

Abstract
A developmental reduction in the radial transmission of synaptic activity has been proposed to underlie the end of the critical period for experience-dependent modification in layers II/III of the visual cortex. Using paired-pulse stimulation, we investigated in visual cortical slices how the propagation of synaptic activity to the superficial layers changes during development and how this process is affected by sensory experience. The results can be summarized as follows. (1) Layers II/III responses to repetitive stimulation of the white matter become increasingly depressed between the third and sixth week of postnatal development, a time course that parallels the end of the critical period. (2) Paired-pulse depression is reduced after dark rearing and also by blocking inhibitory synaptic transmission. (3) Paired-pulse depression and its regulation by age and sensory experience is more pronounced when stimulation is applied to the white matter than when applied to layer IV. Together, these results are consistent with the idea that the maturation of intracortical inhibition reduces the capability of the cortex to relay incoming high-frequency patterns of activity to the supragranular layers.
Keywords: critical period, plasticity, GABA, paired-pulse depression, neocortex, layer IV
Sensory input is crucial for the maturation of connectivity within visual cortex, but only during an early postnatal period (Hubel and Wiesel, 1970). In rodents, the critical period peaks by the third postnatal week. At this age, a simple manipulation, such as monocular deprivation, shifts visual cortical responsiveness toward the nondeprived eye. As cortical circuits mature, cortical plasticity diminishes rapidly. Thus, by the fifth week of age, monocular deprivation has hardly any effect (Fagiolini et al., 1994; Gordon et al., 1996). The factors that restrict experience-dependent plasticity to a short critical period remain unclear.
It is believed that visual cortical plasticity involves some form of use-dependent synaptic modification (Bear et al., 1987; Singer, 1995;Katz and Shatz, 1996). Intracortical inhibition is ideally poised to restrict these activity-dependent mechanisms to a short critical period. By reducing excitation, synaptic inhibition can limit activity-dependent plasticity of synaptic transmission. For example in neocortex in vitro, recruitment of GABAergic inhibition can limit induction of long-term potentiation (LTP), currently the best understood model of synaptic modification (Artola and Singer, 1987;Kirkwood and Bear, 1994). Also, anatomical (Blue and Parnavelas, 1983;Guo et al., 1997) and physiological (Komatsu, 1983; Luhmann and Prince, 1991) findings indicate that synaptic inhibition matures later than excitatory transmission within neocortex. This mismatch in the maturation of excitation and inhibition may provide a window of opportunity, early in postnatal life, for activity-dependent plasticity to occur. Consistent with this idea, dark rearing, which prolongs the critical period (Cynader and Mitchel, 1980; Mower et al., 1985; Guire et al., 1999), delays the anatomical maturation of inhibition within visual cortex (Blue and Parnavelas, 1983).
We have proposed that inhibitory circuits gate the induction of plasticity in superficial cortical layers by constraining the types of activity patterns that reach this region (Kirkwood and Bear, 1994;Kirkwood et al., 1995). Synapses in layers II/III, which can be modified by experience during a well defined critical period (Daw, 1994), receive retinal input primarily via excitatory stellate cells in layer IV that are the major target of thalamocortical axons conveying visual activity. We hypothesized that recruitment of GABAergic circuits inhibits stellate cells in layer IV and hence reduces the transfer of geniculocortical activity to layers II/III. According to this idea, early in development, when inhibitory circuits are not fully formed, patterned activity is faithfully relayed by layer IV to layers II/III, where it can trigger plasticity. Later on, as the efficacy of inhibition increases, activity is filtered out at the level of layer IV, preventing activity-dependent modification of layers II/III synapses. Thus, although layers II/III synapses remain modifiable for a considerable time, the activity requirements for plasticity may be reached only during a short critical period. To further investigate this hypothesis, we examined responses of layers II/III to paired-pulse stimulation (PPS) applied to the white matter and layer IV. Our results indicate that during development, coincident with the decline of the critical period, there is an increasing attenuation in the response of layers II/III to repetitive activation of the white matter.
MATERIALS AND METHODS
Slice preparation. Coronal slices (300–400 μm) from the visual cortex of 3- to 8-week-old Long–Evans rats were prepared as described previously (Kirkwood and Bear, 1994). Briefly, after sectioning in ice-cold oxygenated 95% O2/5% CO2 dissection buffer containing (in mm):sucrose 212.7, KCl 5, NaH2PO4 1.25, MgCl2 3, CaCl2 1, NaHCO3 26, dextrose 10, and kynurenate 10, slices were transferred to a storage chamber containing normal artificial CSF (ACSF) for at least 1 hr before recording. Normal ACSF is similar to the dissection ACSF except that sucrose is replaced by 124 mm NaCl, MgCl2 is lowered to 1 mm, CaCl2 is raised to 2 mm, and kynurenate is omitted. Most of the experiments were performed in an interface chamber that was perfused continuously at a rate of 2 ml/min with 30°C ACSF saturated with 95% O2 and 5% CO2.
Slice stimulation. Synaptic responses were evoked with 0.2 msec pulses of 15–300 μA amplitude delivered using a bipolar stimulating electrode (FHC, 200 μm diameter). The visual cortex was stimulated at one of two sites: (1) at the border of the white matter and layer VI, termed “white matter stimulation,” and (2) in the middle of the cortical thickness, approximately equidistant from the pia and the white matter, termed “layer IV stimulation.” The border of layers IV and V in area 17 (OC1) lies halfway between the pia and the white matter (Zilles, 1985). Thus, the site of middle layer stimulation encompassed lower layer IV and superficial layer V, but excluded layer III.
PPS was delivered every 15 sec. In every experiment the interstimulus interval (ISI) was varied systematically from 20 msec up to 2560 msec, in increments that doubled the ISI of the previous stimulus pair. Thus, a complete series consisted of a sequence of paired pulses with ISIs of 20, 40, 80, 160, 320, 640, 1280, and 2560 msec. To compute paired pulse depression (PPD), the amplitude of the response to the second pulse of the pair was divided by the amplitude of the response to the first pulse in the pair. In each slice, at least three consecutive ISI series were run and averaged. For analysis we considered only data from experiments in which the response to the first pulse was stable (<3% variation over the course of 10 min). In pharmacological experiments, comparisons were made between measurements taken immediately before and 30 min after drug application. In control experiments, no net changes in PPD were detected over this period of time (F(7,16) = 0.72; p = 0.9994). Statistical significance was assessed using two-way repeated measures ANOVA followed by the Fisher post hoc test.
Slice recording. Microelectrodes were filled with ACSF (1–2 MΩ) for extracellular recording. The amplitude of the maximum negative-going field potential (FP) recorded in layer III was used as a measure of the evoked population excitatory synaptic response. Changes in the amplitude of the negative-going FP reflect changes in the magnitude of a synaptic current sink in this region (Mitzdorf, 1985;Aizenman et al., 1996) and correlate with changes in the initial slope of EPSPs recorded intracellularly in layer III neurons (Kirkwood and Bear, 1994). Intracellular recordings were performed with microelectrodes filled with 3 m K Acetate (80–120 MΩ). Only cells with resting membrane potentials more negative than −70 mV and input resistances >20 MΩ were studied. For whole-cell voltage clamp, the cells were visually identified with an infrared differential interference contrast Zeiss microscope. The patch pipettes (2–4 MΩ) were filled with internal solution consisting of (in mm): 130 CsF, 8 KCl, 10 EGTA, 10 HEPES, 2 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), 10 QX-314, pH 7.4, 275–285 mOsm. The junction potential (typically <5 mV) was compensated. Only cells having a resting membrane potential more negative than −65 mV, an access resistance <20 MΩ (8–18 MΩ, compensated at 80%), and an input resistance >100 MΩ (130–410 MΩ) were studied. Under these recording conditions, the IPSCs disappeared within 10–20 min, depending on the access resistance (see Fig. 2B). All drugs were purchased from Sigma (St. Louis, MO)/RBI (Natick, MA).
Fig. 2.
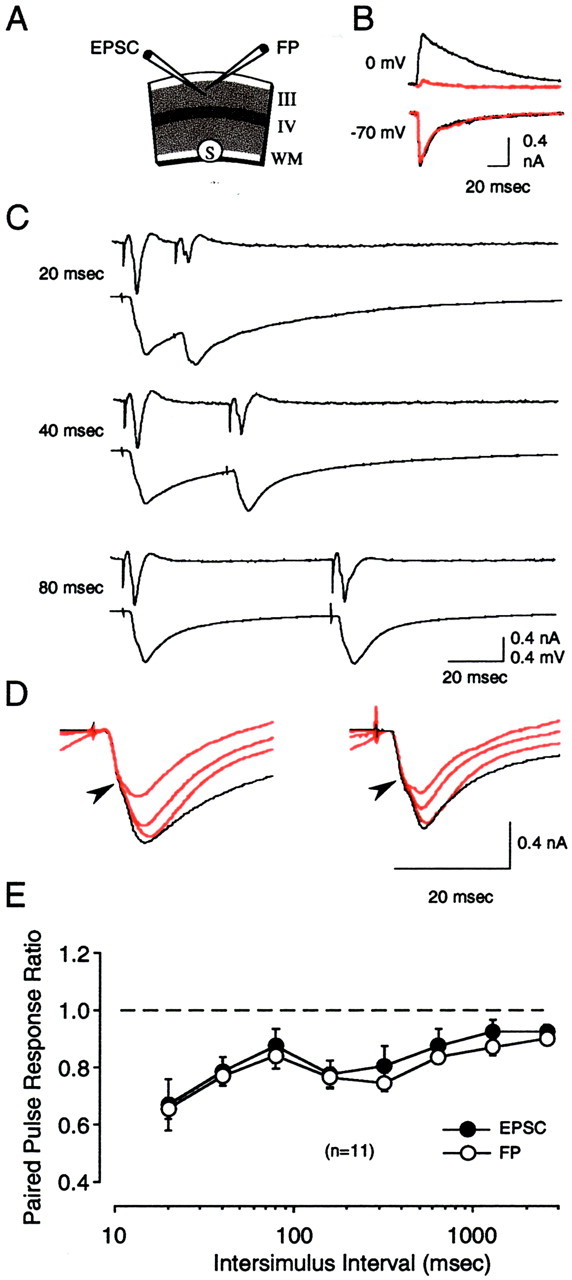
Paired-pulse stimulation produces comparable depression in both EPSCs and FPs. A, Diagram of the experimental conditions. Stimulation was applied in the white matter or in layer IV while isolated EPSCs and FPs were recorded simultaneously in layers II/III. B, IPSCs (recorded at 0 mV) but not EPSCs (recorded at −70 mV) were rapidly blocked in our recording conditions. Traces are synaptic currents immediately (black) and 10 min after (red) going into whole-cell mode. C, Results from a representative experiment showing responses to a series of paired pulses of increasing interstimulus interval (from 20 to 80 msec) applied to the white matter. For each ISI, the average of four consecutive FPs is shown on the top, and the corresponding EPSCs are shown on thebottom. The amplitude of the second response divided by the amplitude of the first response was used to calculate paired-pulse interactions. D, In cells showing complex responses, paired-pulse stimulation preferentially affected the later component (indicated after arrowhead) of the EPSC.Superimposed traces are responses to the first stimulation pulse (black) and the second stimulation pulse (red). For clarity, only responses to short ISIs (20, 40, and 80 msec) are shown. Traces on theright are from the same cell as in C.E, Average results of 10 experiments (with stimulation applied to layer IV) indicate a similar degree of paired-pulse depression in the FP (○) and in the EPSC (●).
Unless stated otherwise, in vitro recordings were performed on visual cortical slices prepared from 5- to 6-week-old rats. Pulsed stimulation was applied to the white matter at an intensity that evoked a response magnitude ∼50% of maximum.
In vivo recordings. Adult (p > 90) male rats were initially anesthetized with Nembutal (65 mg/kg, i.p.) supplemented with Isofluorene (2.0–3.0% in 100% oxygen), tracheotomized, and then placed in a stereotaxic frame. Animals were artificially ventilated (100% O2, 40 bpm) and maintained at 37 ± 0.5°C. Anesthesia was maintained by continuous administration of Nembutal (6–10 mg/hr) through an intraperitoneal catheter. The eyes were covered throughout experimentation, except during positioning of a stimulating electrode into the dorsal lateral geniculate nucleus of the thalamus (dLGN).
A monopolar recording electrode (<1.0 MΩ) was positioned in the primary visual cortex (7.0 mm posterior to bregma; 3.0–4.0 mm lateral to the midline). A screw inserted into the bone overlying the cerebellum served as recording ground. A concentric bipolar electrode (FHC; 75 μm diameter) was positioned 3.8 mm posterior, 3.3 mm lateral to stimulate the dLGN ipsilateral to the visual cortex in which recordings were obtained. To aid in the positioning of the stimulating electrode in the dLGN, visually driven multiunit activity was monitored from this electrode as it was being tracked down through neocortex and overlying hippocampus. The final depth of the stimulating electrode tip was within 100–200 μm of first encountering visually responsive neurons. Histological analysis demonstrated that the stimulating electrode tip was typically positioned within the first 200 μm of the dorsal surface of the dLGN.
Field potentials were elicited with 0.2 msec, 0.3–0.8 mA stimulation pulses, filtered at 0.1 Hz and 3.0 kHz, digitized at 160 kHz, and stored and analyzed using Experimenter's Workbench (DataWave Technologies; Longmont, CO). Before the beginning of each experiment, a full input–output series was performed, and a stimulation intensity yielding an FP amplitude 50–60% of maximum was used for the remainder of experimentation.
Current source density analysis. A roving electrode was tracked down in 100 μm steps from cortical surface to a depth of 2.2–2.5 mm. At each recording depth 10 paired-pulsed stimuli (40 msec ISI) were delivered to the dLGN. After completion of the recording sessions, electrolytic lesions were made along the track to allow for verification of the location and track of the roving electrode. The CSD profile was constructed as described previously (Mitzdorf, 1985;Aizenman et al., 1996), using a spatial differentiation grid of 200 μm.
RESULTS
Probing the gate with paired-pulse stimulation
In visual cortical slices, the induction of LTP in layers II/III depends critically on the site of stimulation and the age of the animal. High-frequency stimulation of the white matter (WM) reliably induces LTP in layers II/III (WM → III LTP) only in slices of immature animals (<4 weeks old), whereas stimulation of layer IV produces LTP (IV → III LTP) throughout postnatal life (Kirkwood et al., 1995). According to the plasticity gate hypothesis, the developmental decrease in the magnitude of WM → III LTP and experience-dependent plasticity results from the maturation of intracortical inhibition, which prevents certain activity patterns from reaching modifiable synapses in layers II/III. To investigate developmental changes in the propagation of neural activity to the upper layers of the visual cortex, we studied the response of layers II/III to PPS. This type of stimulation has been widely used to characterize the frequency response of cortical circuitry in slices (Luhmann and Prince, 1990; Ramoa and Sur, 1996; Castro-Alamancos and Connors, 1997; Buonomano and Merzenich, 1998).
One potential complication in the interpretation of FPs evoked by WM PPS in slices, however, is the possible contribution of antidromically activated cortical circuits, particularly the strong collateral projections of layers V and VI neurons. Previously we used current source density (CSD) analysis to study the spatiotemporal patterns of activity evoked by PPS of the WM (Aizenman et al., 1996). In the present study we repeated this analysis in vivo under conditions in which antidromic cortical activation is minimal, with the aim of assessing the validity of the approach in slices.
In this series of experiments a recording electrode was tracked ventrally through Oc1 in 100 μm steps from the dural surface to well below white matter (≥2000 μm ventral). After each 100 μm advancement of the recording electrode, FPs (n = 10) in response to PPS (40 msec ISI) applied to the dLGN were collected and subjected to CSD analysis. A representative example of the FP profile recorded through Oc1 in response to the first stimulus pulse is presented in Figure 1A. The FP recorded through the depth of Oc1 was consistently observed as a primarily negative-going potential, having a maximum negativity and shortest latency at a depth corresponding to layer IV and deep layer III (∼600–800 μm ventral to the dural surface). The resultant CSD profile for the FP data is presented in Figure 1B. Initial short-latency (∼2.0–5.0 msec) current sinks were observed in layer IV and lower (deep) layer III, with corresponding sources located in adjacent supragranular and infragranular layers. These initial current sinks were followed by sinks in supragranular (depth ∼200–500 μm) and infragranular (depth ∼900–1100 μm) layers, with the latency of these sinks (∼4.0–10.0 msec) increasing with increasing distance from layer IV/deep layer III. This laminar pattern and temporal order of transmembrane currents was consistent across animals (n = 3) and is consistent with previous reports (Mitzdorf, 1985; Vaknin et al., 1988; Brankack et al., 1990; Ferster, 1990; Kenan-Vaknin and Teyler, 1994). As in previous studies performed under similar conditions, there was no indication that layer VI neurons were antidromically activated by this stimulation (Swadlow and Weyand, 1981; Ferster and Lindstrom, 1983; Bourassa and Deschenes, 1995).
Fig. 1.
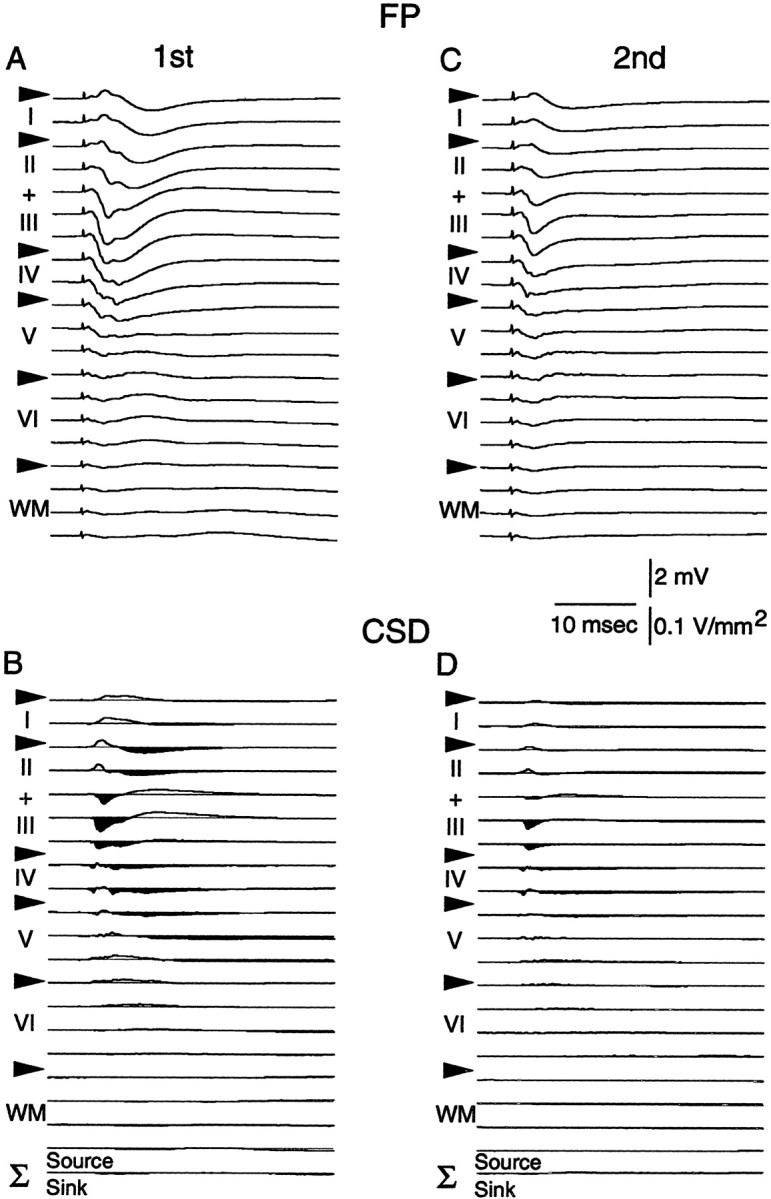
Paired-pulse depression of layers II/III responses evoked by stimulation of the dLGN in vivo.A, C, Field potential profiles recordedin vivo at different cortical depths. A, FPs recorded in response to the first stimulus pulse; C, FPs obtained in response to the second pulse (40 msec ISI).B, D, CSD profiles obtained with the FPs depicted in A and C, respectively. Notice the marked depression of the FPs recorded in upper layers II/III as well as the corresponding decrease in current sinks (downward and in black) in response to the second stimulus pulse. The sum of the CSD profiles is indicated at the bottom in B and D.Arrowheads mark the boundaries of the cortical layers.
Figure 1, C and D, presents the FP and CSD profile obtained in response to the second stimulus pulse. A decrease in the amplitude of the negative-going component of the FP recorded from the cortical surface through to layer IV is apparent in response to the second stimulus pulse (compare FP profiles in A withC). A corresponding decrease in the amplitude of current sinks located in supragranular layers was also observed (depth ∼200–500 μm; response to pulse 2/pulse 1 = 0.50 ± 0.3;n = 3). The qualitative as well as quantitative changes in current sink amplitude observed in the present in vivoexperiments are remarkably similar to those observed when PPS is applied to the WM in vitro (Aizenman et al., 1996, their Fig. 5). This comparison suggests that WM PPS is a useful technique for studying the responses of cortical afferents to orthodromic activation.
Using slices of visual cortex, we performed an extensive characterization of the responses evoked by PPS in which we systematically varied the ISI, stimulation intensity, and stimulation site as well as the postnatal age and visual experience of the animals. Because of the large number of experiments required, synaptic responses were monitored extracellularly as field potentials (FPs), which are relatively easy to record. FPs have been used successfully on many occasions to study synaptic changes; nevertheless, it was desirable to confirm that the effects of PPS on the FPs do correlate with changes in synaptic responses measured intracellularly. Therefore, we conducted a series of experiments in which the responses to PPS were monitored simultaneously using extracellular FPs and isolated EPSCs in visually identified layers II/III pyramidal cells. EPSCs were isolated by recording at −70 mV and omitting ATP and GTP and including DIDS and CsF− in the intracellular recording electrode to block IPSCs (Nelson et al., 1994). In these experiments the stimulation intensity was adjusted to evoke a half-maximal field response, and paired-pulse stimulation was delivered at varying ISIs from 20 to 2560 msec (see Materials and Methods). The results of these experiments are shown in Figure 2. Unlike in hippocampus, where PPS produces a facilitation of the response evoked by the second stimulus pulse, in neocortex PPS of the white matter typically produces a depression of the second response. This PPD was observed in both the FP and EPSC, and it was more pronounced at shorter ISIs. Figure 2C provides an example of the PPD observed in the layers II/III FP and EPSC in response to paired-pulse stimulation (20–80 msec ISI) of the WM. In this and two other experiments, the EPSC had a complex shape with two distinguishable components, possibly reflecting monosynaptic and polysynaptic (Fig.2D, arrowhead) activity. In these three cases the later phase of the EPSC was most noticeably affected by paired-pulse stimulation (Fig. 2D), which is consistent with our previous observation that PPS preferentially affects late polysynaptic inputs onto layers II/III neurons (Aizenman et al., 1996).
A quantitative comparison of the degree of PPD observed in the FP and EPSC was done with data obtained with layer IV stimulation, which evokes similar although smaller PPD as compared with WM (see below). The degree of depression was quantified as the ratio of the second response amplitude to the first response amplitude. As shown in Figure2E, the FP and the EPSC showed comparable degrees of paired-pulse depression at all ISIs (F(8,168) = 0.214; p = 0.988; n = 10), indicating that the depression of the FP response truly reflects a reduction in synaptic activation. Qualitatively similar results were also observed with white matter stimulation (n = 3) (Fig. 2C). Thus, we are confident that measurement of FP amplitude is an adequate approach for studying paired-pulse interactions.
In a final set of control experiments, we asked whether changes in axon excitability contribute to paired-pulse depression. In these experiments we measured the fiber volley (FV) in conjunction with FP amplitudes. As shown in Figure 3, paired-pulse stimulation of the white matter produced a substantial depression of the FPs (F(7,136) = 8.139; p < 0.001) but not the associated FV (F(7,136) = 0.238; p = 0.975). We further confirmed this finding by measuring the FV under excitatory transmission blockade with 10 mmkynurenic acid. Again PPS had no effect on the FV (F(7,112) = 0.991; p = 0.442), whereas the FPs were once again depressed when, before kynurenic acid administration, recordings were performed in normal ACSF (F(7,112) = 11.533; p< 0.001). Together these experiments indicate that paired-pulse depression is not caused by a reduction in axon excitability.
Fig. 3.
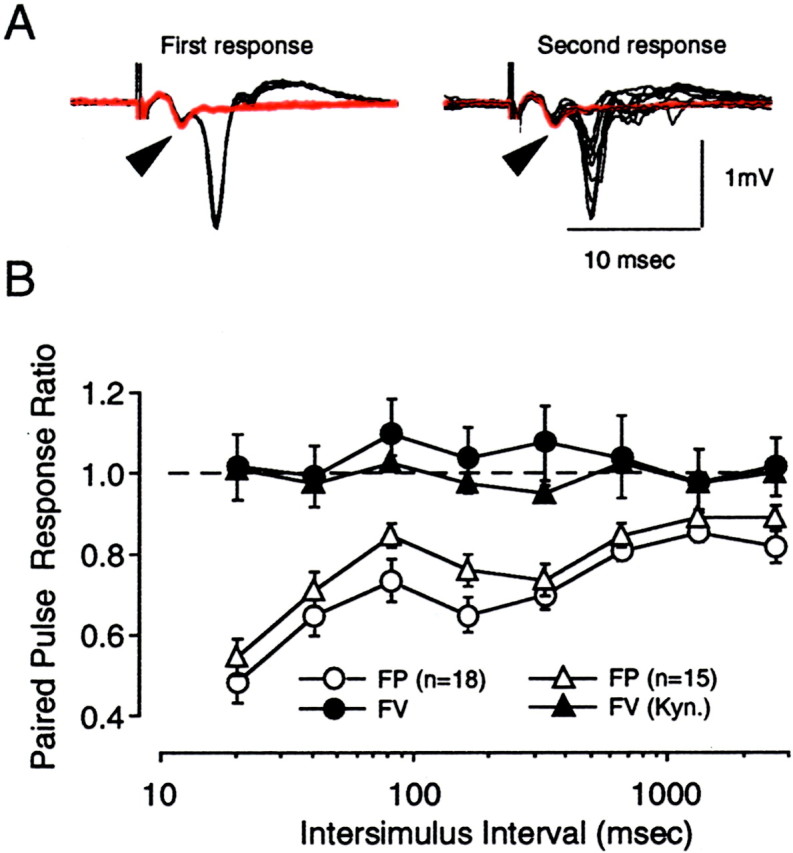
Paired-pulse depression evoked by white matter stimulation is not accompanied by a change in fiber volley amplitude.A, Superimposed traces are responses to the first stimulation pulse (left) and the second stimulation pulse (right) for a complete ISI series (from 20 to 2560 msec). Traces were recorded before (black) and in the presence of 10 mmkynurenic acid (red). Arrowheads indicate the fiber volley. B, Graph summarizing the effects of PPS on the amplitude of the FP (open symbols) and on the FV (filled symbols). The FP and FV were recorded either simultaneously (circles; n = 18) or before and after bath application of kynurenic acid (triangles; n = 15). Stimulation was applied to the white matter.
Involvement of inhibition in PPD evoked with white matter stimulation
Effects of GABA antagonists on PPD
Previous studies suggest an involvement of GABAergic inhibition in cortical PPD (Ramoa and Sur, 1996; Castro-Alamancos and Connors, 1997). In cortex, GABA activates two processes, a fast IPSC mediated by GABAa receptors and a slower IPSC mediated by GABAb receptors. The GABAa response typically peaks at ∼20–30 msec, whereas the GABAb response peaks considerably later, at hundreds of milliseconds (Alger and Nicoll, 1982). We investigated the contribution of these two processes in PPD by studying the effect of selective GABA receptor antagonists on responses evoked using a large range of ISIs (from 20 to 2560 msec). To block GABAa receptors we used the antagonist picrotoxin, which was bath applied at a subsaturating concentration (0.2 μm) to avoid the development of paroxismal discharges. Even at this concentration, picrotoxin increased the FP response. To ensure that the FPs evoked before and after picrotoxin application were matched in size, and to prevent excessive excitation, we reduced stimulation intensity. We studied only those cases in which the final stimulus intensity was >150 μA, in the range at which PPD is independent of stimulus intensity (see Fig. 5). The results of 17 of these experiments are shown in Figure 4A. In control conditions, PPS clearly depressed the second response at all the ISIs tested. The degree of depression was maximal at the shortest ISI (20 msec) and minimal at the longest ISI (2560 msec). Application of picrotoxin consistently reduced the degree of PPD, particularly at the shorter ISIs. A two-factor repeated measures ANOVA revealed a significant interaction between drug and ISI (F(7,32) = 8.124; p = 0.0001), and post hoc tests confirmed significant differences (p < 0.001) at ISIs of 40 and 80 msec (Fig. 4A). Application of the GABAb antagonist saclofen (150 μm) also reduced PPD (F(7,16) = 2.977; p < 0.0067) (Figure 4B), but at longer ISIs. Post hoc analysis demonstrated a statistically significant decrease in PPD at ISIs of 80, 160, and 320 msec. In addition to studying the effects of GABAergic antagonism, we also investigated the effects of increasing the efficacy of inhibition using the benzodiazepine agonist diazepam (40 μm). As shown in Figure4C, diazepam, which enhances the evoked GABAa response, induced a small yet consistent increase in PPD (F(7,34) = 2.31; p = 0.036) at shorter ISIs (20 and 40 msec). Interestingly, picrotoxin, saclofen, and diazepam had no significant effect on PPD evoked at the longest ISI used (2560 msec).
Fig. 5.
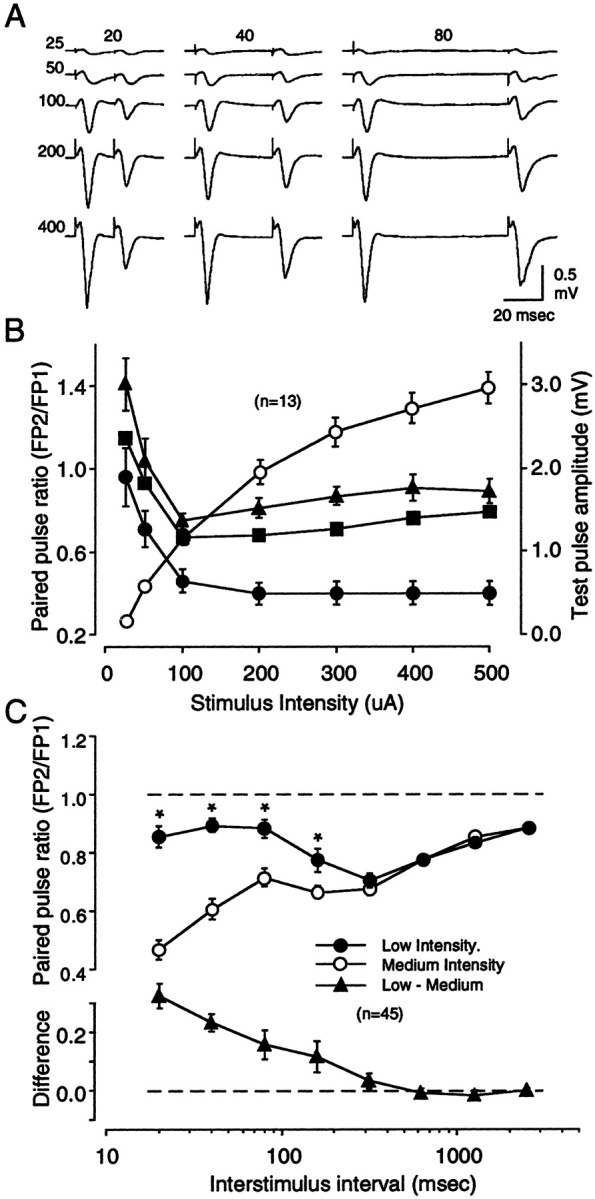
Paired-pulse depression depends on stimulus intensity. FPs were recorded in layers II/III in response to WM stimulation. A, Results from a representative experiment showing FPs in response to a series of paired pulses of increasing intensity (25, 50, 100, 200, and 400 μA; orderedvertically) and increasing ISIs (20, 40, and 80 msec; ordered horizontally). Traces are averages of four consecutive responses. B, Summary of 15 experiments performed as in A. The graphshows the relationship between stimulus intensity and paired-pulse response ratio for the three ISIs tested (left y-axis: ● = 20 msec, ▪ = 40 msec, ▴ = 80 msec). Also shown is the relationship between stimulus intensity and the amplitude of the first response (right y-axis: ○). C, The effects of stimulation intensity on PPD are observed only at shorter ISIs. The data in the top panel compare the average response to PPS of the white matter using low and medium stimulation intensities (see Results). In each slice, a complete ISI series (from 20 to 2560 msec) was first delivered at low stimulation intensity (●) and then delivered at medium stimulation intensity (○). ISIs at which the effect of stimulation intensity was significant (0.01 level; see Results) are labeled with an asterisk. The difference in paired-pulse ratio between the two stimulation intensities was computed for each ISI in each experiment. The average results are shown in the bottom panel.
Fig. 4.

Involvement of inhibitory circuitry in paired-pulse depression evoked with white matter stimulation.A, Bath application of the GABAa antagonist picrotoxin (PTX; 0.2 μm) reduces paired-pulse depression at short ISIs. ISIs at which the effect of PTX was significant (0.01 level; see Results) are labeled with anasterisk. B, Bath application of the GABAb antagonist saclofen (150 μm) reduces paired-pulse depression at intermediate ISIs. C, Bath application of the benzodiazepine agonist Diazepam (40 μm) enhances paired-pulse depression at short ISIs. The average difference in paired-pulse response ratio between control and in the presence of drug is plotted in the bottom panels of A–C. Notice the change in axis scale in A andB. D, Simultaneous intracellular and extracellular recordings reveal a strong similarity between time course of the IPSP and paired-pulse depression of the FP. Top trace, Synaptic response recorded at a depolarized level (−65 mV) reveals an n-shaped hyperpolarizing IPSP. Thistrace is replotted in the bottom panelusing the same logarithmic time scale as the ratio for paired-pulse depression of FPs (solid line).
An involvement of synaptic inhibition in PPD was suggested at the outset by the peculiar n-shape of the PPD profiles, which resembles the time course of the intracellularly recorded IPSP. Like the PPD profiles, a typical IPSP is biphasic, with a fast component mediated by GABAa receptors that peaks early (at ∼20 msec) followed by a later component mediated by GABAb receptors that peaks at ∼150 msec. The similarity in time course between PPD and the IPSC was assessed by performing simultaneous intracellular and extracellular recordings in layers II/III. As is shown in Figure 4D, both the IPSP and the PPD profiles are n-shaped, with peaks and valleys occurring at comparable times.
Effects of stimulation intensity on PPD
Because PPD was evoked using electrical stimulation, it was crucial to determine how PPD varied with stimulus intensity. The effects of increasing stimulation intensity on the response to paired pulses (ISIs of 20, 40, and 80 msec) applied to the white matter is shown in Figure 5. No PPD was observed at low stimulation intensities. On the contrary, there was even some facilitation in the response to the 40 and 80 msec pairs. As stimulation intensity was increased, however, depression developed and reached a stable level at a stimulation intensity that evoked a half-maximal FP (Fig. 5B).
In neocortical slices, IPSPs typically have a higher threshold than EPSPs (Sutor and Hablitz, 1989; Hwa and Avoli, 1992; Ling and Benardo, 1995) (although see Porter et al., 2001). Thus, the enhanced PPD observed at higher stimulus intensities might reflect the recruitment of GABAergic circuits. If so, we would expect greater effects of stimulus intensity at relatively short ISIs, a time in which inhibitory mechanisms are most efficacious. To investigate this possibility, we set out to determine the range of ISIs at which stimulation intensity preferentially affects PPD. In these experiments we used two stimulation intensities across a wide range of ISIs (from 20 to 2560 msec). The first series of ISIs was run at a stimulation intensity that elicited an FP response approximately one-fourth of maximum (0.55 ± 0.02 mV, low intensity); a second series was then run at a stimulus intensity that evoked an FP response between one-half and two-thirds of maximum (1.66 ± 0.1 mV, medium intensity). The PPD profiles thus obtained are shown in Figure 5C. At both intensities, PPD was most pronounced at short ISIs, yet still observed at the longest ISI (2560 msec). As expected, a significantly larger degree of depression was obtained with medium stimulation intensity (F(7,28) = 6.65; p < 0.001). However, the effect of stimulation intensity was clearly restricted to the shorter end of the ISI range. Indeed, a post hoc analysis indicated that a significant difference (p < 0.01) occurred at ISIs <160 msec. Thus, it appears that the degree of PPD strongly depends on the stimulation intensity, but only for stimulus pairs with short ISIs. These results are consistent with the idea that the activation of inhibitory circuits is a major determinant of PPD observed at short ISIs.
White matter stimulation induces larger PPD than layer IV stimulation
We have argued that the critical difference in the ability to induce LTP between WM and layer IV stimulation results from the activation of different patterns of inhibitory synapses after stimulation of these two sites (Aizenman et al., 1996). It was of interest, therefore, to compare the degree of PPD evoked from these two sites. Experiments were conducted in which PPS was first applied to the white matter and then to layer IV in the same slice (Fig.6). WM and layer IV stimulation evoked maximal responses of similar amplitude (WM, 2.83 ± 0.15 mV; layer IV, 3.12 ± 0.16 mV). In each experiment, the stimulus intensity was finely tuned to match the amplitude of the responses evoked with WM with those evoked with layer IV stimulation (1.36 ± 0.05 and 1.36 ± 0.06 mV, respectively). Despite this match in FP amplitude, PPD was consistently and significantly larger (F(7,72) = 11.24; p < 0.0001) when PPS was applied to the white matter than when PPS was applied to layer IV. These differences were restricted to the short end of the ISI spectrum (<360 msec). The differences between WM and layer IV stimulation were reduced when the stimulation intensity was decreased (Fig. 6B) (F(7,48) = 1.514; p = 0.16). These results indicate that the site of cortical stimulation is a strong determinant of the degree of depression obtained at short ISIs. These results are also consistent with the notion that WM and layer IV stimulation recruit inhibition differently.
Fig. 6.
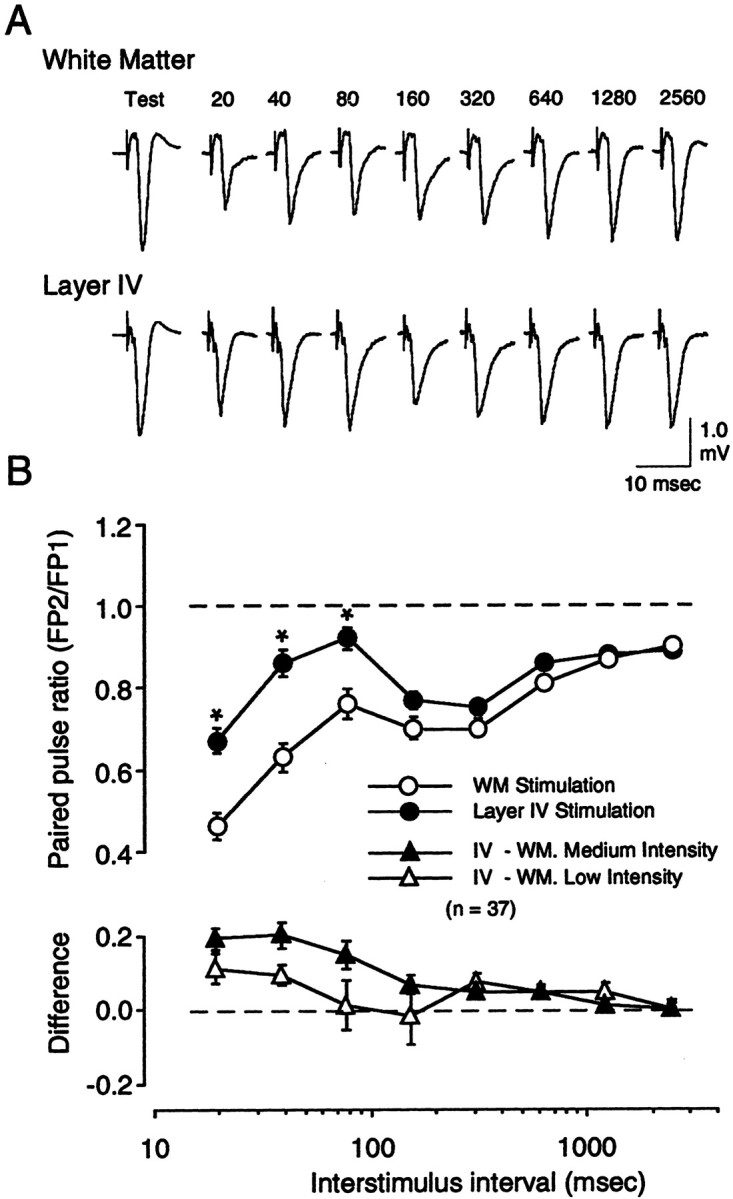
White matter stimulation produces larger paired-pulse depression than layer IV stimulation. A, Example of responses evoked by a complete ISI series (20–2560 msec) applied at medium stimulation intensity first to the white matter (top traces) and subsequently to layer IV (bottom traces). In each case only the second response is shown.B, Average results of 37 experiments performed as inA (the results of 30 of these experiments are also included in Fig. 3). Plotted in the top panel are the paired-pulse response ratios obtained with varying ISIs for both white matter stimulation (○) and layer IV stimulation (●). ISIs at which the effect of stimulation site was significant (0.01 level; see Results) are labeled with an asterisk. The difference in paired-pulse ratio between the two stimulation sites was computed for each ISI in each experiment. The average results are shown in the bottom panel.
Developmental changes in PPD
According to the plasticity gate hypothesis, maturation of inhibition reduces the ability of cortical circuits to relay high-frequency activity to layers II/III and produces a developmental reduction in WM → III LTP. Consistent with this idea, it has been reported that PPD increases in the first postnatal weeks (Ramoa and Sur, 1996). It was of interest therefore to determine whether developmental changes in PPD correlate with changes in WM → III LTP. Thus, we performed an extensive examination of the effects of paired-pulse stimulation applied to the white matter and layer IV at two stimulation intensities (low and medium) in slices prepared from 3-, 4-, 5-, 6-, and 8-week-old rats. Time points of particular interest are the peak of the critical period, which occurs around the third and fourth week of age, and the end of the critical period, which occurs after the fifth week of postnatal life. As shown in Figure7A, white matter stimulation at medium intensity evokes substantially larger PPD in slices from 6-week-old animals than in slices from 4-week-old animals (F(7,30) = 9.52; p = 0.001). The differences between the two PPD profiles were statistically significant at short ISIs: 40, 80, and 160 msec. In contrast to white matter stimulation, PPD evoked with layer IV stimulation was less affected by the age of the animal (Fig. 7B), although the small differences between the two PPD profiles were found to be statistically significant (F(7,28) = 2.8; p = 0.01). At low stimulation intensity, on the other hand, the effect of age was much less pronounced for both white matter stimulation (F(7,30) = 0.879;p = 0.524) and layer IV stimulation (F(7,28) = 0.516; p = 0.822) (data not shown).
Fig. 7.

Postnatal changes in paired-pulse depression. The effects of PPS applied to the white matter and to layer IV were measured for the first 8 weeks of age. A,B, Comparison of the PPD profiles obtained at two different ages (4 and 6 weeks) with white matter stimulation (A) and layer IV stimulation (B). C, Time course of developmental changes in PPD evoked with WM stimulation. Plotted against postnatal age is the average paired-pulse response ratio obtained with short ISIs (20, 40, 80 msec). The numberof experiments included in the analysis is indicated inparentheses. D, Developmental changes in the magnitude of WM → III LTP. Plotted is the average change in response magnitude recorded 20 min after LTP induction at different postnatal ages [redrawn from Kirkwood et al. (1995)].
The developmental changes in PPD evoked with medium intensity stimulation of the white matter were examined in more detail. As shown in Figure 7C, PPD evoked with paired pulses of short ISIs (20, 40, and 80 msec) rapidly increases by the fourth postnatal week and reaches a stable adult level by the sixth week of age. The responses evoked at longer ISIs, on the other hand, were not affected by age (data not shown). The rapid developmental increase in PPD closely matched the developmental reduction in WM → III LTP shown in Figure 7D [replotted from Kirkwood et al. (1995)]. Together, these results indicate that like the induction of LTP, PPD evoked from the white matter is strongly regulated by age, whereas PPD evoked from layer IV is not.
Effects of visual experience on PPD
Like the critical period for visual cortical plasticity, the developmental decline in WM → III LTP can be postponed by rearing animals in the dark. It was important to determine whether dark rearing also arrests the developmental increase in PPD. Therefore, we compared the effects of paired-pulse stimulation in slices prepared from 6-week-old animals reared in the dark from birth and animals reared under a normal light/dark cycle. As shown in Figure8, dark rearing substantially decreased the magnitude of PPD evoked with white matter stimulation (F(7,68) = 15.933; p = 0.0001). As was the case for age, the effects of sensory deprivation were more dramatic at short ISIs and less pronounced when stimulation was applied to layer IV (F(7,41) = 0.612; p = 0.746).
Fig. 8.

Effects of visual deprivation on PPD.A, PPD profiles obtained in response to WM stimulation were determined in slices obtained from 5- to 6-week-old rats reared normally (○) and those reared in the dark (●) and animals exposed for 24 hr to a normal light/dark cycle after dark rearing (▪ anddotted lines). B, PPD profiles for layer IV stimulation in slices obtained from 5- to 6-week-old rats reared normally (○) and reared in the dark (●).
Finally, we also asked whether the effects of visual deprivation could be reversed and found that the degree of PPD was substantially restored in animals that after 6 weeks of dark rearing were exposed to a normal light/dark cycle for a 24 hr period [ANOVA comparing light-reared animals with dark-reared animals exposed to 24 hr light/dark cycle (F(7,41) = 0.612; p = 0.746)]. Thus, like the critical period for the induction of visual cortical plasticity and WM → III LTP, the developmental increases in PPD can be reversibly arrested or reduced by sensory deprivation.
DISCUSSION
Using PPS we confirmed that the relay of excitatory synaptic transmission from WM to layers II/III is more effective in the visual cortex of immature animals than in adults. The developmental increase in WM → III PPD correlates with the decline of WM → III LTP and the critical period for ocular dominance plasticity. Furthermore, rearing animals in the dark reduces this developmental increase in PPD, just as it delays the ending of the critical period and the decline of WM → III LTP. Our results also indicate that WM → III PPD results from the recruitment of intracortical inhibition. Together, these data support the idea that maturation of intracortical inhibition reduces the efficiency in which rapid patterns of synaptic activity are relayed to superficial cortical layers. Such a reduction in the relay of excitatory synaptic transmission might affect the induction of synaptic plasticity and might also be important in determining the end of the critical period.
Mechanisms of PPD in visual cortex
Depression of synaptic responses during repetitive stimulation is typical of many cortical regions (Markram and Tsodyks, 1996;Castro-Alamancos and Connors, 1997; Gil et al., 1997; Reyes et al., 1998). Our data along with previous studies indicate that recruitment of intracortical GABAergic inhibition plays a prominent role in this type of synaptic depression (Ramoa and Sur, 1996; Castro-Alamancos and Connors, 1997; Buonomano and Merzenich, 1998; Ziakopoulos et al., 2000). Clearly, selective blockade of GABAb receptors reduces and even eliminates PPD (Ramoa and Sur, 1996; Buonomano and Merzenich, 1998;Ziakopoulos et al., 2000) (Fig. 3). The role of GABAa receptors has been studied less, partially because of difficulty in adequately blocking these receptors without inducing runaway excitation. We circumvented this problem by demonstrating that partial blockade of GABAa activation is sufficient to revert PPD into facilitation and by demonstrating that enhancing GABAa-mediated events increases PPD (Fig.4). The involvement of inhibition in PPD is also supported by the close similarity between the temporal profile of the paired-pulse interactions and the time course of the inhibitory synaptic response (Fig. 5) (Castro-Alamancos and Connors, 1997; Ziakopoulos et al., 2000). Interestingly, the effects of stimulus intensity, stimulation site (WM vs layer IV), age, and experience on PPD are restricted to ISIs <640 msec, roughly corresponding to the duration of postsynaptic inhibition (Figs. 4, 5).
Although our findings suggest a prominent role of GABAergic circuits in the developmental increase in PPD, changes in the efficacy of excitatory synaptic transmission cannot be excluded. Short-term depression as well as facilitation of excitatory synapses may also contribute to PPD (Markram and Tsodyks, 1996; Stratford et al., 1996;Abbott et al., 1997; Castro-Alamancos and Connors, 1997; Galarreta and Hestrin, 1998; Gil et al., 1999; for review, see Thomson 2000). Short-term depression has been demonstrated at synapses with a high probability of release, such as thalamocortical synapses in layer IV (Stratford et al., 1996). This synaptic depression is thought to be caused primarily by a transient depletion of neurotransmitter during repetitive activation (Markram and Tsodyks, 1996; Abbott et al., 1997) (but see Brody and Yue, 2000; Waldeck et al. 2000). Such a mechanism may account for PPD observed at ISIs longer than the duration of the postsynaptic inhibitory response (>640 msec). Whether changes in excitatory synaptic transmission play a role in the developmental increase in visual cortical PPD, particularly at shorter ISIs, remains unknown. It seems unlikely, however, because in the few cases examined, paired-pulse ratios for unitary glutamatergic responses switch from depression to facilitation during development (Reyes and Sakmann, 1999). Also, in adult cortex, paired-pulse stimulation at short ISIs produces facilitation, not depression, when inhibition is reduced (Castro-Alamancos and Connors, 1997) (Fig. 4). Thus, although other possibilities cannot be ruled out, change in the efficacy or recruitment (Angulo et al., 1999) of synaptic inhibition is the simplest explanation to account for the regulation of WM → III PPD by age and experience.
PPD was observed under voltage clamp, and it was reduced by blocking inhibition. Thus recruitment of inhibition appears to reduce the relay of synaptic activity to layers II/III cells. Activation of GABA receptors might reduce the relay of excitation in at least two ways. Presynaptic GABAb receptors on excitatory axon terminals can reduce glutamate release (Ziakopoulos et al., 2000), whereas postsynaptic GABAa and GABAb receptors can directly reduce excitability through hyperpolarization and shunting (Buonomano and Merzenich, 1998). The relative contribution of these two mechanisms to PPD remains to be determined. Another question concerns the cell type involved in mediating PPD.
GABAergic interneurons are widely diverse in their physiology, connectivity, and molecular makeup. Fast-spiking parvalbumin-containing cells may be important in controlling PPD because they are the largest single type of GABAergic cell within cortex, synapsing primarily onto the soma and proximal dendrites of principal cells (Kawaguchi and Kubota, 1993; Somogyi et al., 1998; Gupta et al., 2000). Thus they are likely to exert a powerful influence on the output of excitatory cells. A final question concerns the laminar location of the inhibitory control of WM → III PPD. We suspect that the locus of this control is below layers II/III because layer IV → III PPD is poorly regulated by age and experience. A likely site is layer IV, where the density of inhibitory contacts is highest within neocortex (Micheva and Beaulieu, 1997). Furthermore, thalamocortical axons, besides synapsing onto excitatory stellate cells, also synapse onto layer IV interneurons (Ahmed et al., 1997; Porter et al., 2001). Such a feedforward GABAergic circuit could inhibit the output of stellate cells and hence the relay of activity to layers II/III. Indeed, in barrel cortex, this powerful feedforward inhibition seemingly prevents stellate cells from firing more than a single action potential in response to incoming thalamic activity (Brumberg et al., 1999). We propose that a similar layer IV frequency filter might also operate in the visual cortex.
Functional significance
Changes in the relay of thalamocortical activity to layers II/III has obvious ramifications for the induction of activity-dependent plasticity in these layers. The close match between the developmental increase in WM → III PPD, the decline of WM → III LTP, and the end of the critical period for ocular dominance plasticity are very suggestive in this regard. Remarkably, these three processes can be delayed by rearing animals in darkness. Anatomical data indicate that maturation of inhibition is also postponed by sensory deprivation. Together, these results are consistent with the notion that increases in the efficacy of inhibition serve to reduce the induction of plasticity in layers II/III. It is worth pointing out that although the plasticity gate hypothesis was initially formulated to account for the developmental decline in WM → II/III LTP, changes in inhibition could affect other forms of synaptic modification. For example, recent work suggests that afferent activity, relayed from the deprived eye, drives mechanisms of synaptic depression in visual cortex that underlie the ocular dominance shift after monocular deprivation (Rittenhouse et al., 1999). Filtering of this afferent activity within layer IV would be expected to render layers II/III neurons insensitive to monocular deprivation.
Besides restricting the radial propagation of excitation, synaptic inhibition might also regulate layers II/III plasticity through other mechanisms. IPSPs decrease not only the firing of action potentials, but also their backpropagation into the dendrites of pyramidal cells (Kim et al., 1995; Tsubokawa and Ross, 1996; Larkum et al., 1999). Recent work suggests that backpropagating spikes are crucial for the induction of LTP in pyramidal cells (Magee and Johnston, 1997). Furthermore, precise timing between backpropagating spikes and synaptic activation determines both the sign and magnitude of synaptic change (Debanne et al., 1994; Markram et al., 1997). Thus, changes in the efficacy of inhibition onto layers II/III cells could affect the induction of plasticity by altering the integrative properties of these cells. Whether a developmental increase in layers II/III inhibition reduces plasticity by such a mechanism remains unanswered.
In summary, an increasing body of evidence indicates an important role of GABAergic circuitry in the timing of the critical period. Studies on genetically altered mice suggest that intracortical inhibition is crucial for both the termination and initiation of the critical period. In GAD 65 (−/−) knockout mice, in which inhibition is constitutively reduced, a brief epoch of monocular deprivation has no effect unless inhibition is pharmacologically enhanced (Hensch et al., 1998). On the other hand, in BDNF-overexpressing mice, in which maturation of inhibition is accelerated, the critical period for monocular deprivation not only ends earlier, but also starts earlier (Huang et al., 1999). Together, these results suggest that experience-dependent plasticity occurs within an optimal range of inhibition. Plasticity will not occur if the balance of inhibition and excitation is below or above that optimal range (Feldman, 2000). Perhaps a minimum of inhibition is required to differentiate cortical activity produced by the open and closed eye. Thus, selective reinforcement of open eye inputs and selective elimination of closed eye inputs might not take place before the beginning of the maturation of inhibition. On the other hand, our data also support the idea that postnatal maturation of inhibition could lead to the closure of the critical period by restricting both the type and amount of activity that reaches modifiable synapses in superficial cortical layers.
Footnotes
This work was supported by grants from the National Institutes of Health and the Sloan Foundation. We thank Dr. H. K. Lee for valuable comments on this manuscript and M. Sullivan for technical assistance.
Correspondence should be addressed to Alfredo Kirkwood, Mind Brain Institute, Johns Hopkins University, 338 Krieger Hall, 3400 N. Charles Street, Baltimore, MD 21218. E-mail:kirkwood@jhu.edu.
REFERENCES
- 1.Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed B, Anderson JC, Martin KA, Nelson JC. Map of the synapses onto layer 4 basket cells of the primary visual cortex of the cat. J Comp Neurol. 1997;380:230–242. [PubMed] [Google Scholar]
- 3.Aizenman C, Kirkwood A, Bear M. Current source density analysis of evoked responses in visual cortex in vitro: implications for the regulation of long-term potentiation. Cereb Cortex. 1996;6:751–758. doi: 10.1093/cercor/6.6.751. [DOI] [PubMed] [Google Scholar]
- 4.Alger BE, Nicoll RA. Pharmachological evidence for two kinds of GABA receptor on rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:125–141. doi: 10.1113/jphysiol.1982.sp014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angulo MC, Staiger JF, Rossier J, Audinat E. Developmental synaptic changes increase the range of integrative capabilities of an identified excitatory neocortical connection. J Neurosci. 1999;19:1566–1576. doi: 10.1523/JNEUROSCI.19-05-01566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artola A, Singer W. Long-term potentiation and NMDA receptors in rat visual cortex. Nature. 1987;330:649–652. doi: 10.1038/330649a0. [DOI] [PubMed] [Google Scholar]
- 7.Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synaptic modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 8.Blue ME, Parnavelas JG. The formation and maturation of synapses in the visual cortex of the rat. II. Quantitative analysis. J Neurocytol. 1983;12:697–712. doi: 10.1007/BF01181531. [DOI] [PubMed] [Google Scholar]
- 9.Bourassa J, Deschenes M. Corticothalamic projections from the primary visual cortex in rats: a single fiber study using biocytin as an anterograde tracer. Neuroscience. 1995;66:253–263. doi: 10.1016/0306-4522(95)00009-8. [DOI] [PubMed] [Google Scholar]
- 10.Brankack J, Schober W, Klingberg F. Different laminar distributions of flash evoked potentials in cortical areas 17 and 18b of freely moving rats. J Hirnforsch. 1990;31:525–533. [PubMed] [Google Scholar]
- 11.Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brumberg JC, Pinto DJ, Simons DJ. Cortical columnar processing in the rat whisker-to-barrel system. J Neurophysiol. 1999;82:1808–1817. doi: 10.1152/jn.1999.82.4.1808. [DOI] [PubMed] [Google Scholar]
- 13.Buonomano DV, Merzenich MM. Net interaction between different forms of short-term synaptic plasticity and slow-IPSPs in the hippocampus and auditory cortex. J Neurophysiol. 1998;80:1765–1774. doi: 10.1152/jn.1998.80.4.1765. [DOI] [PubMed] [Google Scholar]
- 14.Castro-Alamancos MA, Connors BW. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc Natl Acad Sci USA. 1997;94:4161–4166. doi: 10.1073/pnas.94.8.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cynader M, Mitchel DE. Prolonged sensitivity to monocular deprivation in dark-reared cats. J Neurophysiol. 1980;43:1026–1039. doi: 10.1152/jn.1980.43.4.1026. [DOI] [PubMed] [Google Scholar]
- 16.Daw N. Mechanisms of plasticity in the visual cortex. Invest Ophthalmol Vis Sci. 1994;35:4168–4179. [PubMed] [Google Scholar]
- 17.Debanne D, Gähwiler B, Thompson SM. Asynchronous pre- and postsynaptic activity induces associative long-term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci USA. 1994;91:1148–1151. doi: 10.1073/pnas.91.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 1994;34:709–720. doi: 10.1016/0042-6989(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 19.Feldman DE. Inhibition and plasticity. Nat Neurosci. 2000;3:303–304. doi: 10.1038/73849. [DOI] [PubMed] [Google Scholar]
- 20.Ferster D. X- and Y-mediated synaptic potentials in neurons of areas 17 and 18 of cat visual cortex. Vis Neurosci. 1990;4:115–133. doi: 10.1017/s0952523800002285. [DOI] [PubMed] [Google Scholar]
- 21.Ferster D, Lindstrom S. An intracellular analysis of geniculo-cortical connectivity in area 17 of the cat. J Physiol (Lond) 1983;342:181–215. doi: 10.1113/jphysiol.1983.sp014846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat Neurosci. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- 23.Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 24.Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- 25.Gordon JA, Cioffi D, Silva AJ, Stryker MP. Deficient plasticity in the primary visual cortex of alpha-calcium/calmodulin-dependent protein kinase II mutant mice. Neuron. 1996;17:491–499. doi: 10.1016/s0896-6273(00)80181-6. [DOI] [PubMed] [Google Scholar]
- 26.Guire ES, Lickey ME, Gordon B. Critical period for the monocular deprivation effect in rats: assessment with sweep visually evoked potentials. J Neurophysiol. 1999;81:121–128. doi: 10.1152/jn.1999.81.1.121. [DOI] [PubMed] [Google Scholar]
- 27.Guo Y, Kaplan IV, Cooper NG, Mower GD. Expression of two forms of glutamic acid decarboxylase (GAD67 and GAD65) during postnatal development of the cat visual cortex. Dev Brain Res. 1997;103:127–141. doi: 10.1016/s0165-3806(97)81789-0. [DOI] [PubMed] [Google Scholar]
- 28.Gupta A, Wang Y, Markram H. Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science. 2000;287:273–278. doi: 10.1126/science.287.5451.273. [DOI] [PubMed] [Google Scholar]
- 29.Hensch TK, Faglioni M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang JZ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 31.Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol (Lond) 1970;206:419–436. doi: 10.1113/jphysiol.1970.sp009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwa GGC, Avoli M. Excitatory postsynaptic potentials recorded from regular-spiking cells in layer II/III of rat sensorimotor cortex. J Neurophysiol. 1992;67:728–737. doi: 10.1152/jn.1992.67.3.728. [DOI] [PubMed] [Google Scholar]
- 33.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 34.Kawaguchi Y, Kubota Y. Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol. 1993;70:387–396. doi: 10.1152/jn.1993.70.1.387. [DOI] [PubMed] [Google Scholar]
- 35.Kenan-Vaknin G, Teyler TJ. Laminar pattern of synaptic activity in the rat primary visual cortex: comparison of in vivo and in vitro studies employing the current source density analysis. Brain Res. 1994;635:37–48. doi: 10.1016/0006-8993(94)91421-4. [DOI] [PubMed] [Google Scholar]
- 36.Kim HG, Beierlein M, Connors BW. Inhibitory control of excitable dendrites in neocortex. J Neurophysiol. 1995;74:1810–1814. doi: 10.1152/jn.1995.74.4.1810. [DOI] [PubMed] [Google Scholar]
- 37.Kirkwood A, Bear MF. Hebbian synapses in visual cortex. J Neurosci. 1994;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkwood A, Lee H-K, Bear MF. Co-regulation of long-term potentiation and experience-dependent plasticity in visual cortex by age and experience. Nature. 1995;375:328–331. doi: 10.1038/375328a0. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu Y. Development of cortical inhibition in kitten striate cortex investigated by a slice preparation. Dev Brain Res. 1983;8:136–139. [Google Scholar]
- 40.Larkum ME, Kaiser KMM, Sakmann Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci USA. 1999;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling DSF, Benardo LS. Recruitment of GABAa inhibition in rat neocortex is limited and not NMDA dependent. J Neurophysiol. 1995;74:2329–2335. doi: 10.1152/jn.1995.74.6.2329. [DOI] [PubMed] [Google Scholar]
- 42.Luhmann HJ, Prince DA. Control of NMDA receptor-mediated activity by GABAergic mechanisms in mature and developing rat neocortex. Dev Brain Res. 1990;54:287–290. doi: 10.1016/0165-3806(90)90152-o. [DOI] [PubMed] [Google Scholar]
- 43.Luhmann HJ, Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol. 1991;65:247–263. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- 44.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 45.Markram H, Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- 46.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 47.Micheva KD, Beaulieu C. Development and plasticity of the inhibitory neocortical circuitry with an emphasis on the rodent barrel field cortex: a review. Can J Physiol Pharmacol. 1997;75:470–478. [PubMed] [Google Scholar]
- 48.Mitzdorf U. Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol Rev. 1985;65:37–100. doi: 10.1152/physrev.1985.65.1.37. [DOI] [PubMed] [Google Scholar]
- 49.Mower GD, Caplan CJ, Christen WG, Duffy FH. Dark rearing prolongs physiological but not anatomical plasticity of the cat visual cortex. J Comp Neurol. 1985;235:448–466. doi: 10.1002/cne.902350404. [DOI] [PubMed] [Google Scholar]
- 50.Nelson S, Toth L, Sheth B, Sur M. Orientation selectivity of cortical neurons during intracellular blockade of inhibition. Science. 1994;265:774–777. doi: 10.1126/science.8047882. [DOI] [PubMed] [Google Scholar]
- 51.Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramoa AS, Sur M. Short-term plasticity in the visual cortex during development. Cereb Cortex. 1996;6:640–646. doi: 10.1093/cercor/6.4.640. [DOI] [PubMed] [Google Scholar]
- 53.Reyes A, Sakmann B. Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J Neurosci. 1999;19:3827–3835. doi: 10.1523/JNEUROSCI.19-10-03827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- 55.Rittenhouse CD, Shouval HZ, Paradiso MA, Bear MF. Monocular deprivation induces homosynaptic long-term depression in visual cortex. Nature. 1999;397:347–350. doi: 10.1038/16922. [DOI] [PubMed] [Google Scholar]
- 56.Singer W. Development and plasticity of cortical processing archi- tectures. Science. 1995;270:758–759. doi: 10.1126/science.270.5237.758. [DOI] [PubMed] [Google Scholar]
- 57.Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organization in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- 58.Stratford KJ, Tarczy-Hornoch K, Martin KA, Bannister NJ, Jack JJ. Excitatory synaptic inputs to spiny stellate cells in cat visual cortex. Nature. 1996;382:258–261. doi: 10.1038/382258a0. [DOI] [PubMed] [Google Scholar]
- 59.Sutor B, Hablitz JJ. EPSPs in rat neocortical neurons in vitro. I. Electrophysiological evidence for two distinct EPSPs. J Neurophysiol. 1989;61:607–620. doi: 10.1152/jn.1989.61.3.607. [DOI] [PubMed] [Google Scholar]
- 60.Swadlow HA, Weyand TG. Efferent systems of the rabbit visual cortex: laminar distribution of the cells of origin, axonal conduction velocities, and identification of axonal branches. J Comp Neurol. 1981;203:799–822. doi: 10.1002/cne.902030415. [DOI] [PubMed] [Google Scholar]
- 61.Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- 62.Tsubokawa H, Ross WN. IPSPs modulate spike backpropagation and associated (Ca2+)I changes in the dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2896–2906. doi: 10.1152/jn.1996.76.5.2896. [DOI] [PubMed] [Google Scholar]
- 63.Vaknin G, DiScenna PG, Teyler TJ. A method for calculating current source density (CSD) analysis without resorting to recording sites outside the sampling volume. J Neurosci Methods. 1988;24:131–135. doi: 10.1016/0165-0270(88)90056-8. [DOI] [PubMed] [Google Scholar]
- 64.Waldeck RF, Pereda A, Faber DS. Properties of plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5329. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ziakopoulos Z, Brown MW, Bashir ZI. GABAB receptors mediate frequency-dependent depression of excitatory potentials in rat perirhinal cortex in vitro. Eur J Neurosci. 2000;12:803–809. doi: 10.1046/j.1460-9568.2000.00965.x. [DOI] [PubMed] [Google Scholar]
- 66.Zilles K. The cortex of the rat. Springer; Berlin: 1985. [Google Scholar]