
Figure 2.
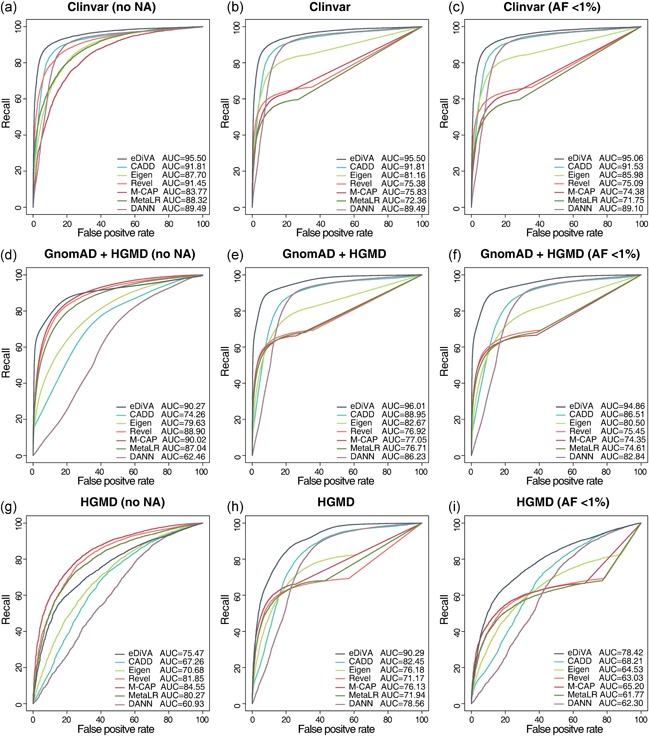
Benchmarking of the pathogenicity classifiers eDiVA‐Score, CADD, Eigen, Revel, and M‐CAP using ROC for (a) set of 10,494 ClinVar pathogenic variants (TP) and 3,887 ClinVar “benign” variants (TN); (b) set of 16,694 ClinVar pathogenic variants (TP) and 19,888 ClinVar “benign” variants (TN), setting missing values to benign, (c) subset of rare variants (AF, <1% from set c); (d) set of 63,712 variants from HGMD (TP) and 100,000 from GnomAD (TN) for which values from all tools are available; (e) set of 96,569 variants from HGMD (TP) and 100,000 from GnomAD (TN), setting missing values to benign; (f) subset of rare variants (AF, <1% from set e); (g) set of 63,712 HGMD variants (“DM” and “DM?”) as TP, and 1,892 HGMD variants (other categories) as TN for which values from all tools are available; (h) set of 96,569 variants from HGMD (“DM” and “DM?”) as TP, and 7,376 HGMD (other categories) as TN, setting missing values to benign; and (i) subset of rare variants (AF, <1% from set h). AF: allele frequency; eDiVA: exome Disease Variant Analysis; M‐CAP: Mendelian clinically applicable pathogenicity; ROC: receiver operating characteristic; TN: true negative; TP: true positive