

Abstract
We describe here a simple and efficient antibody titration approach for cell‐surface markers and intracellular cell signaling targets for mass cytometry. The iterative approach builds upon a well‐characterized backbone panel of antibodies and analysis using bioinformatic tools such as SPADE. Healthy peripheral blood and bone marrow cells are stained with a pre‐optimized “backbone” antibody panel in addition to the progressively diluted (titrated) antibodies. Clustering based on the backbone panel enables the titration of each antibody against a rich hematopoietic background and assures that nonspecific binding and signal spillover can be quantified accurately. Using a slightly expanded backbone panel, antibodies quantifying changes in transcription factors and phosphorylated antigens are titrated on ex vivo stimulated cells to optimize sensitivity and evaluate baseline expression. Based on this information, complex panels of antibodies can be thoroughly optimized for use on healthy whole blood and bone marrow and are easily adaptable to the investigation of samples from for example clinical studies. © 2019 The Authors. Cytometry Part A published by Wiley Periodicals, Inc. on behalf of International Society for Advancement of Cytometry.
Keywords: mass cytometry, panel design, antibody titration, whole blood, bone marrow, CyTOF, phosphoflow
Short abstract

Mass cytometry enables the simultaneous measurement of over 40 antigens on single cells using metal isotope conjugated antibodies, generating highly complex datasets with minimal experimental artifacts 1, 2, 3. As the number of antibodies used to investigate biologically heterogeneous cells increases, so do the demands for an efficient and thorough approach to determine optimal antibody titers. In addition to undesirable signal arising from nonspecific antibody binding, which is an issue in all types of antibody‐based assays, three sources of signal spillover exist in mass cytometry 4, 5: signal overlap of highly abundant metal isotopes into adjacent mass channels (±1 Da), isotope oxidation (+16 Da), and isotopic impurities in the metal isotopes. Although technical approaches to deal with similar experimental artifacts have been well established for conventional flow cytometry 6, mass cytometry has unique requirements 7.
The predictable patterns of signal spillover in mass cytometry are not routinely compensated, as is commonplace in conventional flow cytometry. Although such compensation tools have been developed 8, signal spillover can be significantly reduced by lowering the signal intensities (linearly dependent) and/or by carefully designing antibody panels 9. The former may not allow for sufficient distance between the biologically positive and dim/negative populations, and the latter may introduce unwanted/unnecessary noise in the data. In contrast to conventional flow cytometry the range of “brightness” observed across the mass range of purified metal isotope tags is fairly equal 1, 10. Thus, the choice of isotope may not always provide the staining characteristics needed to capture the biological diversity within the mass cytometers dynamic range, without accepting signal spillover to some degree. Antibody binding to secondary and low affinity epitopes must also be evaluated. This might be a challenging process as the combinatorial possibilities of marker expression quickly exceeds our understanding of the human immune system with increasing numbers of markers. Lastly, we emphasize that determination of optimized antibody titer is application‐specific and is not necessarily transferrable between different biological samples, processing protocols, laboratories, or antibody lots. In addition, we have also observed a variation in the stability of metal conjugated antibodies, potentially changing the optimal titer over time. Taken together, the construction of large antibody panels for mass cytometry is an extremely time consuming, laborious, and demanding undertaking, necessitating an efficient and straightforward approach.
Materials and Methods
Subjects and Samples
Peripheral blood (PB) and Bone marrow (BM) samples were obtained from healthy individuals who provided written informed consent (local ethical committee approval 2012/2247). PB and BM were collected in the presence of heparin. The leukocytes were fixed and erythrocytes lysed using Lyse/Fix buffer (BD Biosciences) within 1 h, and samples were stored at −80°C in physiological saline.
Ex Vivo Stimulation of Peripheral Blood and Bone Marrow
Freshly collected PB and BM from one healthy donor were stimulated ex vivo with IFN‐α (100 ng/ml, 15 min), GM‐CSF (100 ng/ml, 15 min), LPS (10 μg/ml, 15 min), or left untreated. PB and BM cells were fixed, and erythrocytes lysed using the BD Lyse/Fix reagent as above.
Barcoding and Antibody Staining
Fixed leukocytes from PB and BM were barcoded 3 using the Fluidigm 20‐plex metal barcoding kit according to manufacturer's protocol. All antibodies used in this study were either purchased pre‐conjugated from Fluidigm or were conjugated in‐house using the X8 MaxPar conjugation kits according to manufacturer's protocol (Online Tables 1–3). See the online materials for detailed protocols. Briefly, aliquots of 1.5 × 106 cells were first pretreated with heparin (100 IU/ml, 20 min) 11 and then stained with mastermixes of backbone antibody panel mixed with twofold serially diluted panel of antibodies to be titrated in a total staining volume of 50 μl (30 min, room temperature). The dilution of most antibodies started at the concentration recommended by the manufacturer (1 μl antibody per 100 μl cell suspension containing 3 × 106 cells). However, for some antibodies, a pre‐dilution was necessary before a twofold titration was possible. For instance, the 163Dy‐CD56 was diluted by a factor of 10× (Online Fig. 8) before the twofold dilution shown in Supplemental Figure 2. Cells to be stained with intracellular signal transduction antibodies were permeabilized for 10 min on ice with methanol (−20°C, 100%), treated with heparin (100 IU/ml, 20 min) and subsequently stained with progressively titrated (five twofold dilutions) intracellular antibodies (30 min, room temperature). To enable the identification of cells, the DNA was labeled with iridium‐191/193 by incubation in 0.1 nM Ir‐nucleic acid intercalator (Fluidigm) diluted in MaxPar PBS containing 4% PFA (Alfa Aesar, 16% PFA, methanol‐free) overnight at 4°C. Cells that were not permeabilized with methanol (cell surface only) were labeled with iridium‐191/193 by incubating with Ir‐nucleic acid intercalator (0.1 nM) diluted in MaxPar Fix/perm buffer (Fluidigm) overnight at 4°C. Immediately before sample acquisition, cells were washed in MaxPar cell staining buffer and MaxPar water (both from Fluidigm) and left pelleted until analysis on the Helios mass cytometer (Fluidigm). The cells were re‐suspended in MaxPar water supplemented with a 1:10 dilution of the EQ Four Element calibration beads (Fluidigm). The acquisition rate was kept below 400 cells per second to limit the number of acquired cell doublets.
Data Analysis
Machine drift in the data was normalized using the Fluidigm bead normalizer. Cell debris and doublets were manually removed by gating on event length and DNA (Ir‐191/193). The Fluidigm barcode de‐convolution tool was used for de‐barcoding samples. The histogram overlay illustrations were made, and SPADE 12 analysis was performed, in cytobank.org. Sample concatenation and gating was performed in FlowJo (FLOWJO, LLC). For gated populations, the 75th percentile of the dual count in each mass channel was exported for statistics. The heat maps were made using Morpheus (https://software.broadinstitute.org/morpheus/).
Results and Discussion
A graphical illustration of our approach is presented in Figure 1, and a more detailed description is given in the Online Materials. All reagents used in this work can be found in Online Tables 1–3. A backbone panel (Online Table 4) of carefully selected antibodies was established as basis to evaluate the titration of additional antibodies in titration step 1. Titration data for the backbone panel is shown in Figure 1a and Online Figure 1a. The optimal titer for each antibody (red gate) was approximated by contrasting the ability to securely discern positive from negative cells against signal spillover into other mass channels. For example, low‐level spillover of 2–3 dual counts of 145Nd‐CD4 signal into 146Nd‐CD8 can be accepted, as long as co‐expression of CD8 is not of biologic interest. In titration step 2, after optimization of the backbone panel, sample aliquots of metal barcoded 3 and paired PB and BM from two healthy donors were stained with the backbone panel and serially diluted mastermixes of the three “titration panels” (Online Table 6) containing additional cell surface antibodies (Fig. 1b). In these titration panels, all channels theoretically receiving spillover from the included markers were kept empty. For example, 144Nd‐CD38 and 148Nd‐CD16 were placed in different titration panels. We used the SPADE clustering algorithm 12 in Cytobank.org to identify common cell subsets across all files in the experiment based on backbone antigen expression (Fig. 1c and Online Fig. 2). This clustering provided a rich immune‐phenotypic background on which the titration of the additional antibodies could be evaluated (Fig. 1d and Online Fig. 1b–d). In addition to evaluating signal spillover using the cell population with the highest expression (e.g., CD45RO on monocytes) as in step 1, this also allowed for the exact evaluation of the staining pattern in a biologically relevant cell subset (e.g., T helper cells). In this way, staining characteristics can be seen across a wider hematological background, altogether further refining the approximation of optimal titers (indicated in red boxes). In titration step 3, we evaluated antibodies specific for intracellular cell signaling and transcription factors. A metal barcoded pool of ex vivo stimulated PB and BM (IFN‐α [100 ng/ml, 15 min], GM‐CSF [100 ng/ml, 15 min], LPS [10 μg/ml, 15 min]) was stained with a combined backbone panel based on titration steps 1 and 2, and progressively diluted titration panels as described above (Online Table 8). After SPADE clustering on surface antigen expression as above (Online Figs. 3 and 4), we calculated the stimulation‐induced change in cell signaling (Δarcsinh relative to control) for all cell subsets (Fig. 1e and Online Fig. 5). Of note, in our experiment the Δarcsinh after both GM‐CSF and LPS stimulation increased for p‐p38 Y180/T182 in monocytes (CD14+) with increasing dilution of the antibody. Likely, surplus antibody created an increased background, thus masking a drug‐induced regulation in signal transduction after stimulation. This emphasizes the importance of selecting optimal antibody titers using appropriate biological controls. Furthermore, we assessed the signal spillover as a function of drug‐induced alterations in cell signaling. For example, after GM‐CSF stimulation, we could measure spillover signal into the empty 172Yb channel induced by high phosphorylation levels of pERK1/2 Y202/T204 (171Yb) in the myeloid dendritic cell population (mDCs, CD11c+HLA‐DR+). This spillover decreased as a function of antibody titration (Fig. 1e, right panel). The final choice of antibody titers was done by minimizing signal spillover and optimal resolution between positive/stimulated and negative/baseline. We validated our approach by testing the titrated panel (Table 1 and Online Table 1 and 2) on three additional healthy donor PB samples. The staining patterns of both cell surface markers and intracellular signal transduction targets in these additional samples reproduced the antibody titration results, highlighting the usefulness of our approach (See online materials and Online Figs. 6 and 7).
Figure 1.
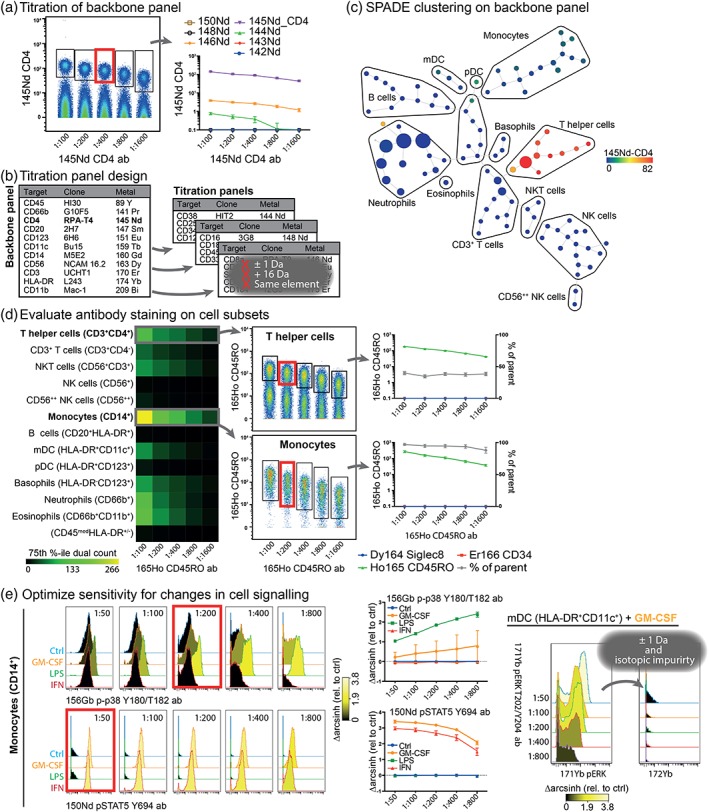
(a) A backbone panel was titrated using PB and BM from two healthy donors. The .fcs files were concatenated to visualize immune staining as a function of antibody concentration and to enable efficient gating of positive cells. The expression of the titrated antigen and all spillovers (±1 Da, +16 Da and channels detecting isotopic impurities) was calculated for the gated cells plotted (75th %‐ile dual counts). Optimal titer (red gate) was chosen by balancing the ability to discern positive from negative cells with the amount of signal overlap generated in other mass channels. (b) Additional cell surface antibodies to be titrated were subdivided into titration panels. Here, all channels receiving spillover were unused in each panel. Cell sample aliquots were stained with the titrated backbone panel and serially diluted mastermixes of the titration panels. (c) A single SPADE clustering was performed to efficiently identify cell subsets in the entire data set. The clustering was based solely on the backbone panel, and cell subsets manually identified. (d) The signal from the titrated antibodies were measured in each of the cell subsets and plotted as a heat map. The data was in selected cell subsets concatenated, and the expression of the titrated antigen and all spillovers calculated for the gated population, as above. The exact staining pattern on a relevant cell subset (i.e., CD45RO expression on T helper cells) could now also be evaluated in addition to signal spillover (i.e., CD45RO expression on monocytes) and panel design. The red gate indicates the chosen antibody titer. The relative abundance of positive cells in the parent cell subset as a function of antibody concentration was also calculated. (e) PB and BM from one healthy donor were stimulated ex vivo with GM‐CSF (100 ng/ml, 15 min), IFN‐α (100 ng/ml, 15 min) or LPS (10 μg/ml, 15 min) or left untreated. The antibodies to be titrated were split into two titration panels, as above. Cells were stained with backbone panel and serially diluted titration panels, and cell subsets identified using SPADE. The phosphorylation level (75th %‐ile) was measured in each population, for all intracellular antibodies and all channels theoretically receiving spillover. Drug‐induced changes in phosphorylation levels were calculated (Δarcsinh relative to ctrl) and plotted as a function of antibody dilution. Lastly, the signal spillover generated by induction of signaling into the empty mass channel was evaluated. Red boxes indicate optimal dilutions of antibodies. Color scales indicate Δarcsinh relative to control.
Table 1.
Antibody panel. (See online Tables 1–4, 6, and 8 in the online materials for more details)
| Specificity | Clone | Isotope | Purpose |
|---|---|---|---|
| CD45 | HI30 | 89 Y | Pan leukocytes |
| CD66b | G10F5 | 141 Pr | Neutrophils |
| Cleaved caspase 3 | D3E9 | 142 Nd | Apoptosis |
| CD38 | HIT2 | 144 Nd | Activation |
| CD4 | RPA‐T4 | 145 Nd | T helper cells |
| CD8a | RPA‐T8 | 146 Nd | Cytotoxic T cells |
| CD20 | 2H7 | 147 Sm | B cells |
| CD16 | 3G8 | 148 Nd | Neutrophils and subsets of NK and monocytes |
| CD25 | 2A3 | 149 Sm | Basophils, Tregs, and activated T helper cells |
| pSTAT5 Y694 | 47 | 150 Nd | Signal transduction |
| CD123 | 6H6 | 151 Eu | Basophils, mDC, and pDC |
| pSTAT1 Y701 | 58D6 | 153 Eu | Signal transduction |
| p‐p38 T180/Y182 | D3F9 | 156 Gd | Signal transduction |
| pSTAT3 Y705 | 4/P‐STAT3 | 158 Gd | Signal transduction |
| CD11c | Bu15 | 159 Tb | Monocytes and mDC |
| CD14 | M5E2 | 160 Gd | Monocytes |
| CD181 (IL‐8RA) | B1 | 161 Dy | Neutrophils |
| FoxP3 | PCH101 | 162 Dy | Tregs |
| CD56 | NCAM 16.2 | 163 Dy | NK cells |
| CD45RO | UCHL1 | 165 Ho | Naïve/memory T cells |
| CD34 | 581 | 166 Er | Hematopoietic stem/progenitor cell |
| CD1c (BDCA‐1) | L161 | 167 Er | Subsets of mDC and B cells |
| CD335 (NKp46) | 9E2 | 169 Tm | NK cells |
| CD3 | UCHT1 | 170 Er | T cells |
| pERK 1/2 T202/Y204 | D1314.E4 | 171 Yb | Signal transduction |
| HLA‐DR | L243 | 174 Yb | Activation, DCs, monocytes, and B cells |
| CD184 (CXCR4) | 12G5 | 175 Lu | Basophils |
| pCREB S133 | 87G3 | 176 Yb | Signal transduction |
| CD11b | Mac‐1 | 209 Bi | Granulocytes, monocytes NK cells, and DCs |
mDC; myeloid dendritic cell; pDC, plasmacytoid dendritic cell; NK, natural killer; Tregs, regulatory T cells.
In summary, we outline here a conceptual framework where we highlight the usefulness of performing iterative antibody titration on cells stained with a backbone panel. We found SPADE to be an excellent tool for automated cell clustering based on the backbone panel. SPADE enabled clustering of cells in a dataset consisting of more than 6 million cells into a single SPADE tree. Using bioinformatic tools, this approach is efficient and straightforward and provides a deeper characterization of each antibody's performance, which is necessary for the demanding task of panel design for mass cytometry assays. Although we have demonstrated the titration of antibodies on healthy PB and BM in this work, this approach can easily be adapted to other sample types for mass cytometry.
Supporting information
Online Table 1 – Commercially available reagents
Online Table 2 – Reagents conjugated in‐house
Online Table 3 – Reagents titrated but not used
Online Table 4 – Antibody backbone panel (Step 1).
Online Table 5 ‐ Titration of Backbone Panel. The table shows the gate statistics for the backbone panel titration (step 1), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the five samples with titrated antibody. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112)
Online Table 6 – Antibody subpanel design for the titration of additional cell surface markers.
Online Table 7 ‐ Titration of additional markers using cells stained with the backbone panel (Step 2). The table shows the gate statistics for the additional antibody titration (step 2), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the five samples x three titration panels. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112). ‘All cells’ refers to all cells included in a “bubble” (population) of the SPADE tree.
Online Table 8 – Antibodies used in titration of intracellular transcription factor and phospho‐specific antibodies
Online Table 9 ‐ Titration of signal transduction antibodies using ex vivo stimulated cells (Step 3). The table shows the gate statistics for the intracellular antibody titration (step 3), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the 5 samples with titrated antibody. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112). All cells refers to all cells included in a “bubble” (population) of the SPADE tree.
Online Figure 1 – Titration of backbone panel and additional cell surface markers (Step 1). a) The density dot plot shows the five titration samples concatenated side‐by‐side for each of the antibodies in the backbone panel (Online table 4). The population staining positive for the antibody of interest is gated. The associated graph shows the 75th %‐ile dual counts of the titrated antibody and the signal spillover into adjacent mass channels as a function of antibody concentration. The red gates denote the chosen titer used in the backbone in later titration experiments. The titrated backbone was subsequently used when titrating additional cell surface markers in Step 2 (Online table 6). The staining characteristics of each titrated cell surface marker is in b), c) and d) visualized as heat maps (75th %‐tile dual counts) on all cell subsets identified by the backbone panel and SPADE clustering (SPADE clustering can be found in Online figure 2). Positive cells in cell subsets of interest are further gated (gating not shown) and the 75th %‐tile dual counts of these populations, and the relative frequency (to negative cells) is calculated and plotted. The red box indicates the chosen titer for the antibodies included in the final panel.
Online Figure 2 – SPADE clustering of immune populations based on backbone panel. All files (5 concentrations x 3 panels x 4 samples) were clustered using the Cytonbank.org implementation of SPADE. Manual annotation of the tree enabled the detection of 13 immune populations. (CD45 was not used for clustering). This data is related to Figure 1‐c and d.
Online Figure 3 – SPADE clustering of immune populations based on the backbone panel used in the titration of signal transduction antibodies. All files (5 concentrations x 2 panels x 4 conditions x 2 tissues) were clustered using the Cytonbank.org implementation of SPADE. Manual annotation of the tree enabled the detection of 16 immune populations. (CD45 was not used for clustering). This data is related to Figure 1‐e.
Online Figure 4 – Gating strategy. The figure shows our manual gating strategy corresponding to the SPADE analysis shown in Online Figure 3 (Step 3).
Online Figure 5 – Histogram overlays for ex vivo stimulated peripheral blood and bone marrow. The figure shows the signal after ex vivo stimulation (IFN‐α: 100 ng/ml, 15 min; GM‐CSF: 100 ng/mL, 15 min; LPS: 10 μg/mL, 15 min; or left untreated) in the indicated immune populations (left to right) and with decreasing concentration of antibody (top to bottom, two‐fold serial dilution range 1:50‐1:800). Panels a‐h) presents data from stimulated peripheral blood. Panels i‐p) presents data from stimulated bone marrow. Color scales indicate Δarcsinh relative to control.
Online Figure 6 – Manual gating of the panel validation samples. The figure shows our manual gating strategy to identify the cell subpopulations used to validate the mass cytometry panel. Only minor adjustments were needed to directly apply the gating used in the titration of signal transduction antibodies using ex vivo stimulated cells (Step 3) on the validation samples
Online Figure 7 – Validation of intracellular markers. The histograms show both the titration of signal transduction antibodies using ex vivo stimulated cells and the three healthy donor validation samples, using the manually gated subpopulations (Online Figure 4 and 6, respectively). The red boxes indicate the titer chosen for the final panel in Online table 1 and 2, while the titer used for the three validation samples is given in red text above the histograms. The color scales indicate the median dual counts.
Online Figure 8 – Pre‐titration of 163 Dy CD56 (NCAM 16.2). The density plot shows a 4‐point, 4‐fold titration, of the CD56 antibody on leukocytes isolated from the peripheral blood using the Lyse/Fix buffer (BD Biosciences) as described above. The broad and unspecific staining profile of the antibody transitioned into an apparently highly specific and bimodal staining of CD56+ leukocytes.
MIFlowCytChecklist
Acknowledgments
This study was supported by The Research Council of Norway (Petromaks program grant #220759), Helse Vest Health Trust (project no. 912160, 912009), and the Norwegian Cancer Society Legacy (Grant no. 104712, 145268, 145269 and 163424) with Solveig & Ole Lunds Legacy. The Helios mass cytometry instrument was a generous gift from the Trond Mohn Foundation.
Literature Cited
- 1. Spitzer MH, Nolan GP. Mass cytometry: Single cells, many features. Cell 2016;165(4):780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Catena R, Ozcan A, Zivanovic N, Bodenmiller B. Enhanced multiplexing in mass cytometry using osmium and ruthenium tetroxide species. Cytometry A 2016;89(5):491–497. [DOI] [PubMed] [Google Scholar]
- 3. Zunder ER, Finck R, Behbehani GK, Amir el AD, Krishnaswamy S, Gonzalez VD, et al. Palladium‐based mass tag cell barcoding with a doublet‐filtering scheme and single‐cell deconvolution algorithm. Nat Protoc 2015;10(2):316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ornatsky OI, Kinach R, Bandura DR, Lou X, Tanner SD, Baranov VI, Nitz M, Winnik MA. Development of analytical methods for multiplex bio‐assay with inductively coupled plasma mass spectrometry. J Anal At Spectrom 2008;23(4):463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler's guide to cytometry. Trends Immunol 2012;33(7):323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med 2007;27(3):469–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takahashi C, Au‐Yeung A, Fuh F, Ramirez‐Montagut T, Bolen C, Mathews W, O'Gorman WE. Mass cytometry panel optimization through the designed distribution of signal interference. Cytometry A 2017;91(1):39–47. [DOI] [PubMed] [Google Scholar]
- 8. Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, Bodenmiller B. Compensation of signal spillover in suspension and imaging mass cytometry. Cell Syst 2018;6(5):612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leipold MD, Newell EW, Maecker HT. Multiparameter phenotyping of human PBMCs using mass cytometry. Methods Mol Biol 2015;1(343):81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tricot S, Meyrand M, Sammicheli C, Elhmouzi‐Younes J, Corneau A, Bertholet S, Malissen M, le Grand R, Nuti S, Luche H, et al. Evaluating the efficiency of isotope transmission for improved panel design and a comparison of the detection sensitivities of mass cytometer instruments. Cytometry A 2015;87(4):357–368. [DOI] [PubMed] [Google Scholar]
- 11.Rahman AH, Tordesillas L, Berin MC. Heparin reduces nonspecific eosinophil staining artifacts in mass cytometry experiments. Cytometry Part A 2016;89(6):601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr, Bruggner RV, Linderman MD, et al. Extracting a cellular hierarchy from high‐dimensional cytometry data with SPADE. Nat Biotechnol 2011;29(10):886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Table 1 – Commercially available reagents
Online Table 2 – Reagents conjugated in‐house
Online Table 3 – Reagents titrated but not used
Online Table 4 – Antibody backbone panel (Step 1).
Online Table 5 ‐ Titration of Backbone Panel. The table shows the gate statistics for the backbone panel titration (step 1), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the five samples with titrated antibody. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112)
Online Table 6 – Antibody subpanel design for the titration of additional cell surface markers.
Online Table 7 ‐ Titration of additional markers using cells stained with the backbone panel (Step 2). The table shows the gate statistics for the additional antibody titration (step 2), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the five samples x three titration panels. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112). ‘All cells’ refers to all cells included in a “bubble” (population) of the SPADE tree.
Online Table 8 – Antibodies used in titration of intracellular transcription factor and phospho‐specific antibodies
Online Table 9 ‐ Titration of signal transduction antibodies using ex vivo stimulated cells (Step 3). The table shows the gate statistics for the intracellular antibody titration (step 3), with percent of total and the standard deviation (SD) and % CV. Averages are made using the data from the 5 samples with titrated antibody. The samples are denoted as bone marrow and peripheral blood from healthy donor 118 (BM118/PB118) and from healthy donor 112 (BM112/PB112). All cells refers to all cells included in a “bubble” (population) of the SPADE tree.
Online Figure 1 – Titration of backbone panel and additional cell surface markers (Step 1). a) The density dot plot shows the five titration samples concatenated side‐by‐side for each of the antibodies in the backbone panel (Online table 4). The population staining positive for the antibody of interest is gated. The associated graph shows the 75th %‐ile dual counts of the titrated antibody and the signal spillover into adjacent mass channels as a function of antibody concentration. The red gates denote the chosen titer used in the backbone in later titration experiments. The titrated backbone was subsequently used when titrating additional cell surface markers in Step 2 (Online table 6). The staining characteristics of each titrated cell surface marker is in b), c) and d) visualized as heat maps (75th %‐tile dual counts) on all cell subsets identified by the backbone panel and SPADE clustering (SPADE clustering can be found in Online figure 2). Positive cells in cell subsets of interest are further gated (gating not shown) and the 75th %‐tile dual counts of these populations, and the relative frequency (to negative cells) is calculated and plotted. The red box indicates the chosen titer for the antibodies included in the final panel.
Online Figure 2 – SPADE clustering of immune populations based on backbone panel. All files (5 concentrations x 3 panels x 4 samples) were clustered using the Cytonbank.org implementation of SPADE. Manual annotation of the tree enabled the detection of 13 immune populations. (CD45 was not used for clustering). This data is related to Figure 1‐c and d.
Online Figure 3 – SPADE clustering of immune populations based on the backbone panel used in the titration of signal transduction antibodies. All files (5 concentrations x 2 panels x 4 conditions x 2 tissues) were clustered using the Cytonbank.org implementation of SPADE. Manual annotation of the tree enabled the detection of 16 immune populations. (CD45 was not used for clustering). This data is related to Figure 1‐e.
Online Figure 4 – Gating strategy. The figure shows our manual gating strategy corresponding to the SPADE analysis shown in Online Figure 3 (Step 3).
Online Figure 5 – Histogram overlays for ex vivo stimulated peripheral blood and bone marrow. The figure shows the signal after ex vivo stimulation (IFN‐α: 100 ng/ml, 15 min; GM‐CSF: 100 ng/mL, 15 min; LPS: 10 μg/mL, 15 min; or left untreated) in the indicated immune populations (left to right) and with decreasing concentration of antibody (top to bottom, two‐fold serial dilution range 1:50‐1:800). Panels a‐h) presents data from stimulated peripheral blood. Panels i‐p) presents data from stimulated bone marrow. Color scales indicate Δarcsinh relative to control.
Online Figure 6 – Manual gating of the panel validation samples. The figure shows our manual gating strategy to identify the cell subpopulations used to validate the mass cytometry panel. Only minor adjustments were needed to directly apply the gating used in the titration of signal transduction antibodies using ex vivo stimulated cells (Step 3) on the validation samples
Online Figure 7 – Validation of intracellular markers. The histograms show both the titration of signal transduction antibodies using ex vivo stimulated cells and the three healthy donor validation samples, using the manually gated subpopulations (Online Figure 4 and 6, respectively). The red boxes indicate the titer chosen for the final panel in Online table 1 and 2, while the titer used for the three validation samples is given in red text above the histograms. The color scales indicate the median dual counts.
Online Figure 8 – Pre‐titration of 163 Dy CD56 (NCAM 16.2). The density plot shows a 4‐point, 4‐fold titration, of the CD56 antibody on leukocytes isolated from the peripheral blood using the Lyse/Fix buffer (BD Biosciences) as described above. The broad and unspecific staining profile of the antibody transitioned into an apparently highly specific and bimodal staining of CD56+ leukocytes.
MIFlowCytChecklist