PTZ, i.p.). Each circle represents a control (open) or a rat 15 weeks after TBI (filled). Convulsions were rated according to a standard scale: no detectable behavioral seizures (class 0); arrest of motion (class I); myoclonic movements (class II); unilateral tonic–clonic seizures (class III); bilateral tonic–clonic seizures (class IV); generalized seizures with loss of postural tone (class V). Generalized seizures were induced in five of eight rats 15 weeks after TBI. In contrast, no generalized seizures were observed in 10 normal rats for 1 hr after PTZ injection.
Persistent hyperexcitability in the DG after TBI
In experimental models of temporal-lobe epilepsy, seizures and hilar cell loss are associated with an enduring propensity for repetitive firing among the dentate granule cells when exposed to GABAA antagonists in vitro (Cronin et al., 1992; Golarai et al., 1992). Thus, we tested the DG for similar changes 2–3 and 15 weeks after TBI. Furthermore, we used optical methods to determine the spatial and temporal organization of these evoked responses in DG after partial isolation, as described below.
First, we tested whether the DG at 2–3 and 14–15 weeks after TBI and in normal slices showed a similar resistance to repetitive firing in normal ACSF (Fig.6A1,B1). Field potentials were evoked by single or paired shocks to the perforant path and recorded in the granule cell layer in coronal slices from septal (∼2.5 mm posterior to bregma) and temporal (∼6.0 mm posterior to bregma) regions from both ipsilateral and contralateral hemispheres. When suprathreshold paired stimuli (50–150 μA, 100 μsec duration, 50–60 msec interval) were delivered in normal ACSF, the pEPSP duration was ∼15–35 msec in slices 2–3 and 14–15 weeks after TBI. Each shock also evoked one or occasionally two negative-going population spikes at latencies of 5–8 msec, reflecting synchronous granule cell firing superimposed on the pEPSP (Fig.6A1,B1, arrows). In normal ACSF, increasing the stimulus intensity did not evoke more than two population spikes in the DG in either TBI or control slices. For between-group comparisons of numbers of evoked population spikes and pEPSP durations, we standardized the intensity of paired stimuli to evoke a first population spike of 80% maximum amplitude.
Fig. 6.

TBI induced a persistent hyperexcitability in the DG that was revealed during disinhibition by GABAAantagonists in hippocampal slices 14–15 weeks after TBI.A1, In slices from normal controls in standard ACSF, each of the paired-shock stimuli (60 msec interpulse interval; 100 μsec shock duration) to the perforant path at 0.05 Hz evoked a typical field response in the granule cell layer, consisting of a pEPSP and a single orthodromic population spike (arrow). A2, In the normal DG, addition of GABAA antagonist picrotoxin (PTX, 50 μm) led to a slightly larger amplitude and longer duration population spike, but not to epileptiform activity.B1, In standard ACSF, and 15 weeks after TBI, field-potential responses to perforant-path stimulation lacked epileptiform activity, despite slightly longer than normal pEPSPs. B2, In post-TBI slices in the presence of GABAA antagonist picrotoxin (PTX, 50 μm), shocks to the perforant path induced burst responses consisting of a prolonged depolarization envelope and multiple population spikes. B3, These epileptiform features were eliminated by bath addition of APV (35 μm).
When subjected to these stimuli, DG in slices tested 2–3 and 14–15 weeks after TBI resembled normal DG in its resistance to repetitive firing in normal ACSF (Fig. 6A1,B1, Table1), as is typical of epileptic DG (Fricke and Prince, 1984; Cronin et al., 1992; Lynch et al., 2000).
Table 1.
TBI induced persistent hyperexcitability in the DG, revealed by disinhibition
| Group | In vitro treatment | Number of population spikes | pEPSP duration | n(rats) | ||
|---|---|---|---|---|---|---|
| Median (minimum, maximum) | Mean ± SE | |||||
| First shock | Second shock | First shock | Second shock | |||
| Normal control | 15 | |||||
| Normal ACSF | 1 (1, 2) | 1 (1, 2) | 14.0 ± 2.6 | 16.6 ± 3.9 | ||
| GABAA blocker | 1 (1, 2) | 1 (1, 2) | 18.0 ± 2.1 | 22.0 ± 4.7 | ||
| 2–3 weeks after TBI | 14 | |||||
| Normal ACSF | 1 (1, 2) | 1 (1, 3) | 19.0 ± 4.7 | 27.9 ± 7.2 | ||
| GABAAblocker | 3 (1, 5) | 4 (1, 8)* | 28.3 ± 2.1 | 41.4 ± 2.1 | ||
| 14–15 weeks after TBI | 8 | |||||
| Normal ACSF | 1 (1, 2) | 1 (1, 2) | 17.5 ± 3.2 | 26.6 ± 6.9 | ||
| GABAAblocker | 3 (1, 6) | 5 (1, 8)* | 30.9 ± 0.5 | 45.0 ± 0.61-160 | ||
Different from normal control during disinhibition;p < 0.01 in Dunn's test with two comparisons.
F1-160: Different from normal control during disinhibition;p < 0.05 in two-tailed t test after correction for four comparisons after Bonferroni's method (including the two comparisons with Dunn's test).
We next examined the effect of GABAA antagonists on the propensity of the DG to fire repetitively during the weeks after TBI, compared with normal controls. Addition of either GABAA receptor blocker picrotoxin (20–50 μm) or bicuculline (50 μm) to ACSF led to stimulus-evoked repetitive firing (“burst responses”) in the DG of the post-TBI slices from the ipsilateral and contralateral hemispheres (Fig. 6B2, Table 1). In contrast, repetitive firing was never seen in normal slices at these standard stimulus intensities (Fig. 6A2). When burst responses were evoked in disinhibited post-TBI slices, our standard paired-pulse stimuli elicited up to six population spikes for the first pulse and eight population spikes for the second pulse (Table 1). Whenever observed, burst responses were sensitive to NMDA receptor antagonist APV, as shown in the example of Figure 6B3. Bath addition of APV (35–50 μm) reduced bursts to one or two population spikes and pEPSP durations to ∼10–20 msec (five of five ipsilateral slices 2–3 weeks after TBI, six of six ipsilateral slices, and four of four contralateral slices 15 weeks after TBI).
Optical localization of burst responses
To localize these burst responses within the DG, we partly isolated the DG with knife cuts at the junction of CA3 and the DG (Fig.7A1,B1) and performed simultaneous field and optical recordings in these slices. As shown in Figure 7B3 (+ Bic.), the bicuculline-induced bursts persisted in post-TBI slices subjected to a knife cut and optical recordings. These optical recordings revealed the spread of depolarization evoked by perforant-path stimulation in slices from normal rats and 2–3 weeks after TBI (n = 10) (Fig. 7A4,B4). Optical signals in Figure7, A4 and B4, depict population depolarizations based on fluorescence changes of the voltage-sensitive dye Di-2-ANEPEQ (see Materials and Methods). In the typical normal and post-TBI slices shown, fibers in the molecular layer of the suprapyramidal blade were activated by single shocks (Fig. 7A1–2,B1–2). Optical signals showed maximal changes in the molecular and granule cell layers of the activated suprapyramidal blade, spreading minimally into the hilus, CA3c, or the infrapyramidal blade in control (Fig.7A4, ACSF) and TBI (Fig. 7B4,ACSF) slices. Thus the spatial patterns of optical responses in the isolated DG in normal and post-TBI slices were similar.
In post-TBI slices exposed to 50 μm bicuculline, single shocks to the perforant path triggered burst responses (Fig.7B3, + Bic.). During these burst responses, optical signals were greater in amplitude at longer post-shock latencies, compared with responses from the same slices in normal ACSF (Fig. 7B4, compare 40–44msec frames in + Bic vs ACSF; also see Fig.8B). However, despite this increase in optical signals during burst responses, depolarization remained predominantly localized in the molecular and granule cell layers, spreading only into a narrow zone in the adjacent infragranular hilar region (Fig. 7B4, + Bic; also see Fig.8B).
Fig. 8.
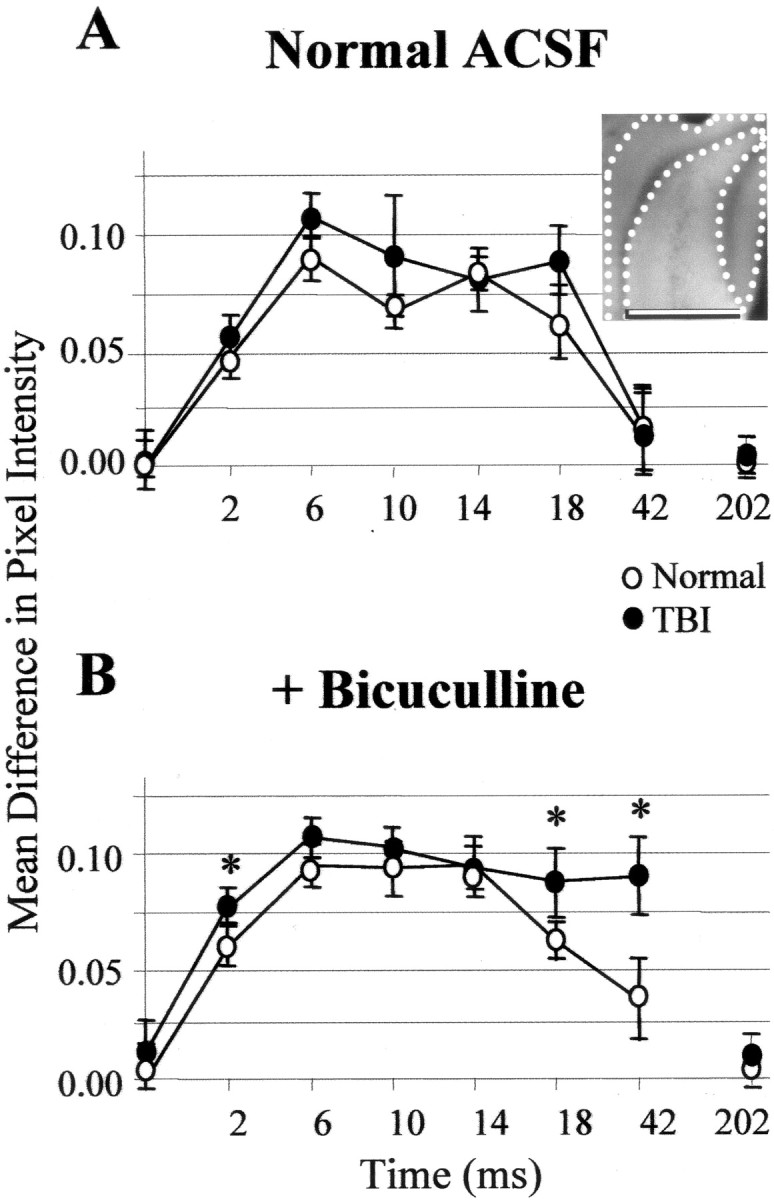
Optically detected depolarization evoked by single shocks to the perforant path was predominantly generated in the granule cell and molecular layers compared with the hilus and CA3c. This difference grew larger at post-shock latencies of 18 and 42 msec (i.e., images recorded 16–20 and 40–44 msec after shock, respectively) during disinhibited burst responses in post-TBI slices relative to normal controls. For a representative slice, the insetshows the regions for which the mean difference in pixel intensity between regions was calculated for inclusion in the group means plotted in A and B. A, For normal (open circles) and TBI (filled circles) slices in ACSF, plotted on the ordinate are the mean pixel intensities in the granule cell and molecular layers (I(GCL + ML); inset: area between dotted lines) minus mean pixel intensities in the hilus and CA3c (I(H + CA3c);inset: area between dotted andsolid lines) for average ratio images at designated times after single shocks. The midpoints of 4 msec recording intervals after single perforant-path shocks are indicated on theabscissa; zero represents no stimulation. The quantity I(GCL + ML) −I(H + CA3c) was greater than zero during the period 0 < time < 44, reflecting greater depolarization in the molecular and granule cell layer than in the hilus and CA3c. B, Same as A, but during disinhibition that induced burst responses in post-TBI (but not in normal) slices. These bursts were associated with signals that were larger in the GCL and ML and significantly greater than controls at 18 msec (i.e., 16–20 msec) and 42 msec (i.e., 40–44 msec) after shock (p < 0.05; Bonferroni two-tailedt test). Error bars represent SD.
Similar results were obtained whether the optical signals were recorded first during ACSF perfusion and then in bicuculline or in the reverse order (i.e., first during bicuculline application and then during washout), thus ruling out significant loss of optical signals attributable to repeated measurements and time (data not shown; four of four slices).
To quantify these regional differences, we used the mean pixel value in a region within an averaged ratio image as a measure of total depolarization. Using this measure, the predominance of depolarization in the molecular and granule cell layers is shown in Figure 8. Here the mean pixel value in the granule cell and molecular layers minus the mean in hilus and CA3c (IGCL + ML −IH + CA3c) is plotted for optical signals evoked by single perforant-path shocks in control and post-TBI slices during exposure to normal ACSF (Fig. 8A) or bicuculline (Fig. 8B). These difference values were significantly above zero during bicuculline exposure in normal and post-TBI slices, consistent with greater depolarization in the granule cell and molecular layers than in the hilus and CA3c. Also, the difference values were significantly higher at 16–20 and 40–44 msec during bicuculline-induced bursts in post-TBI slices relative to controls (Fig. 8B) (p < 0.05;n = 17 controls and 22 post-TBI slices).
Timm evidence for MFSR after TBI
Hilar cell loss and seizure susceptibility in the DG, such as we report above, are associated with MFSR in temporal-lobe epilepsy and experimental models (Lynch et al., 1996; Parent and Lowenstein, 1997;Bausch and McNamara, 1999). Typically, MFSR is first detected within days after experimental lesions or seizures, is well developed by 2 weeks, and is long lasting (Laurberg and Zimmer, 1981; Cavazos et al., 1991). To determine whether TBI induced MFSR in the DG, we compared the distribution of mossy fiber terminals in standardized septal (Fig. 9A) and temporal (Fig. 10A) locations 2–3, 15–16, and 24–27 weeks after TBI relative to normal controls. We visualized mossy fiber terminals using Timm histochemistry, which specifically stains these terminals because of their high content of Zn2+ (Danscher, 1981). In all cases, Timm granules were densely packed in the stratum lucidum of CA3 and in the hilus where mossy fibers normally terminate. In normal cases, Timm granules were sparse or absent in the supragranular region of the DG (Figs. 9B, 10B, arrows) (nine of nine rats), which is occupied mostly by granule cell dendrites and interneuron processes.
Fig. 9.
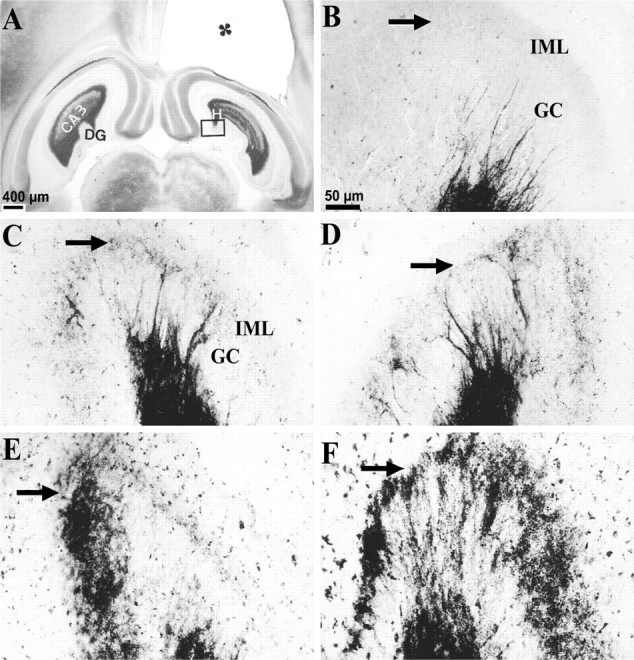
MFSR developed in septal DG over weeks after TBI, as detected by Timm staining. Septal MFSR was detected 2 weeks after TBI and became bilaterally more prominent at 16 and 27 weeks after TBI. MFSR is indicated by Timm granules in the SGL, at the border of the granule cell (GC ) and inner molecular layers (IML). A, A Timm-stained horizontal section at ∼3.6 mm deep with respect to bregma is from a rat 3 weeks after TBI. The cavity in the somatosensory cortex (∗) is caused by TBI. Box corresponds to regions shown at higher magnification in B–F.B, Timm granules are sparse or absent in the SGL (arrow) of a normal rat. C, At 3 weeks after TBI, an abnormal density of Timm granules was detected in the ipsilateral SGL (arrow) from A.D, At 3 weeks after TBI, clusters of Timm granules were found in the contralateral SGL (arrow) fromA. E, At 16 weeks after TBI, Timm granules formed a confluent band in the ipsilateral SGL (arrow), occasionally extending into the IML.F, At 27 weeks after TBI, Timm granules formed a confluent band in the ipsilateral SGL (arrow), extending into the IML. GC, Granule cell layer; H, hilus; IML, inner molecular layer. Scale bar inB applies toB–F.
Fig. 10.
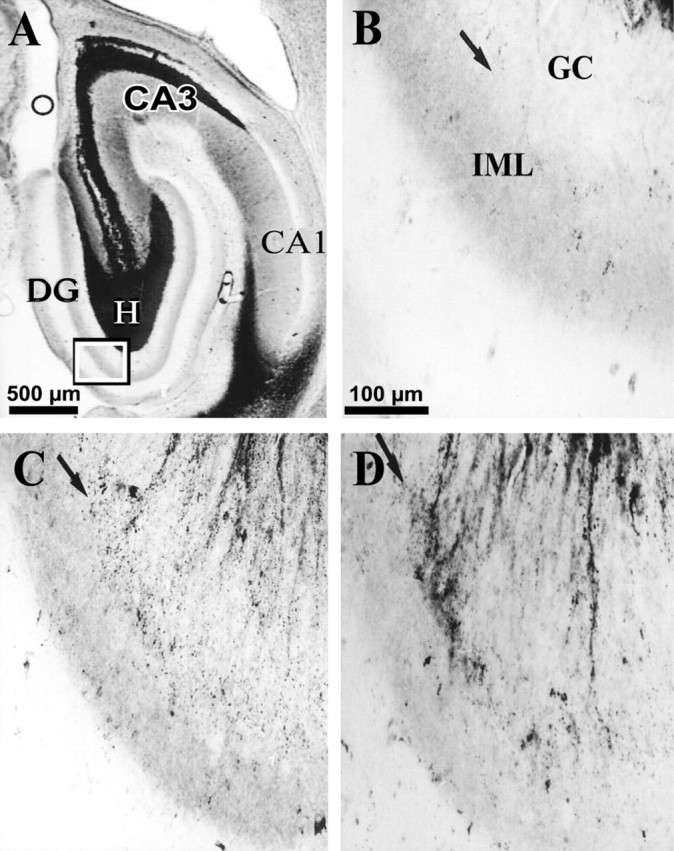
Development of MFSR in the ipsilateral temporal hippocampus weeks after TBI, as detected by Timm staining. Temporal posttraumatic MFSR was first detected 16 weeks after TBI and became more prominent by 27 weeks. MFSR is indicated by Timm granules in the SGL at the border of granule cell (GC) and inner molecular layers (IML). A, A Timm-stained horizontal section ∼4.8 mm posterior to bregma is from a rat 16 weeks after TBI. CA3 cell loss is also apparent in this section (open circle). Box corresponds to regions shown at higher magnification in B–D.B, In a normal rat, Timm granules were sparse or absent in the SGL (arrow) and IML. C, At 16 weeks after TBI, MFSR was detected as Timm granules distributed along the SGL (arrow). D, At 27 weeks after TBI, MFSR intensified (compared with C), as Timm granules formed larger clusters along the SGL (arrow).DG, Dentate gyrus; GC, granule cell layer; H, hilus; IML, inner molecular layer. Scale bar in B applies toB–D.
At 2–3 weeks after TBI, we found evidence for MFSR in the septal regions of both hemispheres (Fig. 9C,D,arrows) but more prominently ipsilateral to the injury (Fig.11, compare A,B). In these post-TBI cases, Timm granules were found in the ipsilateral supragranular layer with a distribution that ranged from occasional clusters (score of 1) to regions of confluent dense laminar bands (score of 5), with a median score of 3. In the contralateral septal DG (Fig. 9D), the distribution of these Timm granules ranged from sparse (score of 1) to occasional regions of confluence (score of 3) with a median score of 2. Although these Timm granules were found in the septal DG of both hemispheres 2–3 weeks after TBI, only the ipsilateral septal DG was statistically different from normal controls at this time point (p < 0.05; Dunn's test) (Fig. 11, compare A, B).
Timm evidence for increased MFSR over weeks after TBI
To assess the permanence of MFSR and to test whether MFSR increased over time after TBI, we examined supragranular Timm staining 15–16 (Fig. 9E) and 24–27 weeks (Fig.9F) after TBI. Notably, 15–16 weeks after TBI (Fig.9E) (n = 13) in the ipsilateral septal DG, we found Timm granules in the supragranular layer with a distribution that ranged from occasional regions of confluence (score of 3) to a dense laminar distribution extending to the inner molecular layer (score of 5), with a median score of 5 (Fig. 11A). The progression of MFSR from 2–3 to 15–16 weeks after TBI in this region was statistically significant (p < 0.05; Dunn's test) (Figs. 9C,E,11A). Similarly, at 15–16 weeks in the contralateral septal DG, the Timm scores were significantly greater than normal (p < 0.01; Dunn's test; median = 3) (Fig.11A). Earlier, at 2–3 weeks, the Timm scores in this region (median = 2) were not significantly different from normal. Suggesting continued bilateral progression, MFSR in the ipsilateral septal region of the DG reached the maximum asymptotic Timm score of 5 in all of three cases at 24–27 weeks after TBI (Figs. 9F,11A), and in the contralateral septal DG, MFSR reached a median score of 3 (Fig. 11B).
MFSR was less prominent in a temporal location
To evaluate MFSR away from the perpendicular axis of weight drop, we examined a temporal location at ∼4.8 mm deep with respect to bregma (Fig. 10A) and also found supragranular Timm granules in this temporal region bilaterally after TBI. At 2–3 weeks after TBI, no significant MFSR had occurred in this temporal region bilaterally (raw data not shown; see summary in Fig.11C,D). However, by 15–16 weeks after TBI, Timm granules were more numerous bilaterally and statistically significant ipsilaterally compared with normal controls (Figs. 10C,11C,D). By 24–27 weeks after TBI, Timm granules formed clusters in the ipsilateral temporal DG (Fig.10D) with a median score of 2 (Fig. 11C). In summary, TBI induced progressive MFSR along the septotemporal axis of the hippocampus that was more dense in the ipsilateral and septal regions than in the contralateral and temporal regions at all time points tested (Fig. 11).
MFSR was not detected after craniotomy or injection of PTZ
Induction of posttraumatic MFSR was critically dependent on impact by weight drop. Specifically, we found no significant MFSR at 2–3 or 16 weeks after a simple craniotomy or 1 d after a single injection of PTZ (n = 8, 3, and 9, respectively; raw data not shown; see summary in Fig. 11). Also, indistinguishable Timm scores were obtained for TBI subjects that did not receive PTZ (n = 5) and for TBI subjects that were tested for seizure susceptibility with one injection of PTZ 18–24 hr before preparation for Timm histology (n = 8) (Fig. 11). These last two groups were pooled for statistical comparisons.
DISCUSSION
In this study, we showed that weight-drop TBI induced a persistent susceptibility to PTZ-evoked behavioral seizures for at least 15 weeks. Similarly, we found DG hyperexcitability during disinhibition in vitro up to 15 weeks after TBI, an important extension on reports of DG hyperexcitability within 1 week after TBI (Reeves et al., 1995,1997; Coulter et al., 1996; Toth et al., 1997). Using optical methods, we localized this hyperexcitability to the granule cell and molecular layers of the DG 2–3 weeks after TBI. Accompanying these functional alterations, we also found MFSR in the septal DG of both hemispheres by 2 weeks after TBI, intensifying by 15 and 27 weeks. These results demonstrate persistent seizure susceptibility after weight-drop TBI in association with functional and structural changes in the DG, including selective cell loss, hyperexcitability, and MFSR, which are implicated in temporal-lobe epilepsy and may be similarly involved in PTE.
Weight-drop TBI as a model of severe human head injury
This well characterized model shares at least three key features with severe human brain injury. First, human PTE often develops after long delays (months to years) after injury and is persistent (Yoshii et al., 1978; Salazar et al., 1985; Asikaninen et al., 1999), as was seizure susceptibility after weight-drop TBI in the present study. Second, human TBI is reported to lead to hippocampal and fornix volume reductions (Bigler et al., 1997; Tate and Bigler, 2000), suggesting hippocampal damage as seen in this and other rat TBI models (Lowenstein et al., 1992; Toth et al., 1997). Third, severe head injuries in humans can lead to lesions and fluid-filled cavities such as observed in this model (Claussen et al., 1977; Koo and LaRoque, 1977; Yang et al., 1997; Asikainen et al., 1999; Pierallini et al., 2000). Although numerous differences between rat and human brains preclude a precise correspondence regarding impact force, the relative extent of cortical and hippocampal injury after TBI, and subsequent epileptogenesis, our findings of persistent MFSR and seizure susceptibility in rat DG after TBI suggest the hypothesis that similar processes may occur in humans.
TBI-induced cell loss resembles hippocampal sclerosis
Our search for seizure susceptibility and MFSR after TBI followed from observations of selective hippocampal cell loss after TBI. Using Fluoro-Jade staining, we confirmed selective ipsilateral cell damage and deafferentation during the week after weight-drop TBI, which slowed subsequently. Our observations were generally consistent with reports of hippocampal atrophy after TBI in humans (Tate and Bigler, 2000) and gradual selective neuron loss in rats after fluid-percussion TBI (Lowenstein et al., 1992; Toth et al., 1998) or kindling (Cavazos et al., 1994).
TBI induced persistent susceptibility to PTZ-evoked seizures and DG hyperexcitability in vitro
A key observation in this study was the persistent susceptibility to PTZ-evoked generalized convulsions 15 weeks after TBI. Such susceptibility is strong evidence for epileptogenesis (Mason and Cooper, 1972; Craig and Colasanti, 1988) and is novel in a brain impact model. We used PTZ to assess seizure susceptibility because longer-term assays such as kindling could induce additional injury and/or MFSR during the required days of testing, precluding measurement of posttraumatic MFSR (Golarai et al., 1992).
We also found evidence for early and persistent hyperexcitability in the DG circuitry in vitro after TBI. At 2–3 and 15–16 weeks after TBI, perforant-path stimulation during block of GABAA inhibition evoked abnormal repetitive firing in the DG that was sensitive to NMDA receptor blocker APV. Using optical recordings in slices of isolated DG, this hyperexcitability was localized mainly to the granule cell and molecular layers with less hilar depolarization. These features of post-TBI hyperexcitability in the DG are consistent with the reported changes in NMDA receptor subunit composition after TBI (Osteen et al., 2000) and are similar to the early, NMDA receptor-dependent hyperexcitability of the granule cells after a single evoked seizure (Lynch et al., 2000) that persists after kindling (Mody et al., 1988) and MFSR (Lynch et al., 2000). Although the causal relations between NMDA receptor activation, MFSR, and hyperexcitability are yet to be found, there is evidence that enhanced NMDA receptor activation promotes structural plasticity such as MFSR (Sutula et al., 1996; Aamodt and Constantine-Paton, 1999), which may in turn facilitate recurrent excitation and seizure susceptibility (Tauck and Nadler, 1985; Sutula et al., 1992b; Dudek et al., 1994) (but see Sloviter, 1992; Kotti et al., 1996). In other models, this hyperexcitability may facilitate epileptogenesis by undermining a putative role of the DG in “damping” high-frequency communication between seizure-prone entorhinal cortex and hippocampus (Fricke and Prince, 1984; Heinemann et al., 1992). Our data are consistent with similar processes in PTE but do not exclude other, for example cortical, mechanisms.
MFSR in posttraumatic and temporal-lobe epilepsies
Posttraumatic MFSR in our study resembled MFSR in human cases and animal models of temporal-lobe epilepsy. Use of Timm scoring enabled us to quantify the regional distribution and time course of MFSR and to compare three of its features with models of temporal-lobe epilepsy that were scored similarly (Cavazos et al., 1991, 1992; Golarai et al., 1992).
First, MFSR developed bilaterally beyond the hemisphere initially injured by weight drop, resembling bilateral MFSR in several epilepsy models (Laurberg and Zimmer, 1981; Stanfield, 1989; Cavazos et al., 1992; Golarai et al., 1992) and consistent with unilateral loss of bilaterally projecting hilar neurons (Seress and Ribak, 1983).
Second, MFSR was persistently more prominent in the hemisphere ipsilateral to TBI, suggesting a persistently dominant role for the injured hemisphere in PTE.
Third, MFSR intensified from 2 to 15 and 27 weeks after TBI. This increase resembles progressive MFSR during kindling and differs from the near maximal MFSR seen 2–3 weeks after a cluster of seizures or the slight decrease in MFSR 3–4 months after termination of kindling stimuli (Cavazos et al., 1991). By analogy to epilepsy models, mutually compatible factors contributing to progressive posttraumatic MFSR may include progressive hilar cell loss (Cavazos et al., 1994) and cumulative DG seizure activity (Cavazos et al., 1991; Golarai et al., 1992). Thus our observations of hilar cell loss, persistent DG hyperexcitability 15 weeks after TBI, and progressive MFSR are consistent with a positive feedback loop between progressive excitotoxic cell death, hyperexcitability, and MFSR in the DG for months after TBI.
Comparison of MFSR progression after TBI and kindling
Posttraumatic MFSR in our experiments may be compared with MFSR during kindling (where the relationship between evoked seizure duration and magnitude of MFSR is well documented) as follows. At 2–3, 15–16, and 27 weeks after TBI, the Timm scores in the ipsilateral–septal DG resembled similar increases above normal controls (on the same scale) in rats that experienced 5, 35, and 100 electrographic seizures, respectively, evoked by perforant-path stimulation (Cavazos et al., 1991). Although our data do not address whether repeated seizures propagate into septal DG during weeks after weight-drop TBI, they permit predictions regarding plausible net duration of such putative seizures. Specifically, the levels of posttraumatic MFSR 2–27 weeks after TBI exceed what the kindling data would predict to arise from the reported cumulative ∼1 min of hippocampal epileptiform activity during the first hours after TBI (Nilsson et al., 1994). Instead, these data are more consistent with repeated focal after-discharges and progressive cell loss over weeks. More studies are needed to test these hypotheses directly.
MFSR and DG hyperexcitability after TBI
The contribution of MFSR to seizure susceptibility has been controversial. MFSR is consistently correlated with temporal-lobe epilepsy in humans and in animal models (Sutula et al., 1992b; Parent and Lowenstein, 1997; Bausch and McNamara, 1999). Also, it is associated with functional reorganization of the DG in vivo(Golarai and Sutula, 1996) and granule cell hyperexcitability in vitro (Cronin et al., 1992; Wuarin and Dudek, 1996; Lynch et al., 2000). However, other work suggests that MFSR increases excitatory drive on both excitatory and inhibitory neurons (Sloviter, 1992; Kotti et al., 1996). Accordingly, our finding that bath application of a GABAA antagonist was necessary to induce repetitive firing in DG after TBI is consistent with a net increase of excitatory drive on a mixed population of postsynaptic targets. As suggested previously, granule cell hyperexcitability and MFSR are likely to facilitate breakdown of the intrinsic seizure resistance of granule cells (Heinemann et al., 1992; Bausch and McNamara, 1999). This loss of normal “filtering” in the DG may be particularly epileptogenic in combination with other TBI-induced changes such as loss of interneurons (Toth et al., 1997), glia dysfunction (D'Ambrosio et al., 1999), and development of seizure foci in other susceptible circuitry, such as among the neocortical neurons near the site of injury (Golarai et al., 1999; ). Furthermore, others have shown neocortical sprouting and seizures after focal injury to correlate positively with epileptogenesis (Jacobs et al., 2000) as well as behavioral recovery (Hernandez and Schallert, 1988; Schallert et al., 1997; Jones et al., 1994; Kolb, 1999). It remains to be seen whether the hippocampal plasticity that we report after impact injury is also associated with cognitive recovery or solely with epileptic pathology.
Footnotes
This work was supported by National Institute of Child Health and Human Development Grant KO1HD01324 and National Institute of Neurological Disorders and Stroke Grant NS-35644. Thanks to Dr. W. R. C. Shuttleworth for participation in Timm scoring, Dr. B. Skipper for advice on statistical analysis, and D. King and D. M. Stilbeck for assistance with surgeries.
Correspondence should be addressed to Dr. Golijeh Golarai, Department of Psychology, Jordan Hall (Building 420), Stanford University, Stanford, CA 94305-2130. E-mail:ggolarai@psych.stanford.edu.
REFERENCES
- 1.Aamodt SM, Constantine-Paton AU. The role of neural activity in synaptic development and its implications for adult brain function. Adv Neurol. 1999;79:133–144. [PubMed] [Google Scholar]
- 2.Annegers JF, Rocca WA, Hauser WA. Causes of epilepsy: contributions of the Rochester epidemiology project. Mayo Clin Proc. 1996;71:570–575. doi: 10.4065/71.6.570. [DOI] [PubMed] [Google Scholar]
- 3.Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- 4.Asikainen I, Kaste M, Sarna S. Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia. 1999;40:584–589. doi: 10.1111/j.1528-1157.1999.tb05560.x. [DOI] [PubMed] [Google Scholar]
- 5.Babb TL. Axonal growth and neosynaptogenesis in human and experimental hippocampal epilepsy. Adv Neurol. 1997;72:45–51. [PubMed] [Google Scholar]
- 6.Bausch SB, McNamara JO. Experimental partial epileptogenesis. Curr Opin Neurol. 1999;12:203–209. doi: 10.1097/00019052-199904000-00012. [DOI] [PubMed] [Google Scholar]
- 7.Bigler ED, Blatter DD, Anderson CV, Johnson SC, Gale SD, Hopkins RO, Burnett B. Hippocampal volume in normal aging and traumatic brain injury. AJNR Am J Neuroradiol. 1997;18:11–23. [PMC free article] [PubMed] [Google Scholar]
- 8. Cavazos JE, Sutula TP. Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res 527 1990. 1 6 [Erratum (1991) 541:179] [DOI] [PubMed] [Google Scholar]
- 9.Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–2803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavazos JE, Golarai G, Sutula TP. Septotemporal variation of the supragranular projection of the mossy fiber pathway in the dentate gyrus of normal and kindled rats. Hippocampus. 1992;2:363–372. doi: 10.1002/hipo.450020404. [DOI] [PubMed] [Google Scholar]
- 11.Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claussen CD, Lohkamp FW, Krastel A. Computed tomography of trauma involving brain and facial skull (craniofacial injuries). J Comput Assist Tomogr. 1977;1:472–481. doi: 10.1097/00004728-197710000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- 14.Cotman CW. Specificity of synaptic growth in brain: remodeling induced by kainic acid lesions. Prog Brain Res. 1979;51:203–215. doi: 10.1016/S0079-6123(08)61306-0. [DOI] [PubMed] [Google Scholar]
- 15.Coulter DA, Rafiq A, Shumate M, Gong QZ, DeLorenzo RJ, Lyeth BG. Brain injury-induced enhanced limbic epileptogenesis: anatomical and physiological parallels to an animal model of temporal lobe epilepsy. Epilepsy Res. 1996;26:81–91. doi: 10.1016/s0920-1211(96)00044-7. [DOI] [PubMed] [Google Scholar]
- 16.Craig CR, Colasanti BK. A study of pentylenetetrazole kindling in rats and mice. Pharmacol Biochem Behav. 1988;31:867–870. doi: 10.1016/0091-3057(88)90397-8. [DOI] [PubMed] [Google Scholar]
- 17.Cronin J, Obenaus A, Houser CR, Dudek FE. Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization of mossy fibers. Brain Res. 1992;573:305–310. doi: 10.1016/0006-8993(92)90777-7. [DOI] [PubMed] [Google Scholar]
- 18.D'Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danscher G. Histochemical demonstration of heavy metals. A revised version of the sulfide silver method suitable for both light and electron microscopy. Histochemistry. 1981;71:1–16. doi: 10.1007/BF00592566. [DOI] [PubMed] [Google Scholar]
- 20.Dudek FE, Obenaus A, Schweitzer JS, Wuarin JP. Functional significance of hippocampal plasticity in epileptic brain: electrophysiological changes of the dentate granule cells associated with mossy fiber sprouting. Hippocampus. 1994;4:259–265. doi: 10.1002/hipo.450040306. [DOI] [PubMed] [Google Scholar]
- 21.Eysel UT. Perilesional cortical dysfunction and reorganization. In: Freund H-J, Witte OW, editors. Advances in neurology. Lippincott-Raven; Philadelphia: 1997. pp. 573–591. [PubMed] [Google Scholar]
- 22.Feeney DM. From laboratory to clinic: noradrenergic enhancement of physical therapy for stroke or trauma patients. Adv Neurol. 1997;73:383–394. [PubMed] [Google Scholar]
- 23.Feeney DM, Walker AE. The prediction of posttraumatic epilepsy. A mathematical approach. Arch Neurol. 1979;36:8–12. doi: 10.1001/archneur.1979.00500370038005. [DOI] [PubMed] [Google Scholar]
- 24.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in rats. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 25.Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217:855–857. doi: 10.1126/science.7100929. [DOI] [PubMed] [Google Scholar]
- 26.Fricke RA, Prince DA. Electrophysiology of dentate gyrus granule cells. J Neurophysiol. 1984;51:195–209. doi: 10.1152/jn.1984.51.2.195. [DOI] [PubMed] [Google Scholar]
- 27.Frotscher M, Zimmer J. Lesion-induced mossy fibers to the molecular layer of the rat fascia dentata: identification of postsynaptic granule cells by the Golgi-EM technique. J Comp Neurol. 1983;215:299–311. doi: 10.1002/cne.902150306. [DOI] [PubMed] [Google Scholar]
- 28.Glantz SA. Biostatistics. McGraw-Hill; New York: 1997. [Google Scholar]
- 29.Golarai G, Sutula TP. Functional alterations in the dentate gyrus after induction of long-term potentiation, kindling, and mossy fiber sprouting. J Neurophysiol. 1996;75:343–353. doi: 10.1152/jn.1996.75.1.343. [DOI] [PubMed] [Google Scholar]
- 30.Golarai G, Cavazos JE, Sutula TP. Activation of the dentate gyrus by pentylenetetrazole evoked seizures induces mossy fiber synaptic reorganization. Brain Res. 1992;593:257–264. doi: 10.1016/0006-8993(92)91316-7. [DOI] [PubMed] [Google Scholar]
- 31.Golarai G, Feeney MD, Connor JA. Epileptiform responses in cortical slices in vitro after traumatic brain injury. Neurosci Abstr. 1999;25:980. [Google Scholar]
- 32.Grafstein BA. Some preliminary electrophysiological studies on chronically neuronally isolated cerebral cortex. Electroencephalogr Clin Neurophysiol. 1957;9:723–725. doi: 10.1016/0013-4694(57)90096-2. [DOI] [PubMed] [Google Scholar]
- 33.Hagemann G, Redecker C, Witte OW. Intact functional inhibition in the surround of experimentally induced focal cortical dysplasias. J Neurophysiol. 2000;84:600–603. doi: 10.1152/jn.2000.84.1.600. [DOI] [PubMed] [Google Scholar]
- 34.Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res [Suppl] 1992;7:273–280. [PubMed] [Google Scholar]
- 35.Hernandez TD, Schallert T. Seizures and recovery from experimental brain damage. Exp Neurol. 1988;102:318–324. doi: 10.1016/0014-4886(88)90226-9. [DOI] [PubMed] [Google Scholar]
- 36.Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- 37.Jacobs KM, Gutnick MJ, Prince DA. Hyperexcitability in a model of cortical maldevelopment. Cereb Cortex. 1996;6:514–523. doi: 10.1093/cercor/6.3.514. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs KM, Graber KD, Kharazia VN, Parada I, Prince DA. Postlesional epilepsy: the ultimate brain plasticity. Epilepsia. 2000;41[Suppl 6]:S153–161. doi: 10.1111/j.1528-1157.2000.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Schallert T. Use-dependent growth of pyramidal neurons after neocortical damage. J Neurosci. 1994;14:2140–2152. doi: 10.1523/JNEUROSCI.14-04-02140.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kolb B. Synaptic plasticity and the organization of behaviour after early and late brain injury. Can J Exp Psychol. 1999;53:62–76. doi: 10.1037/h0087300. [DOI] [PubMed] [Google Scholar]
- 41.Koo AH, LaRoque RL. Evaluation of head trauma by computed tomography. Radiology. 1977;123:345–350. doi: 10.1148/123.2.345. [DOI] [PubMed] [Google Scholar]
- 42.Kotti T, Tapiola T, Riekkinen PJS, Miettinen R. The calretinin-containing mossy cells survive excitotoxic insult in the gerbil dentate gyrus. Comparison of excitotoxicity-induced neuropathological changes in the gerbil and rat. Eur J Neurosci. 1996;8:2371–2378. doi: 10.1111/j.1460-9568.1996.tb01200.x. [DOI] [PubMed] [Google Scholar]
- 43.Krobert KF, Salazar RA, Sutton RL, Feeney DM. Temporal evolution of histopathology and unit activity in rat hippocampal CA3 region after focal cortical contusion. J Neurotrauma. 1992;9:64. [Google Scholar]
- 44.Laurberg S, Zimmer J. Lesion-induced sprouting of hippocampal mossy fiber collaterals to the fascia dentata in developing and adult rats. J Comp Neurol. 1981;200:433–459. doi: 10.1002/cne.902000310. [DOI] [PubMed] [Google Scholar]
- 45.Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch MW, Rutecki PA, Sutula TP. The effects of seizures on the brain. Curr Opin Neurol. 1996;9:97–102. doi: 10.1097/00019052-199604000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Lynch MW, Sayin U, Golarai G, Sutula T. NMDA receptor-dependent plasticity of granule cell spiking in the dentate gyrus of normal and epileptic rats. J Neurophysiol. 2000;84:2868–2879. doi: 10.1152/jn.2000.84.6.2868. [DOI] [PubMed] [Google Scholar]
- 48.Mason CR, Cooper RM. A permanent change in convulsive threshold in normal and brain-damaged rats with repeated small doses of pentylenetetrazole. Epilepsia. 1972;13:663–674. doi: 10.1111/j.1528-1157.1972.tb04401.x. [DOI] [PubMed] [Google Scholar]
- 49.Mitchell TW, Buckmaster PS, Hoover EA, Whalen LR, Dudek FE. Neuron loss and axon reorganization in the dentate gyrus of cats infected with the feline immunodeficiency virus. J Comp Neurol. 1999;411:563–577. [PubMed] [Google Scholar]
- 50.Mody I, Stanton PK, Heinemann U. Activation of N-methyl-d-aspartate receptors parallels changes in cellular and synaptic properties of dentate gyrus granule cells after kindling. J Neurophysiol. 1988;59:1033–1054. doi: 10.1152/jn.1988.59.3.1033. [DOI] [PubMed] [Google Scholar]
- 51.Nilsson P, Ronne-Engström E, Flink R, Ungerstedt U, Carlson H, Hillered L. Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Res. 1994;637:227–232. doi: 10.1016/0006-8993(94)91237-8. [DOI] [PubMed] [Google Scholar]
- 52.Okazaki MM, Evenson DA, Nadler JV. Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats: visualization after retrograde transport of biocytin. J Comp Neurol. 1995;352:515–534. doi: 10.1002/cne.903520404. [DOI] [PubMed] [Google Scholar]
- 53.Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877. [PubMed] [Google Scholar]
- 54.Osteen CL, Giza CC, Hovda DA. Lateral fluid percussion injury induces changes in NMDA receptor subunit composition that may reflect vulnerability or neuroplasticity. J Neurotrauma. 2000;17:958. [Google Scholar]
- 55.Parent JM, Lowenstein DH. Mossy fiber reorganization in the epileptic hippocampus. Curr Opin Neurol. 1997;10:103–109. doi: 10.1097/00019052-199704000-00006. [DOI] [PubMed] [Google Scholar]
- 56.Pierallini A, Pantano P, Fantozzi LM, Bonamini M, Vichi R, Zylberman R, Pisarri F, Cononnese C, Bozzao L. Correlation between MRI findings and long-term outcome in patients with severe brain trauma. Neuroradiology. 2000;42:860–867. doi: 10.1007/s002340000447. [DOI] [PubMed] [Google Scholar]
- 57.Prince DA. Epileptogenic neurons and circuits. Adv Neurol. 1999;79:665–684. [PubMed] [Google Scholar]
- 58.Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- 59.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 60.Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- 61.Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 1997;757:119–132. doi: 10.1016/s0006-8993(97)00170-4. [DOI] [PubMed] [Google Scholar]
- 62.Ribak CE, Seress L, Weber P, Epstein CM, Henry TR, Bakay RA. Alumina gel injections into the temporal lobe of rhesus monkeys cause complex partial seizures and morphological changes found in human temporal lobe epilepsy. J Comp Neurol. 1998;401:266–290. [PubMed] [Google Scholar]
- 63.Sacks J, Glaser NM. Changes in susceptibility to the convulsant action of metrazol. J Pharmacol. 1941;73:289–295. [Google Scholar]
- 64.Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- 65.Schallert T, Kozlowski DA, Humm JL, Cocke RR. Use-dependent structural events in recovery of function. Adv Neurol. 1997;73:229–238. [PubMed] [Google Scholar]
- 66.Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol Pathol. 2000;28:91–99. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- 67.Schmued LC, Albertson C, Slikker WJ. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 68.Seress L, Ribak CE. GABAergic cells in the dentate gyrus appear to be local circuit and projection neurons. Exp Brain Res. 1983;50:173–182. doi: 10.1007/BF00239181. [DOI] [PubMed] [Google Scholar]
- 69.Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- 70.Snedecor GW, Cochran WG. Statistical methods. Iowa State UP; Ames, IA: 1980. [Google Scholar]
- 71.Stanfield BB. Excessive intra- and supragranular mossy fibers in the dentate gyrus of tottering (tg/tg) mice. Brain Res. 1989;480:294–299. doi: 10.1016/0006-8993(89)90195-9. [DOI] [PubMed] [Google Scholar]
- 72.Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- 73.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 74.Sutula T, Cavazos J, Golarai G. Alteration of long-lasting structural and functional effects of kainic acid in the hippocampus by brief treatment with phenobarbital. J Neurosci. 1992a;12:4173–4187. doi: 10.1523/JNEUROSCI.12-11-04173.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sutula TP, Golarai G, Cavazos J. Assessing the functional significance of mossy fiber sprouting. Epilepsy Res [Suppl] 1992b;7:251–259. [PubMed] [Google Scholar]
- 76.Sutula T, Koch J, Golarai G, Watanabe Y, McNamara JO. NMDA receptor dependence of kindling and mossy fiber sprouting: evidence that the NMDA receptor regulates patterning of hippocampal circuits in the adult brain. J Neurosci. 1996;16:7398–7406. doi: 10.1523/JNEUROSCI.16-22-07398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sutula T, Zhang P, Lynch M, Sayin U, Golarai G, Rod R. Synaptic and axonal remodeling of mossy fibers in the hilus and supragranular region of the dentate gyrus in kainate-treated rats. J Comp Neurol. 1998;390:578–594. doi: 10.1002/(sici)1096-9861(19980126)390:4<578::aid-cne9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 78.Tate DF, Bigler ED. Fornix and hippocampus atrophy in traumatic brain injury. Learn Mem. 2000;7:442–446. doi: 10.1101/lm.33000. [DOI] [PubMed] [Google Scholar]
- 79.Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weisend MP, Feeney DM. The relationship between traumatic brain injury-induced changes in brain temperature, behavioral and anatomical outcome. J Neurosurg. 1994;80:120–132. doi: 10.3171/jns.1994.80.1.0120. [DOI] [PubMed] [Google Scholar]
- 83.Wuarin JP, Dudek FE. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J Neurosci. 1996;16:4438–4448. doi: 10.1523/JNEUROSCI.16-14-04438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang DN, Townsend JC, Ilsen PF, Bright DC, Welton TH. Traumatic porencephalic cyst of the brain. J Am Optom Assoc. 1997;68:519–526. [PubMed] [Google Scholar]
- 85.Yoshii N, Samejima H, Sakiyama R, Mizokami T. Posttraumatic epilepsy and CT scan. Neuroradiology. 1978;16:311–313. doi: 10.1007/BF00395285. [DOI] [PubMed] [Google Scholar]