

Abstract
Styrene is an important high production volume chemical used to manufacture polymeric products. In 2018, International Agency for Research on Cancer classified styrene as probably carcinogenic to humans; National Toxicology Program lists styrene as reasonably anticipated to be a human carcinogen. The genotoxicity literature for styrene and its primary metabolite, styrene 7,8‐oxide (SO), begins in the 1970s. Organization of Economic Cooperation and Development (OECD) recently updated most genotoxicity test guidelines, making substantial new recommendations for assay conduct and data evaluation for the standard mutagenicity/clastogenicity assays. Thus, a critical review of the in vitro and in vivo rodent mutagenicity/clastogenicity studies for styrene and SO, based on the latest OECD recommendations, is timely. This critical review considered whether a study was optimally designed, conducted, and interpreted and provides a critical assessment of the evidence for the mutagenicity/clastogenicity of styrene/SO. Information on the ability of styrene/SO to induce other types of genotoxicity endpoints is summarized but not critically reviewed. We conclude that when styrene is metabolized to SO, it can form DNA adducts, and positive in vitro mutagenicity/clastogenicity results can be obtained. SO is mutagenic in bacteria and the in vitro mouse lymphoma gene mutation assay. No rodent in vivo mutation studies were identified. SO is clastogenic in cultured mammalian cells. Although the in vitro assays gave positive responses, styrene/SO is not clastogenic/aneugenic in vivo in rodents. In addition to providing updated information for styrene, this review demonstrates the application of the new OECD guidelines for chemicals with large genetic toxicology databases where published results may or may not be reliable. Environ. Mol. Mutagen. 2019. © 2019 Wiley Periodicals, Inc.
Keywords: styrene oxide, mutagenicity, clastogenicity, chromosome aberrations, DNA adducts, micronucleus, Ames test, gene mutation assays
INTRODUCTION
Styrene (CAS No. 100‐42‐5), a derivative of benzene, is a widely used industrial chemical, obtained for industrial use from petroleum and natural gas by‐products (Helal and Elshafy, 2012). The International Agency for Research on Cancer (IARC) Monograph Volume 82 (IARC, 2002) describes the major uses of styrene in the manufacture of materials, including glass fiber‐reinforced composites, polystyrene products, styrene‐alkyd coatings, and styrene–butadiene synthetic rubber. The wide variety of consumer products that involve styrene in their production includes boats, bathtubs, showers, food packaging, automotive reinforced plastics and body putty, foams and cushioning materials for packaging, disposable tableware, latex paints, and synthetic marble flooring. Human exposure to styrene is primarily occupational with the highest exposures found in industries making consumer products from glass fiber‐reinforced polyester composite plastics (IARC, 2002). Perhaps the greatest nonoccupational exposure source for styrene is from cigarette smoke, with smokers having exposure levels approximately six times higher than nonsmokers (IARC, 2002).
The main metabolite of styrene is styrene 7,8‐oxide (SO; CAS No. 9609‐3) (see Fig. 1), which results from oxidation by cytochromes P450 (Watabe et al., 1978; Vainio et al., 1981, 1984; Hynes et al., 1999; IARC, 2002; Vodicka et al., 2006). SO is hydrolyzed to styrene glycol (CAS 93‐56‐1) by epoxide hydrolase. Additional metabolites of styrene include mandelic acid (MA), phenylglyoxylic acid (PGA), benzyl alcohol, benzoic acid, and hippuric acid (Sugiura and Goto, 1981; Vainio et al., 1981).
Figure 1.
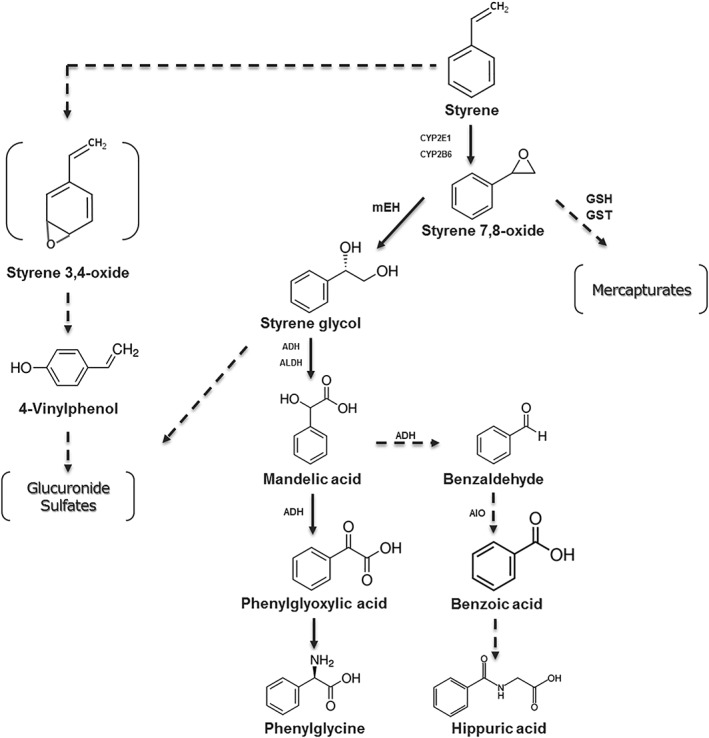
Styrene metabolism: pathways of major interest (modified from NTP [2008]). ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; AlO, aldehyde oxidase; mEH, microsomal epoxide hydrolase; GSH, glutathione; GST, GSH‐S‐transferase.
IARC, the National Toxicology Program (NTP), and the National Academy of Sciences (NAS) have reviewed the available data and assessed the carcinogenicity of styrene. IARC has published three Monographs on the carcinogenicity of styrene (IARC, 1979, 1994, 2002) and one Monograph on SO (IARC, 1985). The 2002 IARC Monograph classifies styrene as possibly carcinogenic to humans (Group 2B) based on the limited evidence in both humans and rodents. At its recent March 2018 meeting, IARC upgraded styrene to be a Group 2A carcinogen, “probably carcinogenic to humans, based on limited evidence in humans and sufficient evidence in experimental animals” (IARC, 2018). In 2008, the NTP published a summary review document as a background information to assist with its styrene carcinogenicity review (NTP, 2008). In 2011, in its 12th Report on Carcinogens, the NTP listed styrene as “reasonably anticipated to be a human carcinogen.” The NAS was tasked with conducting an independent review of this assessment. In 2014, the National Research Council (NRC) of the National Academies Press published a document “Review of the Styrene Assessment in the National Toxicology Program 12th Report on Carcinogens” (NRC, 2014). Based on their review of the available studies, the NRC committee concluded that the NTP classification for the carcinogenicity of styrene was appropriate. This cancer classification was based on limited evidence in humans and sufficient evidence in experimental animals. Because of the relevance of genotoxicity information in the overall weight‐of‐the‐evidence for the mode of action (MOA) for a carcinogen, the IARC Monographs (IARC, 1979, 1985, 1994, 2002), the NTP background document (NTP, 2008), and the NRC report (NRC, 2014) all include summaries of the extensive literature, dating to the 1970s, concerning the genotoxicity of styrene and SO, from in vitro assays and in rodents in vivo. These summaries are based directly on the individual study author determinations and include both positive and negative results.
After these reviews were published, the Organization of Economic Cooperation and Development (OECD) made substantial revisions to the genotoxicity test guidelines (TGs), and these revisions affect the conduct of assays and the interpretation of test results (OECD, 2017; Thybaud et al., 2017). In addition, there have been a few recent publications addressing the potential genotoxicity of styrene/SO. It is, therefore, timely to critically review (based on current OECD recommendations) the evidence as to whether styrene is mutagenic/clastogenic, either in vitro or in rodents in vivo, or both.
It is recognized that many early studies were conducted prior to the development of any TGs and that the results of many of these studies may not be reliable. Although other studies may have met the guidelines in place when they were conducted, experience with the assay may have resulted in new recommendations for assay conduct and interpretation of data. In some cases, this new insight for assay conduct and interpretation means that older results can no longer be interpreted, or results that may have been considered positive (or negative) would no longer be considered definitive results. To provide optimal information concerning the mutagenicity/clastogenicity of a widely studied compound, it is important to critically review the available information and to utilize only high‐quality data in the overall weight‐of‐the‐evidence evaluation. The goal of this critical review is to consider each study and determine whether an individual study was designed and conducted using procedures compliant with the current TGs and whether the published dataset can be interpreted as positive or negative. Based on this curated database, we then provide a critical assessment of the mutagenicity/clastogenicity of styrene and SO. The focus of our critical review is on assays for which there are OECD TGs and for endpoints most directly related to addressing the question as to whether styrene can induce gene mutations. Because they can provide supporting information concerning chemical exposure and the ability of chemicals to cause primary DNA effects, literature information for additional endpoints such as DNA adducts and DNA strand breakage is summarized, but it is not critically reviewed. In addition to providing this updated information for styrene, this review demonstrates how the new OECD guidance can be applied to chemicals that have large (older) genetic toxicology databases, where many of the study results may or may not be reliable.
LITERATURE SEARCH AND SUMMARY OF PREVIOUS REVIEWS
With the goal of conducting a critical review of the available published studies to address the mutagenicity/clastogenicity of styrene and to summarize the information from other genotoxicity endpoints, a PubMed literature search was performed using the following search terms:
(Styrene OR “Styrene oxide” OR “Mandelic acid” OR “4‐vinylphenol” OR “1‐phenylethanol” OR “Phenyl‐gloxylic acid”) AND (“Genetic Damage” OR Genotoxicity OR “Ames Test” OR “Salmonella typhimurium” OR “E. Coli Mutation” OR “Chromosome Aberration” OR “Sister Chromatid Exchange” OR “DNA Adduct” OR “DNA Damage” OR “DNA breakage” OR “DNA breaks” OR “Single Strand breaks” OR Micronucleus OR “Gene Mutation” OR “hprt Mutation” OR “Mouse Lymphoma Assay” OR “Thymidine Kinase Mutation” OR “Transgenic Mutation Assay” OR “Genetic Toxicology” OR Mutation OR Mutagenesis OR Clastogenicity OR Aneuploidy OR Polyploidy OR Mutagenicity OR “Comet Assay” OR Comet OR “Single Cell Gel” OR “Alkaline elution”).
All abstracts returned from the PubMed search were reviewed to identify relevant references for evaluating the genotoxicity of styrene. It was clear from the initial review of the abstracts that many of the papers did not actually contain genetic toxicology information and thus they were easily eliminated from further consideration in this first step. Any genetic toxicology relevant literature cited in the IARC Monographs (IARC, 1985, 1994, 2002) and the NTP Review (NTP, 2008) but not identified in the PubMed search was added to the list of papers to be considered in the review.
The literature search identified several peer‐reviewed journal summary/review articles for styrene. Many of these (particularly the older summaries) simply reported the conclusions (positive/negative calls) of the publication authors with little, if any, critical review of the individual studies (Vainio et al., 1981; Norppa and Vainio, 1983; Norppa et al., 1988; Bond and Bolt, 1989; Barale, 1991). In 2002, The Harvard Center for Risk Analysis conducted a comprehensive (12‐member international expert panel) review of the potential human health effects of styrene exposure (Cohen et al., 2002). The panel concluded that styrene “does not appear to be DNA reactive”, but SO clearly induces mutations in the Ames test. SO binds to both proteins and nucleic acids and forms both stable N2 and O6 guanine adducts in exposed mammalian cells. The panel concluded that the results in animal studies are “less clear cut,” with conflicting results as to whether styrene can cause sister chromatid exchanges (SCEs), chromosome aberrations (CAs), or micronuclei (MN) in rodents.
The first detailed critical review of studies, considering study/data quality and evaluating styrene's cytogenetic effects (in vitro, in vivo, and in occupationally exposed humans), was conducted by Scott and Preston (Scott and Preston, 1994a, 1994b) and includes summary data tables with extensive details. Their review of the in vitro data for styrene and SO resulted in a conclusion that both chemicals can induce CAs and SCEs but that positive results are dependent upon test conditions that favor metabolic activation of styrene to SO over inactivation of SO. They found “no convincing” evidence that in vivo styrene exposure can cause chromosome damage in rodents. For the in vivo studies showing positive results for clastogenicity, they reported that the positive response was only seen at lethal doses and via intraperitoneal (i.p.) injection (now considered to be an inappropriate route, per OECD TG475 [OECD, 2016a]). The positive response was not observed via inhalation in Chinese hamsters or after oral exposure (also an inappropriate substitute for the inhalation route) in mice. SCEs were seen in rodents from in vivo exposure to both styrene and SO but only at very high concentrations.
A decade later, in 2005, Leigh Henderson and Gunter Speit published a critical review of the in vivo rodent genetic toxicology assays. They concluded that there was no clear evidence that styrene induces clastogenic/mutagenic effects in vivo when the test is performed under appropriate test conditions (Speit and Henderson, 2005). They also concluded that “equivocal” results can be observed when the tests were performed using high exposure levels that led to lethality. Also, in 2005, Nestmann et al. (2005) provided an overview of reviews that had been previously conducted. They concluded that rodent studies at exposures up to 1500 mg/m3 (352 ppm) per day showed no evidence of clastogenicity.
STRATEGY FOR CRITICALLY REVIEWING THE MUTAGENICITY/CLASTOGENICITY OF STYRENE
The available literature evaluating the potential mutagenicity/clastogenicity of styrene and SO exposure includes most, but not all, of the standard in vitro mammalian cell and rodent in vivo tests for which there are OECD TGs. We note that in the recent OECD revision process, all the genetic toxicology TGs were considered for revision or deletion (OECD, 2017; Thybaud et al., 2017). The TGs for several assays were archived/deleted. Reasons for deletion included that the assays are rarely used, the underlying mechanisms for the endpoints that they measure are not fully characterized or are no longer considered relevant, or because other assays are considered more relevant for assessing genotoxicity. There are styrene and/or SO data for many of these test systems and this information has been summarized in previous reviews, including the IARC and NTP documents. However, because the results from these tests are no longer considered relevant for new testing for identifying potential human risk, information from these tests was not included in our current review. Table 1 lists the OECD TGs that were deleted.
Table 1.
Deleted OECD TGs
| TG | Title | Date of adoption/deletion |
|---|---|---|
| 472 | Escherichia coli, reverse assay | 1983/1997 |
| 477 | Sex‐linked recessive lethal test in Drosophila melanogaster | 1984/2014 |
| 479 | In vitro SCE assay in mammalian cells | 1986/2014 |
| 480 | Saccharomyces cerevisiae, gene mutation assay | 1986/2014 |
| 481 | S. cerevisiae, mitotic recombination assay | 1986/2014 |
| 482 | DNA damage and repair, UDS in mammalian cells in vitro | 1986/2014 |
| 484 | Mouse spot test | 1986/2014 |
Although the in vitro SCE TG was deleted, and data for the in vitro assay were not included in our review, results from the in vivo SCE assay were included. It should be noted that, in the past, the SCE assay was extensively used in both human and rodent studies and there are data for many different chemical exposures (Latt et al., 1981; Tucker et al., 1993). Extensive research to understand the mechanistic basis, and the significance of SCEs, ultimately led to the conclusion that the two sister chromatids broke and rejoined with one another, physically exchanging DNA segments, but that the process was error free (Wilson and Thompson, 2007). That is, SCEs are not actually reflective of genetic damage. This was the basis for deleting the in vitro SCE OECD TG. Data from the in vivo SCE assay can be useful as a biomarker of exposure, assessing the ability of test materials to reach target tissues. However, positive results observed with the SCE endpoint should not be considered evidence that a chemical is genotoxic.
Weight‐Of‐The‐Evidence Interpretation of the Various Types of Results
There are several endpoints/assays providing useful information for assessing whether a chemical, such as styrene, can cause genetic damage. Endpoints can be classified by the broad term “genotoxicity” or the more restrictive term “mutagenicity.” It is ultimately the ability of a test material to induce mutations, which is the most important. Although many slightly different definitions of the terms genotoxicity and mutagenicity exist, the definitions considered in this critical review are those provided in the OECD Overview to the recent revisions of the TGs (OECD, 2017):
Mutagenicity is a subset of genotoxicity. Mutagenicity results in events that alter the DNA and/or chromosomal number or structure that are irreversible and, therefore, capable of being passed to subsequent cell generations if they are not lethal to the cell in which they occur.
Genotoxicity is a broader term. It includes mutagenicity (described above), and it also includes DNA damage which may be mutagenic, but may also be reversed by DNA repair or other cellular processes, and, thus, which may or may not result in permanent alterations in the structure or information content in a surviving cell or its progeny.
The broader set of genotoxicity tests that do not, however, provide definitive information that the test material is mutagenic include primary DNA damage tests such as unscheduled DNA synthesis (UDS), DNA strand breaks, the Comet assay, and DNA adduct formation. Written to provide background information and a summary of the TG revisions, the OECD Overview document (OECD, 2017) provides a context for these types of assays and clearly states that more weight should be given to the results from tests that measure permanent DNA changes (i.e., mutations) than to DNA damage events that can be reversible. Primary DNA damage tests are useful for preliminary screening and can be useful in some types of mechanistic studies, such as an assessment of oxidative DNA damage and/or MOA assessment for cancer in specific target tissues. They can also serve as an exposure biomarker for in vivo studies to determine whether a test chemical and/or its metabolites can reach a specific target tissue and interact with DNA. Ultimately the goal in evaluating the potential for a test material to induce genetic damage is to determine if the material actually induces mutations. This can only be assessed using gene mutation assays. The chromosomal damage endpoints (CA and MN) do not actually measure mutations, as most of these events are not compatible with cell survival. However, the underlying events that cause microscopically visible chromosome damage and MN also cause mutation.
Relevant Recommendations from the Revised OECD TGs
The recently revised OECD TGs for the standard genetic toxicology assays provide improved guidance concerning assay conduct, acceptance criteria for individual test results, and an overall strategy for interpreting test data (positive/negative). A publication by Thybaud et al. (2017) and an OECD document entitled “Overview of the set of OECD Genetic Toxicology Guidelines and updates performed in 2014–2015” (OECD, 2017) provide details concerning the deliberations upon which the revisions were based and an overview of the most significant changes from previous versions of the TGs. TG471 for the Ames test was not revised and therefore there are no new considerations for the interpretation of data from this assay. While the major revisions occurred in the 2014–2015 versions of the TGs, minor administrative changes were made in 2016; the overview document was adopted in 2016 with minor administrative changes in 2017.
Of particular importance in interpreting previously published data for chemicals such as styrene and SO are the new recommendations for the in vitro mammalian assays concerning the appropriate top level of cytotoxicity (which should not be exceeded), the appropriate top concentration in the absence of cytotoxicity (which should not be exceeded), and the incorporation of the historical range of the spontaneous background levels, in interpreting whether a response is positive or negative. New cytotoxicity and concentration limits were established to ensure that any observed positive responses were biologically relevant, not simply resulting from excess cytotoxicity. In addition, to ensure appropriate statistical power, there are new recommendations for the number of cells that should be scored for the cytogenetic assays, the minimum number of cells that should be treated, maintained during the phenotypic expression, and cloned for mutant enumeration in gene mutation assays. For the cytogenetic assays, the historically scored number of cells (particularly for the older studies) often meant that the actual number of background events was too low to be accurately measured. It was difficult to determine if a small increase in the number of events in the treated group was, in fact, different from the background. Furthermore, random variability might result in an inaccurately high (or low) level of scored events in the background of one or more of the treated groups, thus characterizing responses as positive or negative that were not representative of the actual level of events. For the in vitro mammalian gene mutation assays, in which mutations are rare events (occurring as a few mutants per million cells), it is important to treat, carry, and sample enough cells to obtain an accurate value for the mutant frequency. Most of the cytogenetic and hypoxanthine–guanine phosphoribosyltransferase (hprt) gene mutation studies (particularly the older studies) conducted to evaluate styrene/SO utilized less (often much less) than the currently recommended number of cells.
The new recommendations for determining whether a test chemical is positive or negative bring into consideration the distribution of the historical negative controls. This is done in recognition of the fact that there is biological variability in the background levels of the various endpoints and that the use of even duplicate or triplicate cultures for in vitro assays and five animals per dose point for in vivo assays does not provide a mean value that is the actual mean of the distribution of values that would be obtained had a larger number (10–20 or more) of replicates been technically feasible to use. That is, the full extent of the random variation is not captured in a single experiment. This is especially an issue when the concurrent negative control falls at the low end of the historical control distribution, resulting in random increases in responses being characterized as positive responses. Furthermore, there is a chance (one in 20) when a statistical probability of 0.05% is used that a treated group will be statistically different from the concurrent negative control, when, in fact, there is really no difference. Based on these considerations, the general recommendation in the revised TGs requires that the following three criteria all be met for a clear positive response: (1) “at least one of the test conditions exhibits a statistically significant increase compared to the concurrent negative control,” (2) “the increase is concentration‐related when evaluated with an appropriate trend test,” and (3) “any of the results are outside the distribution of the historical negative control data (e.g., Poisson‐based 95% control limit).” A clear negative response would require that none of these three criteria are met. Of course, applying these criteria (particularly, the third criterion) to published data can be problematic, as there is very rarely any information on historical background ranges in a published paper, but the criteria can serve as a general basis for expert interpretation of data. That is, statistically different, but very small, increases above the concurrent negative control may not actually be reflective of a positive response and should not be considered definitive evidence that the response is positive.
CRITICAL REVIEW OF THE INDIVIDUAL STUDIES FOR STYRENE/SO
In the following sections, a concise description is provided for each of the evaluated endpoints and its relevance to understanding whether styrene and/or SO are mutagenic. For this critical review, individual publications were examined to determine the technical conduct of the assay, to assess whether the approach presented in the publication is consistent with current recommendations, and how the data presented in the publication should be interpreted. An overall weight‐of‐the‐evidence assessment is then provided for the overall database for each endpoint. Starting with the endpoints that are the most relevant for assessing the potential for styrene/SO to induce mutation (gene mutation, CA, and MN), the review details are presented below. As already indicated, the data for endpoints that assess exposure are not critically reviewed but are briefly summarized.
Assays that Assess Gene Mutation
As discussed in the OECD Overview document (OECD, 2017), assays that evaluate the potential for chemicals to cause gene mutation are the most relevant for determining the ability of chemicals to cause genetic damage that will be compatible with cell survival and, therefore, of potential consequence to the whole organism. In vitro gene mutation assays routinely used for hazard identification include the bacterial Ames test and assays using either the Tk or hprt genes in mammalian cells. Data for styrene/SO were identified using all three of these routine in vitro assays. Although in vivo gene mutation assays are available using hprt or transgenes in rodents, no such data were identified for styrene/SO.
Ames Test (TG471B87)
Starting as early as 1976, many investigators have studied the ability of styrene, SO, or other styrene metabolites to induce reverse mutations (point mutations) in a number of different Ames tester strains (OECD, 1997a). Many of these studies were conducted prior to the 1997 (current) OECD TG471 in which specific strains were recommended. Therefore, not all the currently recommended strains were included in these early studies. This has no impact on studies in which the results were positive, as only one strain needs to be positive for the overall result to be positive. However, it does mean that negative results reported for studies using less than the full set of strains should not be considered definitive. In addition, many of the older publications do not have the level of detail that is currently expected or that allows a full evaluation of the methods used. This prevents determining whether the studies were conducted in a way that would be fully compliant with TG471 (OECD, 1997a). Many of the older studies did not routinely include positive control chemicals to demonstrate that the assay, particularly the exogeneous activation system, was working properly. However, the Ames test has generally been conducted according to the methods that were originally published by Bruce Ames (Ames et al., 1975) and, unlike some of the other genetic toxicology tests, the methods and the interpretation of data have not changed substantially over the years. Therefore, unless there were specific reasons to question the methodology of a specific paper, it was assumed for purposes of this evaluation that the methods used in the various publications were generally OECD TG compliant. We considered a response to be positive if there was at least a two‐fold increase (three‐fold for strains with low backgrounds) in the observed number of revertants, above the background.
The majority of Ames test studies used SO as the test material. A concise summary of these individual studies, in chronological order, is provided in Table 2. In addition to the publications which provided Ames test data that could be evaluated for this review, there are publications that include study summaries for large numbers of chemicals and only indicate positive/negative mutagenicity results for styrene and/or SO. Table 3 lists these publications and includes the (limited) summary information presented in the publications.
Table 2.
Summary of Studies Evaluating Styrene Oxide in the Ames Test
| Assay version | Strains | Chemical purity | Range of test concentrations | Range of responses: lowest to highest tested concentration (revertants per plate) | Positive or negative | Reference |
|---|---|---|---|---|---|---|
| Plate incorporation | TA1537, TA1538 | “purest commercially available” | 1–200 μg/plate | No increase over background | Negative | Glatt et al. (1975) |
| Spot test | TA98, TA100, TA1535, TA1537, TA1538 | Not stated | 5 μL of liquid | TA1535: 535 (background = 135) TA100: 2,694 (background = 331) |
Positive | Milvy and Garro (1976) |
| Spotted on agar overlay or petri dish lid | TA100 | Not stated | 50–500 μg/plate | Agar overlay: 379–1,294 Petri dish lid: 294–3,092 |
Positive | Milvy and Garro (1976) |
| Plate incorporation | TA98, TA100, TA1535, TA1537, TA1538 | Not stated | 10−9–10−5 mol/plate | TA1535: 19–357 (without S9) and 18–484 (with S9) TA100: 115–1,111 (without S9) and 108–1,165 (with S9) |
Positive | Vainio et al. (1976) |
| Plate incorporation | TA98, TA100, TA1535, TA1537, TA1538 | 97% purity | 1 nmol/plate to 100 μmol/plate | TA100 and TA1535 (graphical data) | Positive | de Meester et al. (1977) |
| Plate incorporation | TA98, TA100, TA1535, TA1537, TA1538 | Not stated | 50–1,000 μg/plate | TA1535 (graphical data). Clear dose response but not clear that the response reaches two‐fold | Unclear result | Stoltz and Whitey (1977) |
| Suspension (including assessment of toxicity) | TA100 | Not stated | 0.42–2.02 μmol/plate | 268–467 revertants per 109 survivors | Positive | Sugiura et al. (1978) |
| Preincubation | TA98, TA100, TA1535, TA1537, TA1538 | Purified by distillation | 10–500 μg/plate | TA100: 84–268 (without S9) TA1535: 34–195 (without S9) |
Positive | Watabe et al. (1978) |
| Plate incorporation | TA98, TA100, TA1535, TA1537, TA1538 | Not stated | 1–10 μmol/plate | TA100 and TA1535 (graphical data) |
Positive | Busk (1979) |
| Suspension | TA100 | Purified by distillation | 1–6 mM | Graphical data: clear dose response. Response decreased by the addition of S9 | Positive | Yoshikawa et al. (1980) |
| Plate incorporation | TA98, TA100, TA1535, TA1537 | Not stated | 125–1000 μg/plate (4) Without S9 |
TA1535: 46–183 TA100: 246–1,334 TA1537: 6–8 TA98: 31–38 |
Positive (TA1535, TA100) Negative (TA1537, TA98) | El‐Tantawy and Hammock (1980) |
| Suspension | TA100 | Not stated | 0–33 mM/plate (graphical data) | Graphical data, clear dose response (approx. 1,200 at 33 mM) | Positive | Turchi et al. (1981) |
| Plate incorporation (exposure in desiccator) | G46, TA98, TA100, TA1530, TA1535, TA1537, TA1538 | Not stated | 16% and 24% (v/v) styrene oxide in air | TA100, TA1530, TA1535 (graphical data—see discussion) | Positive | de Meester et al. (1981) |
| Plate incorporation | TA98, TA100, TA1535, TA1537, TA1538 | Not stated | 0.1–10 μmol/plate | TA100: 47–488 | Positive | Watabe et al. (1980) |
| Preincubation | TA100 | Not stated (testing included “R” and “S” enantiomers) | 1–5 μmol/tube (higher concentrations gave survival <20%) | Racemic mixture: 387–1,374 | Positive | Pagano et al. (1982) |
| Preincubation | TA100 and TA100 GSH− derivatives | Not stated | 1–5 mM | Graphical data—clear dose response | Positive | Kerklaan et al. (1985) |
| Plate incorporation | TA100 | Not stated | 300 and 600 μg/plate Without S9 (Used as positive control: nine separate experiments) |
0 μg/plate: 91–187 300 μg/plate: 275–435 600 μg/plate: 400–723 |
Positive | Cheh (1986) |
| Preincubation | TA100 | Purified by distillation | 0.01–10 μmol/tube | 108–1,406 | Positive | Rosman et al. (1986) |
| Plate incorporation Preincubation and Tedlar™ Bag | TA100, TA102 | “highest commercially available” | 50–1,000 μg/plate | Plate: TA100: 120–242; TA102: 247–309 Preincubation: TA100: 154–343; TA102: 268–410 Tedlar™ Bag: TA100: 132–122; TA102: 258–276 |
TA100: Positive (the preincubation method was the most sensitive) TA102: Negative |
Hughes et al. (1987) |
| Preincubation | TA100 | >98% purity (testing included “R” and “S” enantiomers) | 50–800 μg/plate | Graphical data. Clear dose response. The “R” enantiomer is more potent | Positive | Seiler (1990) |
| Plate incorporation | TA100 | “Pure” | 150–1,500 μg/plate (10) Without S9 (seven repeat experiments/two labs) |
186–1,450 | Positive | Claxton et al. (1991) |
| Preincubation | TA98, TA100 | 98% purity | 33–666 μg/plate without S9 33–3,333 μg/plate with S9 |
TA98: 16–34 TA100: 182–628 (without S9—response lower with S9) |
Positive: TA100 Negative: TA98 |
Zeiger et al. (1992) |
| Plate incorporation | TA100, TA104 | Not stated | 0.5–8 μM 0.5–32 μM Without S9 (two separate experiments) |
TA100: 144–2,532 TA104: 421–1,501 |
Positive | Einisto et al. (1993) |
| Plate incorporation | TA100 | Purified by chromatography (testing included “R” and “S” enantiomers) | 1–8 μmol/plate | “R”: 216–1,146 “S”: 180–717 |
Positive | Sinsheimer et al. (1993) |
Table 3.
Publications Providing Overall Positive/Negative Calls for the Ames Test But No Data
| Assay version/strains | Chemical information | Results | Reference |
|---|---|---|---|
| Styrene | |||
| Plate incorporation TA98, TA100, TA1535, TA1537, TA1538 | Commercial source, highest available purity | Negative (with and without S9) | Simmon et al. (1977) |
| Preincubation Tubes were capped for volatiles TA97, TA98, TA100, TA1535, TA1537 |
Label purity: 99+% Analyzed purity: 99+% |
Negative (with and without S9) | Dunkel and Simmon (1980), Dunkel et al. (1985), Zeiger (1987), and Zeiger et al. (1988) |
| Preincubation TA98, TA100, TA1537 | No information on purity or source | Negative | Ishidate et al. (1981) |
| Plate incorporation TA98, TA100, TA1535, TA1537, TA1538 | Reagent grade pure | Negative (with and without S9) | De Flora et al. (1984) |
| Plate incorporation TA97, TA98, TA100 | Commercial source, purity 99% | Positive in TA97 and TA100 (without S9; tested between 100–1,000 μg/plate). Note that the paper has conflicting statements as to whether their results were positive or negative | Brams et al. (1987) |
| Styrene Oxide | |||
| Plate incorporation TA1535 | No information on purity or source | Positive (described as a weak positive; without S9) | McCann et al. (1975) and McCann and Ames (1976) |
| Plate incorporation TA100, TA1535 | 97% purity | Positive | Wade et al. (1978) |
| Suspension TA100, E. coli WP2uvrA | Analytical grade | Positive in both strains (without S9) | Hemminki and Falck (1979) |
| Plate incorporation G46, TA98, TA100, TA1530, TA1535, TA1538 | Commercial source, no information on purity | Positive in TA100 (without S9) | Bartsch et al. (1980) |
| Preincubation | No information on purity or source | Positive | Ishidate et al. (1981) |
| Plate incorporation TA98, TA100 | Analytical grade | Positive (without S9; tested between 30–1,000 μg/plate) | Glatt et al. (1983) |
| Plate incorporation TA98, TA100, TA1535, TA1538, TA1537 | Reagent grade pure | Positive in TA100 and TA1537 Negative in TA1535, TA1537 and TA1538 | De Flora et al. (1984) |
| Plate incorporation TA98, TA100, TA1535 | Not provided | Positive (without S9) | Khudoley et al. (1987) |
| Plate incorporation TA97, TA98, TA100 | Commercial source, no information on purity | Positive in TA97 and TA100 (without S9; tested between 600–6,000 μg/plate) | Brams et al. (1987) |
| Plate incorporation TA98, TA100, TA1535, TA1538, TA1537 | Styrene‐7,8‐glycol Reagent grade pure |
Negative (with and without S9) | De Flora et al. (1984) |
Only a small subset of the Ames test studies used styrene as the test material. Many of these studies were designed to address questions such as metabolism, the mutagenicity of styrene metabolites, and/or the methods for exposure, and the methods used were nonstandard. These studies are summarized below in chronological order.
Vainio et al. (1976) evaluated styrene in strains TA98, TA100, TA1535, TA1537, and TA1538, both with and without S9, prepared from male Sprague Dawley rat liver, following induction with Clophen C. A data table from a preliminary screening, using concentrations 0, 10−8, 10−7, and 10−6 mol/plate, indicates that the number of revertants in TA1535 (with S9) were 16, 112, 84, and 16, respectively; in TA100 (without S9) were 83, 115, 56, and 197, respectively; and in TA100 (with S9) were 97, 163, 78, and 206, respectively. A more detailed presentation of results is graphically provided in the publication, for the evaluation in TA1535 (with S9). Eight concentrations between 10−9 and 10−6 were used and there is clearly a dose‐response increase that exceeds the background by at least two‐fold, in the 10−9 to 10−8 concentration range, followed by a drop in the number of revertants per plate, likely due to toxicity. Based on this publication, styrene (both with and without S9) is weakly mutagenic in TA100 and clearly mutagenic (with S9) in TA1535. Styrene glycol was also evaluated (with S9) and was nonmutagenic.
Milvy and Garro (1976) evaluated a series of styrene metabolites (SO, styrene glycol, d‐ and l‐MA, PGA, benzyl alcohol, benzoic acid, and hippuric acid) using the spot test version of the Ames test. The test was conducted using TA98, TA100, TA1535, TA1537, and TA1538. In this screening assay, only SO was positive, and these positive results were seen in TA100 and TA1535.
de Meester et al. (1977) provided data indicating that styrene is not mutagenic (strains TA98, TA100, TA1535, TA1537, and TA1538) without S9 activation at concentrations between 1 and 100 μM/plate. A positive response was observed in test strain TA1535 with Aroclor‐1254‐induced male Wistar rat liver S9. Fifteen different concentrations of styrene from 1 to 15 μM/plate were evaluated. The assay was conducted using a protocol that determined cytotoxicity based on % bacterial survival, thus demonstrating a clear dose‐responsive increase in cytotoxicity. The number of revertants per plate increased from a background of 14 ± 3 to 136 ± 19 at a concentration of 11 μM/plate. It should be noted that this top concentration resulted in a 5% survival for the bacteria; however, a positive response was observed at multiple lower, and less cytotoxic, concentrations. These authors also investigated whether the mutagenicity of SO would be altered with S9. Without S9, SO was mutagenic (in strains TA100 and TA1535) within a range of 100 nM/plate to 20 μM/plate. With S9, higher SO concentrations were required (from 4 to 60 μM/plate) to observe a positive response, suggesting a decrease in mutagenicity related to SO metabolism. Although the data for styrene were tabulated, the data for SO were only presented graphically; however, it was clear that the SO response exceeded two‐fold above background and demonstrated a dose‐response. The authors speculated that the lower response for SO exposure with S9 might be due to further metabolism of SO to styrene glycol which is known to be nonmutagenic in TA1535.
Using both the spot test and the plate incorporation method, Stoltz and Whitey (1977) evaluated styrene, with Aroclor 1254‐induced rat and hamster liver S9, in strains TA98, TA100, TA1535, TA1537, and TA1538. They used styrene concentrations up to one milligram per plate and found the results to be negative.
Watabe et al. (1978) presented a series of experiments investigating the mutagenicity of styrene in strains TA98, TA100, TA1535, TA1537, and TA1538. As their primary goal was to investigate the metabolism of styrene, they used S9 mixes prepared from Wistar rat livers that were induced using several different inducing agents (3‐methylcholanthrene [3‐MC], phenobarbital [PB], and polychlorinated biphenyls [PCBs]). Gas chromatography (GC) was used to evaluate the conversion of styrene to SO and styrene glycol with the different S9 mixes under the same conditions that were used for the mutation experiments but without the presence of the bacteria. The authors tested styrene in the presence of PCB‐induced S9 up to a concentration of 500 μg/plate in the five strains and found no increase in the number of revertants for any of the strains. From the data presented in Table 1 of the publication, it appears that the 500 μg/plate concentration was cytotoxic to all strains except for TA100. To further investigate the potential mutagenicity of styrene, experiments were conducted using S9 mixes prepared from all the above listed inducers and in the presence of 1,1,1‐trichloro‐2,3‐propene oxide (TCPO), an inhibitor of epoxide hydratase which would be expected to prolong the half‐life of any SO that was produced by the S9 metabolic activation. For these experiments, the Ames test was conducted using TA100. To obtain a mutant frequency, the number of revertants were expressed per surviving bacteria rather than as the number of revertants per plate. Only two concentrations of styrene (three and six millimolar) were used. There was no increase in the mutant frequency for the uninduced S9 or the PCB‐induced S9. The six millimolar styrene concentration coupled with PB‐induced or 3‐MC‐induced S9 caused an increase in the mutant frequency (83 and 477 induced mutants per 108 surviving bacteria, respectively). In both cases, however, this increase in mutant frequency occurred at very high levels of cytotoxicity (9% and 6% survival, respectively). It should be noted that the Ames test is not normally conducted in a way to quantify the level of cytotoxicity and, therefore, there is no recommended maximum level of cytotoxicity. If one were to apply the 10% level recommended for the mammalian cell gene mutation assays, then it would be concluded that these results were not biologically relevant. To further investigate the response with the 3‐MC‐induced S9, the authors conducted additional experiments using TA100 and four millimolar of both styrene and SO, with and without the addition of TCPO. With 3‐MC S9 and TCPO, the four millimolar styrene exposure yielded a survival of 30% and an induced mutant frequency of 91 per 108 surviving bacteria. SO, conversely, showed an induced mutant frequency of 235 per 108 surviving bacteria (at 51% survival), which was not observed with the addition of 3‐MC S9 alone (87% survival) or the addition of both 3‐MC S9 and TCPO (84% survival). These last two treatment conditions showed induced mutant frequencies of 17 and 54 per 108 surviving bacteria, respectively. The GC analysis showed that the SO formed during the styrene treatment was rapidly inactivated to styrene glycol. The addition of TCPO prolonged the half‐life of SO, as expected. When the analysis was conducted using the variously induced S9 mixes, it was determined that the highest accumulation of SO occurred in the presence of the 3‐MC‐induced S9 plus TCPO and that the lowest accumulation was with the PCB‐induced S9 plus TCPO.
Watabe et al. (1978) also evaluated the mutagenicity of 1‐vinylbenzene 3,4‐oxide (styrene 3,4‐oxide) in TA100, T98, and TA1537. Although they only used four concentrations with S9 (the highest of which was cytotoxic based on a downturn in the dose‐response curve), there was an overall positive dose‐response and at least a doubling of the number of revertants per plate in TA100.
Busk (1979) evaluated styrene using strains TA98, TA100, TA1535, TA1537, and TA1538, and a concentration range of 10−3–15 μM/plate, with and without S9, and found styrene to be negative. With the goal of modifying the metabolic activation for styrene, S9 preparations were made from the livers of Aroclor 1254‐ and Clophen C‐induced rats. In addition, two metabolic inhibitors, including TCPO and diethyl maleate (to inhibit glutathione conjugation), were added to the S9/styrene exposure. This strategy was an attempt to prevent further metabolism of any SO that might be formed by the S9 metabolic activation system. In all cases, styrene was found to be nonmutagenic.
de Meester et al. (1981) speculated that poor solubility and the volatility of styrene may contribute to the divergent results that had been reported for styrene. They conducted a study in which plates containing the bacteria and S9 were placed in a desiccator filled with either styrene or SO mixed with air for a 24‐hr exposure. Strains TA100, TA1530, and TA1535, exposed to 24% (v/v) styrene in air, all showed an increase in the number of revertants per plate that, based on a visual analysis of the graphic data, appear to be at least twice the background number of revertants. Graphical dose‐response data are presented for TA1530 for five concentrations (2666–13,330 ppm) of styrene, and there does appear to be a positive dose‐response, which plateaus (at 8000 ppm) and then declines at 13,300 ppm. Exposure to 24% (v/v) SO in air was also mutagenic to the same three strains with a similar system. The presence of S9 lowered the number of revertants per plate compared to what was observed with the SO exposure alone.
Considering all the available Ames test data, including the very large number of SO studies, it is clear that styrene via SO metabolism is mutagenic, in vitro, in the Ames test.
In Vitro Mammalian Gene Mutation Assays
The in vitro mammalian cell gene mutation assays for which either styrene or SO data are available include the mouse lymphoma thymidine kinase (Tk) assay (often called the mouse lymphoma assay [MLA]) and the hprt assay. hprt assay data were identified using exposure to V79 cells, two human lymphoblastoid cell lines, and primary human T‐lymphocytes. Brief details of the publications including a summary of the methods and results as well as our critical review and overall interpretation of the individual study data are provided in Table 4. A description of both of the in vitro mammalian gene mutation assays and weight‐of‐the‐evidence conclusions as to whether styrene/SO can induce mutations in mammalian cells are as follows.
Table 4.
Summary of Studies Evaluating Styrene/SO in in vitro Gene Mutation Assays
| Assay | Methods | Results | Comments | Reference |
|---|---|---|---|---|
| Styrene | ||||
| HPRT‐V79 cells | Methods poorly described. One hour treatment. 8.5 and 17 mM styrene. Three expression periods (72, 90, and 114 hr). 8‐azaguanine selection. | Styrene, even at 17 mM showed little cytotoxicity and little increase in MF. | Not OECD TG476 compliant. Unclear whether sufficient cells were used. Expression period too short. Does not appear that cells were subcultured during expression. Very low number of viable cells reported in the cultures (Table 2 of the publication). Uninterpretable. | Loprieno et al. (1976) |
| HPRT‐V79 cells | Both liver perfusion system and S9 experiments. 240 and 480 μM styrene used in perfusion study. 250, 480, and 960 μM styrene used with and without S9. Concentrations of styrene and SO evaluated in the perfusion mixture. Unclear how many cells treated, but only 4 × 105 cells carried during expression. 6‐thioguanine selection. | The exposed cultures (both methods) showed little cytotoxicity. While there was some increase in the MF with exposure in the liver perfusion, there was substantial variability. The with and without S9 showed little to no increase in MF and there was substantial variability. The styrene was metabolized within two hour. During this time, the SO was measured at only 2%–4% of the styrene concentration at the beginning of the experiments. | Not OECD TG476 compliant. Uninterpretable. | Beije and Jenssen (1982) |
| Styrene Oxide | ||||
| Mouse Lymphoma‐TK TK+/− 3.7.2C cells | Agar version. Three hour treatment with and without S9. Without S9: nine doses (0–103.6 μg/mL). With S9: nine doses (0–10.5 μg/mL). Two‐day expression. TFT selection. Automatic counter set to count only large colonies. | Without S9: MF from 40 to 590 × 106 at RTGs above 10%. With S9: MF from 58 to 94 × 106 at RTGs above 10%. | Not OECD TG490 compliant. Did not count the small colony mutants; however, the result was positive without S9 (showed a dose response and exceeded the GEF) based solely on the large colony MF. With S9, result was uninterpretable. | Amacher and Turner (1982) |
| HPRT‐V79 cells | Methods poorly described. One hour treatment. 4.25, 8.5, 17, and 25 mM SO. Three expression periods (72, 90, and 114 hr). 8‐azaguanine selection. | SO was cytotoxic at 17 and 25 mM and there was an increase in MF. | Not OECD TG476 compliant. Unclear whether sufficient cells were used. Expression period too short. Does not appear that cells were subcultured during expression. Very low number of viable cells reported in the cultures (Table 2). Uninterpretable. | Loprieno et al. (1976) |
| HPRT‐V79 cells | Methods indicated as that of Loprieno et al. (1976). The focus of the study was to examine expression time. 8.5 and 17 mM with one hour exposure. Different expression times (0, 66, 90, 114, 162, and 210 hr). EMS as a positive control, but no negative control. | The observed MF increases and then decreases with increasing expression time in some experiments, but not in others. There are some cultures with apparently “high” MFs, but no negative control and insufficient number of concentrations to assess. | Not OECD TG476 compliant. No negative control. Cloning efficiencies for some cultures that appear to be positive are too low (8–25%). Three of the five experiments used concentrations (17 mM) that are substantially above the recommended maximum (10 mM). Uninterpretable. | Bonatti et al. (1978) |
| HPRT‐V79 cells | Cytotoxicity and mutagenicity assessed in different treated cultures. Unclear how many cells treated, but does not appear to be sufficient. Four hour treatment. Eight‐day expression. Six TG selection. | Data presented graphically, not possible to evaluate response. | Not OECD TG476 compliant. Data only presented graphically and could not be evaluated. Uninterpretable. | Sugiura et al. (1979) |
| HPRT‐V79 cells | One hour treatment. Different expression times up to 210 hr. Six TG selection. | Data presented graphically, and the SO data presented appear to be from another publication. | Not OECD TG476 compliant. SO data not clearly presented. Uninterpretable. | Turchi et al. (1981) |
| HPRT‐V79 cells | Both liver perfusion system and S9 experiments. 240 μM SO used in perfusion study. 104 and 208 μM SO used without S9. Unclear how many cells treated, but only 4 × 105 cells carried during expression. | In the liver perfusion study, there was little cytotoxicity and no increase in MF. The SO was totally metabolized within 30 min. Without activation, the SO induced cytotoxicity, but little increase in MF. | Not OECD TG476 compliant. Uninterpretable. | Beije and Jenssen (1982) |
| HPRT‐V79 cells | Insufficient detail for the methods. This was a 40 compound study. Unclear how many cells were used but appears not to be sufficient. Six‐day expression, six TG selection. | Data presented graphically and impossible to interpret. | Not OECD TG476 compliant. Uninterpretable. | Nishi et al. (1984a) |
| HPRT‐V79 cells | Insufficient detail for the methods. Focus was the association between the induction of SCEs and mutation. Multiple chemicals tested—basically the same set as in Nishi et al. (1984a). Three hour treatment. Six‐day expression, six TG selection. Unclear how many cells were used but appears to be insufficient. | Data presented graphically comparing the number of induced SCEs/cell vs. the number of induced mutants per 106 viable cells. Although in the text, the authors indicate that SO was a weak inducer of mutation, it is impossible, in the graph, to see any real increase in the number of mutants, particularly when compared with the mutant frequencies that are clearly induced by other known mutagens such as methyl nitrosourea, ethyl nitrosourea, ethyl methanesulfonate, ICR 191 and ICR 170. | Not OECD TG476 compliant. Uninterpretable. | Nishi et al. (1984b) |
| Human T‐lymphocytes | Exposed to SO for 24 hr or 6 days and 8 days for expression. Nine different experiments with nine different donors. | While the authors indicate that increases in the hprt mutant frequency were observed, a close look at the data indicates that most of the cell cultures had very poor (less than 20%) survival after the end of treatment. Furthermore, the number of cell doublings during the eight‐day expression time was very low, ranging from 1.2 to 3.7 (primarily in the untreated cultures). | Not OECD TG476 compliant. Appears that the number of cells used was inadequate. Very poor cell survival/growth. Uninterpretable. |
Bastlová et al. (1995) |
| Two human B‐cell lymphoblastoid cell lines differing in glutathione‐S‐transferase gene status | The methods in one paper (Shield and Sanderson, 2004) were reasonably well‐detailed and it was possible to determine that sufficient cells were exposed and an appropriate expression time and selective agent were used. In the second paper (Shield and Sanderson, 2001), an insufficient number of cells were treated. | In both studies, based on the graphical presentation of the data, the cell lines deficient in GSTM1 showed a greater degree of cytotoxicity (assessed by % population growth over eight days) and higher mutant frequency than the cell lines expressing GSTM1 activity. | Not OECD TG476 compliant. The recommended cytotoxicity method for assessing the appropriateness of concentration selection was not used in either study making it impossible to determine if appropriate concentrations (per TG476) were used. Uninterpretable. | Shield and Sanderson (2001, 2004) |
MLA using the Tk gene (TG490)
The MLA, using L5178Y TK+/− 3.7.2C cells, detects a wide spectrum of genetic events, including both single gene mutations and viable chromosomal events (including chromosomal rearrangements, deletions, and mitotic recombination; OECD, 2016b). To evaluate the full array of mutational events, careful attention must be paid to the optimal quantification of all the mutants, both small and large colony mutants. The MLA has historically been a part of OECD TG476 (OECD, 2016c). A new TG (TG490) was written to incorporate specific internationally agreed upon recommendations for use in the conduct and interpretation of data from this assay (OECD, 2016b). These recommendations include criteria for defining an acceptable assay based on ensuring optimal mutant colony growth, as well as the use of a global evaluation factor in the interpretation of data (positive/negative). For styrene, no studies using the MLA were identified. Amacher and Turner (1982) evaluated SO for its ability to induce mutation in the MLA, with and without S9 metabolic activation. The protocol was not compliant with OECD TG490, because the methods used to grow and count the mutant colonies were suboptimal. That is, only a subset of the mutants (the large colony mutants) was counted, thus losing the ability to detect the small colony mutants. However, without S9, SO was clearly positive (using current data interpretation criteria [the global evaluation factor]) based solely on the large colony Tk mutant response. With S9, the response is clearly different from the response without S9 and was not positive. The cytotoxicity of the SO was much lower without S9 than with S9; substantially higher concentrations of SO were needed to attain a cytotoxicity level (based on relative total growth) between 10% and 20% without S9. Despite the deficiencies in the assay conduct, we concluded that SO without S9 was mutagenic in the MLA.
Gene mutation assays using the hprt gene (TG476)
The hprt assay detects primarily gene mutations, including point mutations and small‐scale deletions; however, there is evidence that some larger deletions can be detected (OECD, 2016c). New recommendations for conducting the assay include ensuring that a sufficient number of cells are used for appropriate statistical power and to reduce variability that can result from using insufficient cell numbers to quantify rare events. The new recommendation is that 20 million cells be treated and no less than 2 million be present during expression and mutant selection. Two studies evaluating styrene and nine studies evaluating SO for its ability to induce hprt gene mutation in cultured mammalian cells were reviewed (see Table 4). Most studies were published prior to the mid‐1980s. Many of the publications did not provide an adequate description of the methods. For many papers, it was not possible to determine how many cells were used. No publications were identified that used methods consistent with the current recommendations. Several of the publications presented data only in graphic form that was impossible to evaluate. Deficiencies (at least two) were identified in every study and therefore, based on this published information, it is not possible to determine whether styrene or SO is mutagenic in the in vitro hprt assay.
Weight‐Of‐The‐Evidence: Does Styrene/SO Induce Gene Mutations In Vitro?
For the Ames test, there are studies for styrene exposure that give positive results and studies that give negative results. Some investigators attributed at least part of the differences to the technical aspects of evaluating a volatile chemical. However, the underlying cause for these differences may simply be based on differences in the metabolic system(s) that were used in the various studies. Based on the literature, it is unclear whether unmetabolized styrene is mutagenic. However, it is clear that SO is mutagenic in the Ames test. Overall, the Ames test results indicate that when styrene is metabolized to SO, it is positive in the Ames test. Most of the positive results for SO exposure were obtained without exogenous metabolic activation and, thus, the SO is not further metabolized. There is a balance between the activation of styrene to SO and the detoxification of SO, and the end result for a particular study depends upon this balance (Vainio et al., 1981; Norppa and Vainio, 1983). In the section above, several studies designed to provide insight into styrene metabolism and Ames test results are described.
For the in vitro mammalian cell gene mutation studies, none of the assays using the hprt locus were interpretable, based on at least two deficiencies in every study. Although the single MLA study had technical shortcomings, it was possible to identify SO as mutagenic. Thus, based on this single study, we conclude that SO is capable of inducing gene mutations in mammalian cells in culture. As there were no interpretable studies on styrene, no conclusion can be drawn as to whether unmetabolized styrene can cause gene mutation in mammalian cells in culture.
Although there are several methods and an OECD TG for evaluating in vivo induction of gene mutation, there were no studies identified for rodents treated in vivo with either styrene or SO and evaluated for the induction of gene mutation.
Assays that Assess Chromosomal Damage
There were several publications identified in which the authors evaluated styrene or SO for the ability to induce CA or MN, with either in vitro or in vivo exposures. Unfortunately, many of the in vitro studies were conducted and published prior to the recognition that excessive cytotoxicity can result in biologically irrelevant positive responses for the CA and MN assays (Lorge et al., 2008; OECD, 2017), and the introduction of the requirement to include an appropriate measure of cytotoxicity in the experimental design.
CA Assays
CAs are microscopically visible alterations to chromosomes. They include visible breaks, deletions, and rearranged sections of chromosomes. There are standard recommendations as to how they should be scored and reported. Gaps (defined as a nonstaining region of the chromatid with minimal misalignment of that chromatid) should be scored and reported but not considered in the decision as to whether the test chemical can induce chromosome breakage. It is important to note that some of the older studies included gaps or other nonstandard events in the reported values.
CAs in cultured mammalian cells (TG473)
As previously discussed, the revised TG for this assay incorporates new recommendations for appropriate measures of cytotoxicity, maximum levels of cytotoxicity, top concentration in the absence of cytotoxicity, and data interpretation (OECD, 2016d). In addition, the number of metaphases recommended for scoring (per test concentration) has been increased to 300 from the numbers recommended in previous TGs. Four studies for styrene and seven studies for SO have been reported and are summarized chronologically in Table 5. Only one of the studies met the criteria of the revised TG which would allow for its interpretation as a positive or negative study. The Jantunen et al. (1986) study shows that when there is metabolic capability, styrene can induce CAs in mammalian cells in culture. The details and rationale for judging this study as positive are as follows.
Table 5.
Summary of Studies Evaluating Styrene/SO in the in vitro Chromosome Aberration Assay
| Cell type | Methods | Results | Comments | Reference |
|---|---|---|---|---|
| Styrene | ||||
| Human lymphocytes | Single concentration: 0.03% v/v. Cytotoxicity measure: mitotic index. Chromosomal abnormalities, including interphase cells with MN, nuclear bridges, aneuploidy, polyploidy, breaks, and pulverized chromatids. | Mitotic index data not presented. | Not OECD TG473 compliant. The lack of an appropriate concurrent measure of cytotoxicity, scoring of events that are not currently normally scored and the use of a single concentration make this experiment uninterpretable. | Linnainmaa et al. (1978a, 1978b) |
| CHL cells | Methods not fully described. Scoring included polyploid cells in addition to structural aberrations. | The authors reported that styrene did not induce chromosomal damage; however, no data to evaluate. | Not OECD TG473 compliant. Insufficient data to evaluate. Uninterpretable. | Ishidate et al. (1981) |
| Human lymphocytes | Peripheral blood lymphocytes from a 29‐year‐old healthy female donor. Treated with concentrations between 5 × 10−4 and 5 × 10−6 mol/mL for 24 hr. No S9. When possible 200 metaphases scored per culture. | Data presented graphically. Authors report aberration percentage of 5% at the highest concentration (compared to 1.5% in the untreated control); however, there does not appear to be a dose‐related response and only the top concentration appears different than the untreated control. | Not OECD TG473 compliant. No measure of cytotoxicity. Uninterpretable. | Pohlová et al. (1984) |
| Human lymphocytes | Cultures with and without erythrocytes. Five concentrations between 0.5 and 6 mM styrene. Mitotic index used for cytotoxicity. 200 cells scored per culture. | Full tabulation of data. With erythrocytes: 4 mM culture had an appropriate level of cytotoxicity and was clearly positive (19 ± 3.0 aberrations per 100 cells compared to the untreated control which contained 2.0 ± 0 aberrations per 100 cells). Without erythrocytes: two concentrations (1 and 2 mM) of styrene with acceptable levels of cytotoxicity. Both concentrations appear to be positive (4.5 ± 0.5 and 7.0 ± 5.0 aberrations per 100 cells compared to the untreated control 1.5 ± 0.5). | Positive. | Jantunen et al. (1986) |
| Styrene Oxide | ||||
| Human whole blood cultures | Methods not fully described. Two concentrations: 0.1 and 0.5 mM. 100 cells from control and 200 from each of two treated cultures scored. Gaps were included. | When gaps are excluded, the negative control had no aberrations, the low concentration had five aberrations and the top concentration had seven aberrations. | Not OECD TG473 compliant. Because there was no measure of cytotoxicity, only two test concentrations and insufficient technical detail, the study is considered uninterpretable. | Fabry et al. (1978) |
| Human lymphocytes | Single concentration: 0.008% v/v. Cytotoxicity measure: mitotic index. Chromosomal abnormalities, including interphase cells with MN, nuclear bridges, aneuploidy, polyploidy, breaks, and pulverized chromatids. | Mitotic index data not presented. | Not OECD TG473 compliant. The lack of an appropriate concurrent measure of cytotoxicity, scoring of events that are not currently normally scored and the use of a single concentration make this experiment uninterpretable. | Linnainmaa et al. (1978a, 1978b) |
| CHL cells | A single concentration (2.4 mM or 0.25 mg/mL) for styrene treatment was presented. No measure of cytotoxicity. |
While no measure of cytotoxicity was used in the study, which evaluated a number of chemicals, it is clear from the text that concentrations were used for many of the test materials that resulted in no metaphases. Although the incidence of CAs was clearly much higher (and clearly positive) with S9 activation than without, it is not possible to determine the level of cytotoxicity attained in this culture. | Not OECD TG473 compliant. The lack of an appropriate concurrent measure of cytotoxicity, and only a single concentration make the experiment uninterpretable. | Matsuoka et al. (1979) |
| CHL cells | More than 400 chemicals were tested. Treatment times of 24 and 48 hr. Both with and without S9. Otherwise methods not fully described and no data provided. | No data included. | Not OECD TG473 compliant. No data. Uninterpretable. | Ishidate and Yoshikawa (1980) |
| CHL cells | Methods not fully described. Scoring included polyploid cells in addition to structural aberrations. | Reported data only a calculation of the concentration in which 20% of the cells had aberrations. For SO, this concentration was reported to be 0.057 mg/mL. | Not OECD TG473 compliant. Insufficient data to evaluate. Uninterpretable. | Ishidate et al. (1981) |
| Human lymphocytes | PHA‐stimulated lymphocytes from peripheral blood of healthy male donor. Three concentrations (0.05, 0.20, and 0.40 mM), 48 hr exposure. When possible 200 cells scored per culture. No measure of cytotoxicity. | The top concentration used for styrene resulted in no metaphases that could be scored. Although the middle concentration yielded a response that would clearly be positive, in the absence of an appropriate cytotoxicity measure it is not possible to determine if that concentration was excessively cytotoxic. | Not OECD TG473 compliant. No measure of cytotoxicity. Uninterpretable. | Norppa et al. (1981) |
| Human lymphocytes | Peripheral blood lymphocytes from a 29‐year‐old healthy female donor. Treated with concentrations between 1 × 10−3 and 5 × 10−6 mol/mL for 24 hr. No S9. When possible 200 metaphases scored per culture. | Data presented graphically. Authors report aberration percentage of 13.8% at the highest concentration (compared to 2% in the untreated control); there does appear to be a dose‐related response. | Not OECD TG473 compliant. No measure of cytotoxicity. Uninterpretable. | Pohlová et al. (1984) |
Jantunen et al. (1986) used cultured human lymphocytes (both with and without erythrocytes) to evaluate the ability of styrene to induce CAs. The erythrocytes were added to provide metabolic capability. The response with styrene was clearly positive (a clear dose‐dependent increase) in the whole blood cultures with a weaker positive response in the lymphocyte cultures that did not include erythrocytes. As a measure of cytotoxicity, the authors used mitotic index which is acceptable for use with lymphocytes in primary cultures (per the 2016 version of TG 473). The data are fully tabulated in the publication, thus allowing for a full review of the information. Five concentrations ranging between 0.5 and 6 mM styrene were used for the study. The top concentration was clearly too cytotoxic to give reliable results, but the four millimolar culture had an appropriate level of cytotoxicity and was clearly positive (19 ± 3.0 aberrations per 100 cells compared to the untreated control, which contained 2.0 ± 0 aberrations per 100 cells). In the experiment investigating exposure to lymphocytes without erythrocytes, there were only two concentrations (one and two millimolar) of styrene with acceptable levels of cytotoxicity. The one millimolar concentration was positive (4.5 ± 0.5 aberrations per 100 cells compared to the untreated control 1.5 ± 0.5). The higher two millimolar styrene concentration showed substantial variability and, therefore, it is less clear as to whether, without the erythrocytes, there is a positive response. It should be noted that the authors only scored 200 cells per concentration compared to the currently recommended 300 cells per test concentration. The increase from 200 to 300 cells for scoring was made to increase statistical power and to decrease statistical variability; the fact that the authors scored only 200 cells may have contributed to the high variability in the two millimolar, without erythrocytes, result. Given that the response in the whole blood cultures is clearly well above the concurrent background, it can be concluded that styrene is clastogenic in vitro.
In vivo rodent CAs (TG475)
CAs are generally evaluated in bone marrow cells of exposed rodents (OECD, 2016a). There are some studies for styrene in which lymphocytes were evaluated; however, lymphocytes are technically more difficult and historically have been used much less frequently than bone marrow cells. The new OECD recommendation is that 200 metaphases be evaluated per animal. There were eight studies identified that included CA evaluation following exposure to styrene and three studies for SO exposure. The key details for these studies are summarized in Table 6. In evaluating the relevance of a specific study, route of exposure is important. Inhalation is the most relevant exposure route for both styrene and SO. Six inhalation studies were identified for styrene and one for SO. For styrene, there was one study that used oral exposure and one study that used i.p. injection. For two of the SO studies, the route of exposure was i.p. injection. It should be noted that the new OECD TGs for in vivo genetic toxicology studies consider i.p. to be an irrelevant route of exposure. Of the 11 styrene/SO studies, 9 were uninterpretable because of noncompliance with OECD TG475 (OECD, 2016a) with all having multiple deficiencies (see Table 6). Reasons for noncompliance included using less than the recommended three treatment doses, fewer than the recommended number of animals per treatment group, no assessment of toxicity, insufficient number of cells scored, and the use of the i.p. route of exposure. For the two remaining studies (both using repeated inhalation exposure to styrene), the only deficiency was that less than the recommended 200 metaphases per animal were scored. Overall, these two studies were negative. Thus, based on these two studies, there is no evidence that inhalation exposure to styrene can induce CAs in vivo in rodents.
Table 6.
Rodent studies evaluating chromosome aberrations following exposure to styrene or styrene oxide
| Species/gender | Route | Doses and duration | Sampling times | Methods | Results | Comments | Reference |
|---|---|---|---|---|---|---|---|
| Styrene | |||||||
| Rat Wister/male | Inhalation, whole body | 300 ppm, 5 days/week, 6 hr/day, and 3 months. | Weekly at 2, 4, 6, 7, 8, 9, 10, and 11 weeks. | Bone marrow, one to five animals for each time point. Hundred metaphases evaluated. | Aberrant cells (minus gaps) ranged from one to nine in the treated animals and from zero to four in the controls. | Not OECD TG compliant. Only a single dose, no assessment of toxicity, unacceptably low number of animals at most time points. | Meretoja et al. (1978) |
| Chinese Hamster/male | Inhalation, whole body | 300 ppm, 4 days (6 hr/day and 3 hr on fourth day) and 21 days (6 hr/day and 3 hr on the last day). | Immediately after last exposure. | Bone marrow. Three controls. Four treated. 100–150 metaphases evaluated. | Four‐day treatment: no aberrations in either group. Twenty‐one‐day treatment: control: 0.7% and styrene treated: 0.5%. | Not OECD TG compliant. Only a single dose, no assessment of toxicity, unacceptably low number of animals per group. Insufficient cells scored to avoid 0%. | Norppa et al. (1980) |
| Mouse CD1/male | Oral | Four daily oral doses of 500 mg/kg and a 70‐day course of daily oral 200 mg/kg. | Twenty‐four hours after last exposure. | Bone marrow. Three controls. Six treated for the 500 mg/kg group and seven treated for the 200 mg/kg group. Generally, 100 metaphases scored. | Four‐day treatment—controls: 2.33%, treated: 2.0%. Seventy‐day treatment—controls: 3.33%, treated: 1.66%. Mitotic index was not affected by treatment. Cyclophosphamide positive control used. Note that urinary styrene metabolites were evaluated. | Not OECD TG compliant. Only a single dose used for the two treatment times, unacceptably low number of animals per group. | Sbrana et al. (1983) |
| Rats Sprague–Dawley/male and female | Inhalation, whole body | 600 and 1,000 ppm, 6 hr/day, 5 days/week, 12 months. | After last exposure. | Bone marrow. Four animals per group. Hundred metaphases photographed but only 73–94 cells were scored. | The treated animals did not have more aberrations than the controls. Males: four control and three treated animals (per exposure group) scored. Only two (control) animals had any cells with aberrations when gaps were excluded. Females: four animals scored per group. No animals had aberrations when gaps were excluded. |
Not OECD TG compliant. Insufficient cells scored to avoid 0%. Only two treatment groups. Unacceptably low number of animals per group. | Sinha et al. (1983) |
| Mouse C57B1/6/male | i.p. | 50, 250, 750, and 1,000 mg/kg. Single injection. | 16 hr after BUdRa implant and styrene injection. | Bone marrow Implanted BUdR tablet, i.p. injection of styrene 30 min after tablet implant. Four animals per group, except in the top two dose groups where death resulted in three and one animal scored. One hundred first division cells scored. First division cells identified by the BUdR incorporation. |
% aberrant cells: Negative control (no BUdR): 0.75 ± 0.96. Negative control (with BUdR): 0.5 ± 0.58 Solvent control: 0.25 ± 0.5. There was no increase in the 5 aberrant cells in the treated animals and the top dose animal had 0% aberrant cells. Mitotic index was severely decreased at the top two doses in the animals that survived (and were scored). |
Not OECD TG compliant. Number of animals per group is below the OECD recommended (five), particularly in the top dose groups. i.p. is no longer considered a relevant route of exposure. The use of BUdR is not standard. | Sharief et al. (1986) |
| Mouse B6C3F1/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Mononuclear leucocytes from the blood or spleen from six animals were cultured with BUdR. The Lung cells isolated and cultured and BUdR added (from eight animals). Where possible 100 first division metaphases scored. | There was no evidence of a positive response. Spleen: the controls ranged from 0% to 2%. The treated animals ranged from 0% to 3%. One animal in the top dose group had too few cells to score. Lung: the controls ranged from 3% to 6%. The treated animals ranged from 1% to 13%. The 13% was observed in a single middle dose animal; 9% was the next highest observed value. | Reasonably OECD TG compliant for the mononuclear leucocytes studies. Did not score the recommended 200 cells. The evaluation of CAs in lung cells does not have a standard method and therefore not easy to interpret. The use of BUdR is not standard. Overall a negative response. | Kligerman et al. (1992, 1993) |
| Rats Fischer 344/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Peripheral blood lymphocytes with BUdR. Five animals per group. 100 first division metaphases scored. | Controls and treated animals were not statistically different. Control: 1.4% ± 1.7% and top dose‐treated group: 2.0% ± 1.2%. | Reasonably OECD TG compliant. Did not score the recommended 200 cells. The use of BUdR is not standard. Overall a negative response. | Kligerman et al. (1993) |
| Rats Fischer 344/male | Inhalation, whole body | 150, 500, and 1,000 ppm, 6 hr/day, 5 days/week for 4 weeks. | 1, 2, 3, 4 weeks and 4 weeks after last exposure. | Peripheral blood lymphocytes. Cultured with BUdR added 24 hr after culture initiation. Cells harvested 68 hr after culture initiation. A minimum of 25 cells scored. Four to six animals per group. Positive control: ethylene oxide. | No increase in the treated vs. controls. The controls ranged from 0.06% to 3.4%. The treated ranged from 0% to 3.2%. | Not OECD TG compliant. Insufficient cells scored. Several dosed groups had 0% aberrant cells in all animals. Some groups had less than five animals. The use of BUdR is not standard. These deficiencies make the results uninterpretable. | Preston and Abernethy (1993) |
| Styrene Oxide | |||||||
| Mouse BALB/c/male | i.p. | 125 or 250 mg/kg. Single injection. | 1, 2, 6, and 13 days. | Bone marrow, 157–200 cells scored, number of animals unclear. No data presented for the negative control. | One chromatid break was observed in 757 cells analyzed. | Uninterpretable—Methods not well‐described. Noted that “most of the mice treated with 250 or 125 mg/kg remained alive for several days.” Number of animals unclear. No negative control data. | Fabry et al. (1978) |
| Chinese hamster/male | Inhalation Whole Body and i.p. |
25, 50, 75, and 100 ppm. 9 hr (6 hr Day 1 and 3 hr Day 2), and 21 hr (6 hr daily for 3 days and 3 hr Day 4) (except for the top dose only 9 hr exposure; the animals showed signs of poisoning). The low dose also used for a 3‐week exposure (6 hr daily, 5 days/week). Single injection of 500 mg/kg (i.p.). | After last exposure for the inhalation and for i.p., 24 hr after injection. | Two to three animals in each treatment group; four animals in the low dose 3‐week exposure; five animals in the i.p. group (three died). Ten animals in the negative control. Two animals in the olive oil (vehicle control). Bone marrow. One hundred cells scored. | Negative control: 0.8% aberrations. Inhalation treated: 0%–1%, except for the 100 ppm which was 2.0%. Olive oil control for i.p. treatment: 2.0%. 500 mg/kg: two animals that were alive: 1.5% and the three animals that died: 12.3%. | Not OECD TG compliant. Insufficient number of animals in each dose group. Insufficient number of cells scored. The top doses used, particularly for i.p. were clearly too toxic. i.p. is no longer considered a relevant route of exposure. | Norppa et al. (1979) |
| Mouse CD1/male | i.p. | 100 mg/kg. Single injection. Both S and R enantiomers. | 24 hr after injection. | Bone marrow. Four animals per group. One hundred metaphases scored. Positive control DMBA.b | The R enantiomer was negative (control: 1.00% ± 0.82% and treated: 1.75% ± 0.96%). The S enantiomer was statistically different (control: 1.00% ± 0.82% and treated 2.75% ± 0.50%). | Not OECD TG compliant. A single‐dose level. Insufficient number of cells scored. Four animals per group. i.p. is no longer considered a relevant route of exposure. | Sinsheimer et al. (1993) |
BUdR, bromodeoxyuridine.
DMBA, 2,4‐dimethoxybenzaldehyde.
Weight‐of‐the‐evidence: Does styrene/SO induces CAs?
Considering all the published studies for styrene/SO, in which CA induction was evaluated as an endpoint, one in vitro mammalian study indicates that, when there is metabolic capability included, styrene can induce CAs in mammalian cells in culture. The weight‐of‐the‐evidence from the in vivo rodent studies indicates that repeated inhalation exposure to styrene does not induce CA.
MN Assays
The MN endpoint can be used to evaluate the ability of a test chemical to induce both chromosome breakage and aneuploidy. Historically, MN have been evaluated by microscopic analysis and most of the studies conducted with styrene/SO (in vitro and in vivo) have used manual microscopic scoring. More recently, new automated techniques have been developed using image analysis or flow cytometry to score MN which provide an easy way to evaluate more cells for higher statistical power, providing a more rigorous evaluation of the test material. One of the in vivo SO inhalation studies (Gate et al., 2012) used the flow cytometric scoring method.
MN in cultured mammalian cells (TG487)
As previously discussed, the revised TG for this assay incorporates new recommendations for new methods of analysis, appropriate measures of cytotoxicity, maximum levels of cytotoxicity, top concentration in the absence of cytotoxicity, and data interpretation (OECD, 2016e). No studies were identified using styrene exposures, and four studies were identified for SO exposure (summarized chronologically in Table 7). There were two studies that provided interpretable data, and both studies provide evidence that SO can induce MN in cultured mammalian cells. These two studies and the basis for our conclusion are described below.
Table 7.
Summary of Studies Evaluating Styrene/SO in the in vitro MN Assay
| Cell line | Methods | Results | Comments | Reference |
|---|---|---|---|---|
| Styrene | ||||
| No identified data | ||||
| Styrene Oxide | ||||
| V79 cells | Only a single concentration (0.75 mM). Three harvest times, 25, 40, and 50 hr. Both a cloning efficiency and a mitotic index. | The number of MN appears to be substantially higher in the single treated culture than in the control culture. | Not OECD TG487 compliant. Although the number of MN in this culture appears to be substantially higher than the number of MN in the control culture, the lack of an appropriate cytotoxicity measure and the fact that only one concentration is presented make the study uninterpretable. | Turchi et al. (1981) |
| Whole blood cultures from two male and two female donors | Cytokinesis‐block method. Cells were exposed to 10, 20, 50, 100, and 200 μM SO. Scoring for the presence of MN was accomplished using 1,000 binucleated cells per individual per test concentration. Cytotoxicity was assessed using CBPI (an OECD TG487 compliant method for cytotoxicity using lymphocytes). | All of the cultures were within the acceptable cytotoxicity range. The background frequency varied from 3 to 18 MN/1,000 cells and in all cases a clear induction of MN was observed in the cultures treated with SO. | OECD TG487 reasonably compliant (number of cells scored less than the current recommendation). Positive. | Laffon et al. (2001) |
| Whole blood cultures from 20 individuals | Whole blood cultures were exposed to SO (0.1, 0.2, or 0.3 mM), and 1,000 cells were scored for the presence of MN. | No difference in the MN frequency controls and treated. | Not OECD TG487 compliant. Because there was no measure of cytotoxicity the results are uninterpretable. | Godderis et al. (2006) |
| Whole blood cultures (from health male or female nonsmokers) | 500, 600, and 750 μM SO exposure. Cytokinesis‐block method. The study was designed to compare the addition of CytoB 2 hr after the initiation of exposure to whole blood cultures and at the start of exposure 1,000 cells scored per culture. The nucleus division index (i.e., the replicative index) was used as measure of cytotoxicity. | Data presented graphically. Clearly positive when tested using the standard in vitro assay. | OECD TG487 compliant. Positive. | Speit et al. (2012) |
Laffon et al. (2001) used whole blood cultures from two male and two female human donors to evaluate SO induction of MN using the cytokinesis‐block method. Cells were exposed to 10, 20, 50, 100, or 200 μM SO. Scoring for the presence of MN was accomplished using 1000 binucleated cells per individual per test concentration. Cytotoxicity was assessed using cytokinesis‐blocked proliferation index (CBPI) (an OECD TG487 compliant method for cytotoxicity using lymphocytes) and all cultures were within the acceptable cytotoxicity range. The background frequency varied from 3 to 18 MN/1000 cells and in all cases a clear induction of MN was observed in the cultures treated with SO. From this experiment, it can be concluded that SO can induce MN in vitro.
Speit et al. (2012) used the CBPI MN assay and whole blood cultures to evaluate the clastogenicity/aneugenicity of SO in human lymphocytes. As the study was conducted after the approval of TG487, the methods used for the experiments were based on the recommendations in that guideline. No exogenous metabolic activation system was used. Based on the graphical data presented in the publication, it can be concluded that SO was positive for the induction of MN.
In vivo rodent MN (TG474)
As with CAs, MN have historically been evaluated in bone marrow cells of exposed rodents, typically mouse (OECD, 2016f). Lymphocytes can also be evaluated but are technically more difficult and historically have been used much less frequently than bone marrow cells. There are two studies (Kligerman et al., 1992, 1993) for styrene in which lymphocytes were evaluated. More recently, peripheral blood erythrocytes have been evaluated using the flow cytometric method as described above, thus providing more statistical power and in addition allowing rats to serve as test animals. The new OECD recommendation is that 4000 cells be scored per animal.
There were 10 studies identified that included MN evaluation following in vivo exposure to styrene and four studies for in vivo SO exposure. The key details for these studies are summarized in Table 8. Of the 14 total studies, 10 were considered uninterpretable because of noncompliance with OECD TG474 (OECD, 2016f). Reasons for noncompliance included less than the recommended number of exposure levels, fewer than the recommended number of animals per treatment group, insufficient number of cells scored, and the use of the i.p. route of exposure. Studies that were judged to be noncompliant included multiple deficiencies. For inhalation, the most relevant route of exposure for both styrene and for SO, three studies reasonably compliant with OECD guidelines were identified for styrene exposure and one study identified for SO exposure. For the two Kligerman styrene studies (Kligerman et al., 1992, 1993), the one deficiency was that less than the currently recommended 4000 cells per animal were scored. Overall these two studies were negative. One inhalation study using both styrene and SO exposures (Gate et al., 2012) was well conducted using flow cytometric scoring of 20,000 cells, and it was possible to conclude that neither styrene nor SO exposure induced MN. Thus, we conclude that there is no evidence that styrene/SO is clastogenic/aneugenic in vivo in rodents via inhalation exposure.
Table 8.
Rodent Studies Evaluating MN Following Exposure to Styrene/SO
| Species/gender | Route | Doses and duration | Sampling times | Methods | Results | Comments | Reference |
|---|---|---|---|---|---|---|---|
| Styrene | |||||||
| Chinese hamster/gender not stated | i.p. | 1 g/kg. Single injection. | 30 hr after treatment. | Four treated and two controls. Bone marrow. Thousand PCEa and 1,000 NCEb scored. The ratio of PCE/NCE determined for one animal. | Control—PCEs: 1.75 ± 0.75, NCEs: 0.75 ± 0.48 MN/1,000 cells. Styrene treated—PCEs: 2.0 ± 0.38, NCEs: 1.50 ± 0.57 MN/1,000 cells. These were not statistically different. | Not OECD TG compliant. Only a single dose. Insufficient number of animals per group. Insufficient number of cells scored. i.p. is no longer considered a relevant route of exposure. | Penttila et al. (1980) |
| Mouse C57BL/6/male | i.p. | 250, 500, 1,000, and 1,500 mg/kg. Single injection. | 30 hr after treatment. | Four animals per treated group and 13 controls. Bone marrow. Two thousand PCE and 2,000 NCE scored. | Authors indicated that the response for PCEs was positive, however, statistically significant increases were seen at the 250, and 1,000 ppm exposure levels (and not the other doses), thus there was no dose response. The NCE response was negative. The 1,500 mg/kg was lethal to 2 of 4 animals. The ratio of PCEs/NCEs was decreased (dose response). | Not OECD TG compliant. i.p. is no longer considered a relevant route of exposure. Insufficient number of cells scored (however, 2,000 is better than the 1,000 that were generally used in this timeframe). The number of animals was below the recommendation. There was no dose response for the result that the authors called positive. | Norppa (1981) |
| Mouse LACA Swiss/male | i.p. | 150, 300, 450, and 600 mg/kg. Single injection. | 30, 48, and 72 hr posttreatment. | 5–10 animals per group. Bone marrow. Thousand PCEs scored. Ratio of PCE/NCE determined. | Detailed results presented in publication. The authors reported a statistically significant increase in the top dose group but only at the 48 hr time point. MN/1,000 PCEs: controls (1.3 ± 0.3) 600 mg/kg (4.2 ± 1.0). They note that this dose level was lethal to 50% of the mice. | Not OECD TG compliant. i.p. is no longer considered a relevant route of exposure. Insufficient number of cells scored. | Simula and Priestly (1992) |
| Rat Porton/male | i.p. | 300, 750, 1,500, and 3,000 mg/kg. Single injection. | 30, 48, and 72 hr post‐treatment. | Four to nine animals per group. Bone marrow. Thousand PCEs scored. Ratio of PCE/NCE determined. | No increase in the frequency of MN in the treated vs. controls for any dose or sampling time. MN/1,000 PCEs: controls varied from 1.2 to 1.3 and the treated varied from 0.9 to 2.0. | Not OECD TG compliant. i.p. is no longer considered a relevant route of exposure. Insufficient number of cells scored. | Simula and Priestly (1992) |
| Mouse B6C3F1/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Whole blood smears to score NCEs. Spleen cell cultures treated with cytochalasin B. Thousand binucleated splenocytes and 2,000 NCEs scored. | There was no difference between the controls and treated. Spleen: Controls ranged from 3 to 9 MN per 1,000 cells and top dose ranged between 5 and 11 MN per 1,000 cells (there were too few cells to score in one animal). Peripheral blood: Controls ranged from 1 to 4 MN per 1,000 NCEs and the top dose treated ranged between 1 and 6 MN per 1,000 NCEs. | Reasonably OECD TG compliant. Did not score the recommended 4,000 cells. Overall negative response. | Kligerman et al. (1992, 1993) |
| Rats Fischer 344/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Peripheral blood lymphocytes with cytochalasin B. Five animals per group. Bone marrow scored NCEs. About 1,000–2,000 NCEs and 1,000 splenocytes scored. | Controls and treated animals were not statistically different. Control: 2.4 ± 2.8 and top dose treated 2.0 ± 3.1 MN per 1,000 NCEs. Data for the splenocytes not presented but stated to be negative. | Reasonably OECD TG compliant. Did not score the recommended 4,000 cells. Overall a negative response. | Kligerman et al. (1993) |
| Mouse B6C3F1/male | Inhalation, whole body | 50 ppm, 8 hr. | 24 hr. | Seven animals, two used for blood styrene evaluation. Cells from the blood and bone marrow scored for MN using flow cytometry; 50,000 events collected for each sample. Positive control: benzene. | No increase in the frequency of MN in the treated vs. controls. SO was detected in the blood. | Not OECD TG compliant. Only a single dose. Goal of the study was to evaluate combined exposure to butadiene and styrene, not specifically for styrene alone. Overall, a well‐conducted study for the question addressed. | Leavens et al. (1997) |
| Mouse NMRI/male | Inhalation, whole body | 750 and 1,500 mg/m3, 6 hr/day, 7 days/week for 1, 3, 7, and 21 days. | Directly after last exposure. | Bone marrow. Six animals per group. Unclear how many cells scored. No positive control. | Data presented graphically. The high‐dose group had a statistically significant increase in MN at 7 days exposure (treated: 10.4 ± 2.5 per 1,000 cell, which was stated to be twice the control level) but not at 21 days exposure. | Not OECD TG compliant. Only two doses. Number of cells scored unclear, but likely to be 1,000 (which is less than the currently recommended number). The fact that the 7‐day exposure to the top dose was positive but not the 21 day exposure makes the result suspect. Note that styrene specific DNA adducts (7‐guanine and 1‐adenine) were detected in the lungs at all time points. | Vodicka et al. (2001) |
| Mouse NMRI/male | Inhalation, whole body | 750 and 1,500 mg/m3, 6 hr/day for 1, 3, 7, 14, or 21 days. | Shortly after last exposure. | Bone marrow. Five animals per group. Two separate labs independently evaluated the slides. Two thousand PCEs scored. Positive control: cyclophosphamide injected i.p. |
No increase in frequency of MN in the treated vs. controls. Laboratory 1: The control group means varied from 1.3%–3.0% and the treated varied from 1.7% to 3.2%. Laboratory 2: the control group means varied from 2.1% to 4.4% and the treated varied from 1.8% to 4.6%. | Not OECD TG compliant. Only two treatment groups. The top dose caused lethality in 7 of 35 animals. Fewer than the currently recommended 4,000 cells scored by each lab; however, the individual animal data shows no animals with 0%. Taken together, the two labs did score the recommended 4,000 cells. Overall negative result. | Engelhardt et al. (2003) |
| Rats Fisher 344/male | Inhalation, whole body | 300 and 1,000 ppm, 6 hr/day, 5 days/week for 4 weeks. | Day 3 and Day 20 of exposure period. | Six animals per group. Flow cytometry for analysis. Blood samples taken from the tail vein. Twenty thousand cells scored. Positive control ENU. Blood concentrations of SO were measured. Counted the number of leukocytes as a measure of toxicity. |
No difference in the control and treated groups at either time point. The 3‐day controls: mean 0.23% ± 0.11% and the top dose: mean 0.16% ± 0.14% reticulocytes with MN. At 20 days: control mean: 0.15% ± 0.05% and the top dose: 0.18% ± 0.11% reticulocytes with MN. Toxicity: 3 days—the % reticulocytes in the control: mean 3.92 ± 0.11 and top dose: mean 2.65 ± 0.14. 20 days—control mean 2.19 ± 0.05 and top dose: mean 2.41 ± 0.11. The % reticulocytes decreased with increasing dose at the Day 3 but not the Day 20 time points. (However, the control was much lower at 20 than 3 days.) | The study was designed according to the 1997 version of TG474. Three exposure groups were used but MN data are reported for only two exposure groups. These are clearly high doses. Therefore, the study is not totally OECD TG compliant. Because the flow cytometric method can score a large number of cells (20,000 vs. the currently recommended 4,000 cells) there is more statistical power. Overall, given the high doses used, the study is negative. | Gate et al. (2012) |
| Styrene Oxide | |||||||
| Mouse BALB/c/male | i.p. | 125 or 250 mg/kg. Single injection. | 1, 2, 6, and 13 days. | PCEs evaluated 30 hr after injection. Ten controls, seven treated animals. Thousand cells scored for each animal. | 0.27% in the controls and 0.43% in the treated. Authors indicated this was negative. | Uninterpretable—Methods not well‐described. Noted that “most of the mice treated with 250 or 125 mg/kg remained alive for several days.” i.p. is no longer considered a relevant route of exposure. | Fabry et al. (1978) |
| Chinese hamster | i.p. | 250 mg/kg. Single injection. | 30 hr after treatment. | Four treated and two controls. Bone marrow. Thousand PCE and 1,000 NCE scored. The ratio of PCE/NCE determined for one animal. | Control: PCEs— 1.75 ± 0.75, NCEs—0.75 ± 0.48 MN/1,000 cells. SO treated: PCEs—2.0 ± 0.54, NCEs—1.13 ± 0.35 MN/1,000 cells. These were not statistically different. | Not OECD TG compliant. Only a single dose. Insufficient number of animals per group. Insufficient number of cells scored. i.p. is no longer considered a relevant route of exposure. | Penttila et al. (1980) |
| Mouse CD1/male | i.p. | 100, 200, and 400 mg/kg. Single injection. | 0, 24, 48, and 72 hr after treatment. | Collaborative trial involving four labs, sampling times varied in the different labs. At least five animals per group. At least 1,000 cells scored. | Eight different analyses for across the four labs. All responses were negative. | Not OECD TG compliant. i.p. is no longer considered a relevant route of exposure. Did not score the recommended 4,000 cells. | Morita et al. (1997) |
| Rats Fisher 344/male | Inhalation, whole body | 25, 50, and 75 ppm, 6 hr/day, 5 days/week for 4 weeks. | Day 3 and Day 20 of exposure period. | Six animals per group. Flow cytometry for analysis. Blood samples taken from the tail vein. Number of cells scored unclear. Positive control ethylnitrosourea. Blood concentrations of SO were measured. Counted the number of leukocytes as a measure of toxicity. |
No difference in the control and treated groups at either time point. The 3 day controls: mean 0.23% ± 0.11% and the top dose: mean 0.25% ± 0.07% reticulocytes with MN. At 20 days: control mean: 0.15% ± 0.05% and the top dose: 0.10% ± 0.05% reticulocytes with MN. Toxicity: 3 days—the % reticulocytes in the control: mean 3.92 ± 0.11 and top dose: mean 2.30 ± 0.07. 20 days—control mean 2.19 ± 0.05 and top dose: mean 1.41 ± 0.05. The % reticulocytes decreased with increasing dose at both time points. | Negative: The study was conducted according to the 1997 version of TG474. | Gate et al. (2012) |
PCEs, polychromatic or immature erythrocytes.
NCEs, normochromatic or mature erythrocytes.
Weight‐of‐the‐evidence: Does styrene/SO induce MN?
There were no interpretable in vitro studies in which styrene itself was evaluated. The Laffon et al. (2001) and the recent study by Speit et al. (2012), conducted using the OECD TG487 (OECD, 2016e), provides definitive evidence that SO can induce MN in vitro in mammalian cells. Although the currently recommended number of cells were not scored, two studies (Kligerman et al., 1992, 1993) using inhalation exposure to styrene provide evidence that it is negative in vivo in rodents. The recent inhalation study conducted by Gate et al. (2012) provides definitive evidence that neither styrene nor SO induces MN in vivo in rats.
Assays that Detect Exposure
There are several assays/endpoints (SCE, adducts, UDS, and DNA strand breakage) that are useful to detect exposure, but do not provide any definitive proof that a test chemical can induce mutations. With the exception of the in vivo UDS (TG486; OECD, 1997b) and the in vivo Comet assay (TG489; OECD, 2016g), there are no OECD guidelines for these endpoints, thus there are no standard, internationally agreed, approaches for measurement of these biomarkers of exposure. These assays and the available information for styrene/SO are described briefly below.
In Vivo Rodent SCE
No OECD TG for the in vivo SCE assay was ever drafted and approved. A few general principles from the other in vivo TGs can, however, be applied, such as number of animals per test group, number of exposure levels, number of cells scored, and considering the i.p. route of exposure to not be relevant. The reviewed studies are briefly summarized in Table 9. Eight studies were identified and reviewed for styrene exposure. Of these eight, five were conducted using inhalation exposure and three using i.p. Only one of the studies (Kligerman et al., 1993) was found to be well‐conducted and to provide evidence that inhalation exposure to styrene can cause the induction of SCEs in peripheral blood lymphocytes in rats. Of the three studies conducted for SO, two used the inhalation route of exposure and one used i.p. All three studies had multiple deficiencies which made the results uninterpretable.
Table 9.
Rodent Studies Evaluating SCEs Following Exposure to Styrene or Styrene Oxide
| Species/gender | Route | Doses and duration | Sampling times | Methods | Results | Comments | Reference |
|---|---|---|---|---|---|---|---|
| Styrene | |||||||
| Mouse C57B1/6/male | i.p. | 50, 100, 250, 500, 750, and 1,000 mg/kg. Single injection. | 24 hr after BUdRa implant and styrene injection. | Bone marrow. Implanted BUdR tablet, i.p. injection of styrene 30 min after tablet implant. Three to four animals per group, except in the top two dose groups where death resulted in two and one animal scored; 30 sec division cells scored. | SCEs/cell Negative control: 4.3 ± 0.70. Solvent control: 3.9 ± 0.68. Authors report a statistically significant increase in SCEs; however, the highest frequency was seen at the 500 mg/kg level (6.0 ± 0.97) and declined at the higher doses. Replicative index was not impacted in the animals that survived (and were scored). |
Number of animals per group is below the OECD recommended (five), particularly in the top dose groups. i.p. is no longer considered a relevant route of exposure. | Sharief et al. (1986) |
| Mouse BDF1/male | Inhalation, head only | 565 ± 18.5 ppm; 6 hr/day for 4 days. | Next day following last exposure. | Bone marrow and liver cells (from hepatectomized animals). BUdR on last day of styrene exposure. 4 controls and 4 treated (with and without hepatectomy). 30 cells scored. | The treated animals had a much higher (and statistically significant) SCE frequency than the controls. SCEs/cell in bone marrow: Controls: mean 3.0 ± 1.9 (hepatectomized) and 3.3 ± 1.9 (non‐hepatectomized). Treated: 11.9 ± 3.4 (hepatectomized) and 11.0 ± 4.0 (non‐hepatectomized). Liver: control mean 3.8 ± 2.0 and treated 12.2 ± 3.7. | Only a single dose. The number of animals per group is below the OECD recommended 5. The liver SCE is not a standard method. Small number of cells scored. The large difference between the controls and treated is difficult to dismiss based on study deficiencies. However. It would not be appropriate to call the response positive based on a single dose. | Conner et al., 1979 |
| Mouse BDF1/male | Inhalation, head only | 104, 387, 591, and 922 ppm, 6 hr/day for 4 days and 922 ppm, 6 hr/day for 1 or 2 days. | Next day following last exposure. | Bone marrow, alveolar macrophages. Two sets of animals (one hepatectomized and one not). Regenerating liver in hepatectomized animals. BUdR injection immediately after last treatment. Fifteen animals in the controls. The treated groups ranged from three to four animals. The number of second division cells scored ranged from 23–62. | The authors report exposure related increases in SCEs. The low dose groups (for all three cell types) were not different than controls. The other dose groups were significantly higher than the controls, with the exception that the single day exposure (top dose) was not different than the controls. The controls ranged from 2.9 to 3.5 SCEs/cell. The highest response was seen in the top dose/4‐day treatment where the SCEs/cell ranged from 8.0 to 11.0 SCEs/cell. | The number of animals in treated groups is below the OECD recommended (five). The liver SCE is not a standard method. The large difference between the controls and top dose treated is difficult to dismiss based on study deficiencies. It is noted that earlier study by Conner et al. (1979) used a single dose of 565 ppm. The SCE/cell values observed in that study were similar to the values obtained in this study at the 922 ppm dose. This highlights the variability between the two studies. | Conner et al. (1980) |
| Mouse LACA Swiss/male | i.p. | 75, 150, 300, and 450 mg/kg. Single injection. | 24 hr postinjection. | Five animals per group (one died in the top dose group). Splenocytes isolated and cultured for 40 hr. Twenty metaphases scored. Replicative index determined. | A statistically significant increase at the top dose. SCEs/chromosome: control (0.23 ± 0.02), 450 mg/kg (0.27 ± 0.02). This dose was lethal in 1 of 5 animals. | Insufficient number of cells scored. i.p. is no longer considered a relevant route of exposure. Top dose was too toxic. | Simula and Priestly (1992) |
| Rat Porton/male | i.p. | 375, 750, 1,500, and 3,000 mg/kg. Single injection. | 48 hr postinjection. | Five animals per group (four died in the top dose group). Splenocytes isolated and cultured for 62 hr. Twenty metaphases scored. Replicative index determined. | A statistically significant increase in the top three doses (however, the top dose lost four animals). SCEs/chromosome: control (0.21 ± 0.01), 1,500 mg/kg (0.45 ± 0.04). | Insufficient number of cells scored. i.p. is no longer considered a relevant route of exposure. Top dose was too toxic (and above the 2,000 mg/kg top dose recommended in the OECD CA and MN TGs). | Simula and Priestly (1992) |
| Mouse B6C3F1/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Mononuclear leucocytes from blood or spleen from six animals were cultured with BUdR. Lung cells isolated and cultured and BUdR added (from eight animals). Peripheral blood lymphocytes cultured with BUdR. Where possible 50 s division metaphases scored. For the peripheral blood, 25 sec division metaphase scored (where possible). | The authors reported the SCE response in the splenic lymphocytes, peripheral blood lymphocytes, and lung cells to be positive. Spleen: Controls varied from 9.8 to 10.9 SCEs/cell and the top dose group varied from 12.4 to 13.3 SCEs/cell (with one animal having too few cells to score). Peripheral blood: Controls varied from 9.6 to 10.1 SCEs/cell and the top dose group varied from 10.9 to 12.1 SCEs/cell. There were fewer than six animals evaluated in each group for peripheral blood. Lung: Controls varied from 8.0 to 11 SCEs/cell and the top dose group varied from 10.3 to 12.7 SCEs/cell. | Overall a positive response. It should be noted that the lung cell SCE is not a standardly conducted method. There were fewer than five animals in each group for the peripheral blood SCE analysis. | Kligerman et al. (1992, 1993) |
| Rats Fischer 344/female | Inhalation, whole body | 125, 250, and 500 ppm, 6 hr/day for 14 days. | One day after last exposure. | Peripheral blood lymphocytes with BUdR. Five animals per group; 50 sec division metaphases scored. | Authors report a statistically significant positive response. Control: 11.3 ± 0.7 SCEs/cell. Top dose: 14.3 ± 2.1 SCEs/cell. | A positive response. | Kligerman et al. (1993) |
| Rats Fischer 344/male | Inhalation, whole body | 150, 500, and 1,000 ppm, 6 hr/day, 5 days/week for 4 weeks. | 1, 2, 3, and 4 weeks and 4 weeks after last exposure. | Peripheral blood lymphocytes. Cultured with BUdR added 24 hr after culture initiation. Cells harvested 68 hr after culture initiation. A minimum of 25 sec division cells scored. Four to six animals per group. Positive control: ethylene oxide. | No increase in the treated vs. controls. The control means ranged from 5.36–6.6 SCEs/cell. The treated means ranged from 5.02–6.8 SCEs/cell. | Small number of cells scored. Some groups may have had less than five animals (number unclear). Overall a negative response, but not definitive. | Preston and Abernethy (1993) |
| Styrene Oxide | |||||||
| Chinese hamster/male | Inhalation, whole body and i.p. | 25, 50, 75, and 100 ppm. Nine hours (6 hr Day 1 and 3 hr Day 2) and 21 hr (6 hr daily for 3 days and 3 hr Day 4) (except for the top dose only 9 hr exposure; the animals showed signs of poisoning). The low dose also used for a 3‐week exposure (6 hr daily, 5 days/week). Single dose of 500 mg/kg (i.p.). | After last exposure for the inhalation and for i.p. 7 hr after injection. | 2–3 animals in each treatment group; 6 animals in the i.p. group. 13 animals in the negative control. 4 animals in the olive oil (vehicle control). Bone marrow cells, labeled with BUdR in vitro. Number of cells scored varied from 10–104 in the SO treated animals. MMS used as a positive control. | Negative control: 7.9 SCEs/cell. The SO treated animals varied from 6.4–7.8 SCEs/cell. The 500 mg/kg i.p. treated animals were 9.4 SCEs/cell, which was statistically different than the control. The MMS positive control: 17.5 SCEs/cell. | Insufficient number of animals in each dose group. Insufficient number of cells scored. The top doses used, particularly for i.p. were clearly too toxic. i.p. is no longer considered a relevant route of exposure. | Norppa et al. (1979) |
| Mouse | Inhalation | 50 and 70 ppm, 5 h. | Unclear. | Bone marrow, alveolar macrophages and regenerating liver cells. Three animals in the control and 50 ppm groups and one to two animals in the 70 ppm group. Twenty cells scored. | The authors report this a preliminary experiment; with a slight increase in SCEs in the alveolar macrophages and regenerating liver cells at 50 ppm but not 70 ppm. However, the top group had only one to two animals and there was a “dramatic reduction in total as well as second division metaphase yields.” | Uninterpretable. Methods are not detailed. Only two doses the top dose being very toxic. Too few cells scored. | Conner et al. (1982) |
| Mouse CD1/male | i.p. | 100 mg/kg, single injection. Both S and R enantiomers. | 24 hr after injection. | Bone marrow. Four animals per group; 30 s division cells scored. Positive control DMBA.b | The R enantiomer was negative (control: 3.55 ± 0.48 and treated: 3.61 ± 0.25 SCEs/cell). The S enantiomer was statistically different (control: 3.55 ± 0.48 and treated 5.06 ± 0.49 SCEs/cell). | A single dose. Insufficient number of cells scored. Four animals per group. i.p. is no longer considered a relevant route of exposure. | Sinsheimer et al. (1993) |
BUdR, bromodeoxyuridine.
DMBA, 2,4‐dimethoxybenzaldehyde.
As already discussed, the induction of SCEs is not indicative of the induction of genetic damage but rather an indication of exposure.
Information from Assays that Detect Primary DNA Interaction/Damage
Assays for primary DNA interaction/damage, including DNA adducts, can be conducted using noncellular approaches, cells in culture, and in vivo rodents (or other animals). These endpoints demonstrate exposure of the analyzed tissue to the chemical of interest, provided there is no endogenous/background source. While human relevance increases as one moves from the noncellular to in vitro, to in vivo, the biological relevance of the DNA‐related endpoints stays the same. They all provide an indication that a test material can interact with the DNA of the analyzed tissue, but because the various primary DNA interactions/damage can be repaired, these tests/endpoints do not provide any definitive proof that the test material actually induces mutations (OECD, 2017). In fact, specific DNA adducts have different properties and different potential for effects (Jarabek et al., 2009; Pottenger et al., 2014). Indeed, certain DNA adducts have been demonstrated as nonmutagenic (Philippin et al., 2014; Pottenger et al., 2018). Thus, DNA adducts, DNA strand breakage, and UDS are all, like the SCE endpoint, biomarkers of exposure. Protein adducts, although not indicative of DNA exposure, are useful systemic exposure biomarkers. The in vitro UDS OECD TG was eliminated in 2014, and the in vivo UDS assay was strongly considered for deletion but ultimately was retained (but not revised), because there are some jurisdictions that recommend it as a follow‐up assay for positive results in the standard in vitro assays. However, the recommendation to retain this TG was primarily made because of limited alternatives (OECD, 2017).
Protein adducts
SO has been shown to react with nucleophilic sites on plasma proteins and hemoglobin (Hb) to form SO–albumin and SO–Hb adducts; styrene will not react with these moieties without activation. Many groups have developed methods to measure alkylated Hb and other plasma proteins as biomarkers of exposure to styrene/SO; these methods are now fairly routine and are used to conduct biomonitoring of workers. Although the Hb adducts from SO will be present as α and β structural isomers (see explanation below), there has not been much reported on this aspect for Hb adducts from SO exposure; thus, this aspect is not addressed in the following brief summary of data.
Styrene and SO exposure results in alkylation of the amino acid residues at the N‐terminal of proteins. Generally, cysteine is the most reactive amino acid and results in formation of S‐(2‐hydroxy‐1‐phenylethyl) cysteine in albumin. Alkylation of Hb by styrene/SO results in formation of N′‐(2‐hydroxy‐1‐phenylethyl) valine and N′‐(1‐hydroxy‐2‐phenylethyl) valine adducts in Hb (Hemminki, 1986; Basile et al., 2002). Rappaport et al. (1993) found that reaction rates of SO with Hb and albumin were higher in the rat than in humans. In rodents, both styrene and SO exposures result in the formation of cysteine adducts in albumin and Hb (Ting et al., 1990; Rappaport et al., 1993), adducts with side chain carboxylic acid residues in Hb (Sepai et al., 1993) and N‐terminal valine adducts in Hb (Osterman‐Golkar et al., 1995). Bergmark et al. (1990) identified N‐terminal valine and carboxylic acid phenylethyl adducts in purified human Hb treated in vitro with [3H]SO. Rappaport and Yeowell‐O'Connell (1999) found that SO was more effective at producing albumin adducts than styrene after inhalation exposure. Derivatized SO–Hb adducts were measured using GC/MS techniques in Hb obtained from male Fischer rats repeatedly exposed to styrene via inhalation (Latriano et al., 1991). A clear linear dose‐response has been shown in both animal and human studies (Pauwels et al., 1996; Liu et al., 2001) at styrene and SO concentrations that do not overload the detoxification process (Osterman‐Golkar et al., 1995). It should be noted that the formation of protein adducts is not an assay for genotoxicity, nor evidence for mutation.
DNA adducts
DNA adducts represent chemically bound moieties to the DNA of cells or tissues. They are detected as modified DNA bases, deoxynucleosides, or oligonucleotides. All four DNA bases can form adducts; Figure 2 depicts the styrene oxide‐related binding sites for the nucleic acids. Although some DNA adducts are specific to individual chemicals, others are not, such as those formed from reactive oxygen species (e.g., 8‐hydroxy‐deoxyguanosine [8‐OHdG]). DNA repair, which actively occurs with most DNA adducts, affects the life‐span of adducts as does their chemistry. For example, chemically unstable adducts (e.g., N3‐adenine and N7‐guanine adducts) have half‐lives from hours to about three to five days. Following their depurination (spontaneous removal of the unstable adducted base), an apurinic (AP) site is left that can be mutagenic but generally is very rapidly repaired (Rios‐Blanco et al., 2000; Rusyn et al., 2005; Swenberg et al., 2008).
Figure 2.
Styrene oxide DNA binding sites.
Three useful reviews on styrene‐related DNA adducts are available. Phillips and Farmer (1994) focused on styrene and SO, while in 2000, Koskinen and Plná (2000) published a “mini‐review” of specific DNA adducts that are induced by mono‐substituted epoxides, including styrene. The more recent Vodicka et al. (2002) review provides a thorough discussion of SO‐induced DNA adducts and information on their potential effects. Because of the unique chemistry of styrene/SO, the DNA adducts formed present a complicated array. This section focuses on the adducts formed by binding of the metabolite SO, the predominant reactive metabolite of styrene, which can bind to DNA via either the 1‐phenylethyl or the 2‐phenylethyl position, giving rise to either alpha‐ or beta‐structural isomers (Fig. 3). Theoretically, each of these isomers can form at any of the nucleophilic sites on DNA bases (Fig. 2). The chemistry is further complicated by the presence of the chiral carbon in SO, which results in either the R or S enantiomer. Thus, at each nucleic acid binding site available for adduction on DNA, the SO moiety is either α or β, and each of those isomers is bound as either R or S, which is not always determined. Figure 3 shows how some of these structures differ. The nomenclature can vary considerably among authors, also depending on the analyte being measured (e.g., nucleic acid vs. nucleoside vs. nucleotide). In order to simplify for this brief review, the following streamlined nomenclature will be used: the structural isomer will be identified, if known (e.g., α or β), preceded by the enantiomeric designation, if known (R or S); the atom bound will be named with its corresponding nucleic acid base atom number (e.g., N7)—endocyclic atoms are italicized and exocyclic atom numbers are superscripted (e.g., O6); SO to represent the 7,8‐styrene oxide moiety; and the letter for the particular nucleic acid base (e.g., G, A, C, or T). A few examples are shown below:
Figure 3.
Some representative DNA adducts of SO. Structural isomers are shown for endocyclic N7‐SO‐deoxyguanine (N7‐SO‐G) adducts and exocyclic O6‐SO deoxyguanine (O6‐SO‐G) adducts.
N7‐(2‐hydroxy‐1‐phenylethyl)guanine = αN7‐SO‐G;
N7‐(1‐hydroxy‐2‐phenylethyl)guanine = βN7‐SO‐G;
O6‐(2‐hydroxy‐1‐phenylethyl)guanine = αO6‐SO‐G;
N1‐(2‐hydroxy‐1‐phenylethyl)adenine = αN1‐SO‐A.
The amount of information obtained on specific SO‐induced adducts depends on the methods used to generate the samples and the methods used to analyze the samples. The former affects the proportion of each specific adduct formed as the availability of the binding sites (therefore the resulting adduct distribution profile) depends in part on what form of DNA is the target: naked DNA being reacted directly with SO (noncellular), or DNA of cells in culture (in vitro), or DNA in tissues from whole animal exposures to styrene or SO (in vivo). A wider variety of adducts are found for SO‐treated naked DNA, whereas tissues from in vivo exposures result in much lower adduction rates and reduced numbers of sites adducted. Another layer of complexity depends on the methods used to identify the adducts. The two most common methods are (1) 32P‐postlabeling, a method that enzymatically attaches a 32P label to the 5′‐position of a nucleotide‐3′‐phosphate (both modified and nonmodified), which is then separated either by thin layer chromatography (TLC) or by high‐pressure liquid chromatography (HPLC), and chromatographed with standards to identify what adducts are present; and (2) chromatography (often HPLC) coupled with tandem mass spectrometry (MS/MS), which provides structural information to confirm the adduct structure, and allows for quantification. Although 32P‐postlabeling can be very sensitive, it requires special modifications to include the predominant DNA adduct, N7‐SO‐G (Vodicka and Hemminki, 1988a, 1991a; Kumar et al., 1997), which is a significant drawback; this method does not provide structural confirmation information. The capability to obtain important structural confirmation is only available with techniques that rely on MS/MS.
The kind of structural detail and adduct distribution profiles described above can be critical in consideration of MOA, as different adducts have different capabilities vis‐à‐vis genotoxic effect, ranging from nonmutagenic (e.g., low‐molecular–weight N7‐alkyl/hydroxyalkyl‐G adducts) (Philippin et al., 2014; Pottenger et al., 2018) to promutagenic if not repaired (e.g., O6‐alkyl‐G adducts) (ibid). Of course, formation of an adduct does not provide any definitive evidence for mutation. Indeed, the adduct represents the first step in a MOA for a mutation that has many steps and off‐routes, such as requiring cell replication to permanently incorporate a change in base sequence, which may or may not result in a change in amino acid sequence upon translation (Pottenger and Gollapudi, 2010). In fact, there is a ubiquitous background of endogenous DNA adducts, including pro‐mutagenic ones, present in every cell (Nakamura et al., 2014), although no specific SO adducts have been identified as endogenous to date.
The numerous available studies using noncellular systems demonstrate convincingly that SO reacts with DNA mainly to form guanine adducts, typically in the following order quantitatively: N7‐SO‐G >> N2‐SO‐G > O6‐SO‐G adducts (Savela et al., 1986; Pongracz et al., 1989, 1992; Kumar et al., 1997; Siethoff et al., 1999); Vodicka and Hemminki, 1988a, 1988b, 1991ba,b; Yang et al., 2005). These have also been identified in animal and human cells/tissues, with in vitro and in vivo studies. Although N7‐SO‐G is the major adduct formed in the in vitro reaction of SO with DNA, due to the high nucleophilicity of the N7 position, SO adducts have been identified from all the nucleic acid bases. Savela et al. (1986) reported that N7‐SO‐G adducts accounted for 82% of the SO‐induced DNA adducts formed in naked DNA treated with SO. Several in vitro studies (Qian and Dipple, 1995; Koskinen and Hemminki, 1999; Kim et al., 2000) all found N1‐ and N6‐SO‐A adducts. In a study designed to assess both mutation induction and DNA adducts, Bastlová et al. (1995) found that although there was a significant correlation between SO concentrations and formation of DNA adduct levels (32P‐postlabeling), there was no correlation between SO‐induced hprt gene mutations in human T‐lymphocytes and levels of DNA adducts measured. However, it should be noted that there were deficiencies in the hprt evaluation (see gene mutation section above).
As the most relevant exposure route, in vivo styrene inhalation exposure studies evaluating DNA adducts in rodents confirm the formation of many of the same SO‐induced DNA adducts identified with in vitro and noncellular systems; several studies are briefly summarized below. As expected, the highly nucleophilic N7‐G site clearly predominates with in vivo formation of N7‐SO‐G. SO‐induced DNA adducts have been detected in animal blood, urine, lung, liver, and other tissues of styrene‐exposed animals. Latriano et al. (1991) found N7‐, N2‐, and O6‐SO‐G in DNA isolated from the lung and livers of styrene‐exposed rats using 32P‐postlabeling. Also with 32P‐postlabeling, coupled with TLC and extraction techniques, Otteneder et al. (1999) confirmed the formation of α and βO6‐SO‐G adducts in the livers of female rats chronically exposed via inhalation to styrene. Following a single inhalation exposure of mice and rats to [14C]styrene and using HPLC/LSC, Boogaard et al. (2000) found adducts in whole liver (α and βN7‐SO‐G, N2‐SO‐G, and O6‐SO‐G), whole lung, and lung Clara cells (mainly N7‐SO‐G), which varied between mice and rats. Koskinen et al. (2001a, 2001b) reported βN1‐SO‐A DNA adduct formation after repeated inhalation exposure of male NMRI mice to styrene by 32P‐postlabeling. Vodicka et al. (2001, 2006) confirmed the formation of N7‐SO‐G and βN1‐SO‐A DNA adducts in mouse lung and liver with 32P‐postlabeling and HPLC analyses after repeated exposure to styrene via inhalation. Using HPLC, TLC, and 32P‐postlabeling, Otteneder et al. (2002) evaluated O6‐SO‐G adduct loads in mouse and rat lung and liver (rat only) tissues following repeated inhalation exposure to styrene (chronic and shorter term). The authors identified α and βO6‐SO‐G adducts in liver DNA, but levels in lung tissue were below detection. The researchers concluded that the levels of DNA adducts were detectable in rat liver (nontarget organ), but not in the mouse lung (target organ), thus did not correspond with tumor incidence. Mikeš et al. (2009) confirmed formation of N7‐SO‐G and α and βN3‐SO‐A DNA adducts in mouse urine after repeated inhalation exposure to styrene, concluding these adducts were likely rapidly depurinated and eliminated.
UDS (TG486)
The UDS assay (OECD, 1997b) is used for the detection of DNA damage/repair activity. The in vivo rodent UDS is generally conducted in the liver and the methodology is well‐established (measurement of incorporation of [3H]‐labeled thymidine during damage‐induced DNA synthesis). There are a few publications involving exposure to styrene or SO and assessment using the UDS assay either in vitro or in vivo; these are summarized here. Brouns et al. (1979) exposed freshly isolated rat hepatocytes to SO and found no evidence of DNA repair activity in the cells. Pero et al. (1982) exposed human lymphocytes in culture to styrene (10–750 μM for 15 min) and concluded that the UDS response was negative. In 1989, Williams et al. (1989) reported positive results for styrene exposure from a UDS validation study on 300 chemicals using rat hepatocytes treated in vitro and assessed for DNA repair to evaluate genotoxicity. Clay (2004) exposed mice by inhalation to concentrations of styrene ranging from 160 to 1000 ppm and evaluated DNA repair in the liver. He concluded that the observed negative response was consistent with a nongenotoxic MOA for mouse lung tumors. One criticism of this Clay study is that MOA evaluations should be conducted using the target tissue, which would be mouse lung in this case.
DNA strand breakage
There are several techniques that have been used to detect DNA strand breakage. These include alkaline elution, DNA unwinding, and the Comet assay. Of these techniques, only the in vivo alkaline Comet assay has an OECD TG (TG489; OECD, 2016g); there is no TG for the in vitro Comet assay. Although not included in the TG, and therefore without any standard procedure or interpretation, the addition of formamidopyrimidine (FAPy) DNA glycosylase (FPG) allows for the detection of certain oxidative adducts such as 8‐OHdG; the FAPy‐G and FAPy‐A adducts can also be detected.
Several studies were identified that evaluated the ability of SO to induce DNA strand breakage. The in vitro studies are summarized as follows. As a part of their development of an alkaline elution assay using rat hepatocytes, Sina et al. (1983) used SO as one of their test chemicals. They reported that SO‐induced single‐strand breaks at concentrations resulting in less than 30% cytotoxicity. SO in vitro exposure caused single‐strand DNA breaks (SSBs) in testicular cells both from Wistar rats and from human organ transplant donors using the alkaline elution technique (Bjørge et al., 1996). Herrero et al. (1997) reported that SO induces SSBs in V79 Chinese hamster cells. Köhlerová and Stetina (2003) treated isolated human peripheral lymphocytes with SO and, based on the Comet assay, determined that DNA strand breaks were induced. They further investigated the removal of these breaks and found that their half‐life was about two to four hours. In 2009, Cemeli et al. used lymphocytes from 18 healthy volunteers and treated them with SO (50, 100, and 200 μM; Cemeli et al., 2009). The Comet assay was performed on the treated cells, and a positive dose‐response effect was observed. Fabiani et al. (2012) treated both freshly isolated peripheral blood mononuclear cells and promyelocytic leukemia cells (HL60) with SO and found DNA breakage based on a positive response in the Comet assay. Godderis et al. (2006) treated peripheral blood mononuclear cells from 20 individuals with SO and, using the Comet assay found a positive response. Isolated peripheral blood mononuclear cells or T‐cells, exposed to SO, were positive for the Comet assay (Bausinger and Speit, 2014). Thus, it seems clear that when mammalian cells are exposed in culture to SO directly, DNA strand breakage can be detected.
We briefly describe two in vivo studies. Vaghef and Hellman (1998) used a single i.p. injection of styrene or SO to expose female B57BL/6 mice. Primary DNA damage was evaluated in several organs using the alkaline Comet assay. The authors reported that both chemicals induced DNA damage in the lymphocytes, liver, bone marrow, and kidney, when the animals were sacrificed and tissue evaluated four hours after exposure. A second time point (16 hr post‐treatment) was also evaluated, and the level of DNA damage was decreased in all tissues except the bone marrow cells. This study was conducted prior to the approval of the OECD TG for the in vivo Comet assay, and not all of the details needed to determine if the study was compliant with the TG were included in the publication. However, the fact that the route of exposure was i.p. restricts the utility of the study.
Vodicka et al. (2001) exposed male NMRI mice by inhalation using 750 and 1500 mg/m3 styrene. Mice were exposed six hours per day, seven days per week, for 1, 3, 7, and 21 days. The Comet assay (conducted to measure both breaks and oxidative damage) was used to evaluate double strand DNA breaks (DSBs) in the peripheral lymphocytes, liver, and bone marrow cells. A slight increase in the number of breaks was found in the bone marrow after seven days of inhalation exposure, while a significant increase in the endonuclease III‐sensitive sites (a marker for AP sites) was only observed after 21 days. In the liver, no increases were seen after 21 days of inhalation exposure for either strand breaks or endonuclease III‐sensitive sites. There was an increase in the lymphocyte strand breaks at 7, but not 21 days.
Weight‐of‐the‐evidence information from primary DNA interactions/damage assays
Styrene/SO exposure results in the formation of both protein and DNA adducts, some of which are chemical‐specific and, therefore, are particularly useful to evaluate exposure to styrene. This ability to form adducts has been demonstrated in vitro and in vivo. However, as already emphasized, this does not mean that these adducts result in the induction of gene mutations.
DNA breakage can be evaluated using several techniques. In recent years, the Comet assay has become the most widely used. It is clear that SO exposure induces DNA strand breaks in cultured mammalian cells. The in vivo results for DNA strand breaks following inhalation exposure to styrene are not as clear. Based on the sole inhalation study, DSBs were mostly negative (lymphocytes and liver) and statistically slightly increased only for a single duration and dose (seven‐day, high dose) in lymphocytes but not after 1, 3, or 21 days of exposure.
DISCUSSION
The styrene genotoxicity literature originates in the 1970s and includes several hundred publications. These studies coincided with the development of the field of genetic toxicology, and the methods used in the investigations were generally in line with those used in the timeframe in which they were conducted. Many of the early styrene/SO reviews for the genotoxicity literature simply reported the positive/negative calls of the publication authors. This resulted in review authors referencing a large number of studies as positive, providing a perception that there is an extensive literature to indicate that styrene/SO is mutagenic/genotoxic. In addition to providing more weight to positive studies than to negative studies, these reviews included many studies that were conducted using now outdated methods or inadequately described ones.
Critical reviews of the literature, previously provided by several genetic toxicology experts (Scott and Preston, 1994a, 1994b; Cohen et al., 2002; Speit and Henderson, 2005), have all concluded that, with the exception of in vitro studies where there is clear evidence that SO is mutagenic/clastogenic, there is no convincing evidence that styrene/SO is mutagenic/clastogenic in vivo in rodents.
In revisiting the mutagenicity/clastogenicity of styrene, we have critically reviewed, based on the recent OECD TG revisions, the literature for the Ames test, gene mutation in mammalian cells, CAs in mammalian cells, MN in mammalian cells, and in vivo rodent studies for CA and MN. Because of changes in methods and data interpretation for the standard genetic toxicology tests, we found much of the published styrene/SO data to be uninterpretable.
We organized our review based on the relative weight that the individual endpoints contribute to the evaluation as to whether a test material can induce mutation. The downside of this approach is that studies conducted using multiple endpoints are divided by endpoint. We think that it is important to highlight a recent study conducted by Gate et al. (2012) that demonstrates an integrated approach to evaluating effects of repeated (four weeks) inhalation (whole body) exposure to styrene and to SO in Fischer 344 rats. Although a discussion of all the details of the paper is beyond the scope of this review, the key results from this study are important to the overall interpretation of the available data on the mutagenicity/clastogenicity of styrene/SO. Rats were exposed to three concentrations of either styrene (75, 300, or 1000 ppm) or SO (25, 50, or 75 ppm). The frequency of MN in peripheral blood reticulocytes was evaluated by flow cytometry at the end of Day 3 and Day 20 of exposure. This method uses a small sample of tail vein blood, thus allowing for sequential sampling of the same animals (contrary to bone marrow MN analysis). Gate et al. (2012) scored 20,000 cells per animal, resulting in a very high statistical power, whereas the current OECD TG474 (OECD, 2016f) recommends scoring a minimum of 4000 cells, and most older studies scored 1000 to 2000 cells. The Comet assay (both the alkaline and FPG‐oxidative damage versions) was also performed on the 3‐ and 20‐day blood samples, which evaluted100 cells (leukocytes) with the Comet Assay IV software and reported percentage of DNA in the Comet tail as the endpoint. The MN assay was clearly negative for all the exposure levels and time points for both the styrene and SO exposures. The alkaline Comet assay evaluating DNA strand breaks in leukocytes found no increases at either time point at any exposure level for both styrene and SO. For the FPG Comet, which assesses oxidative damage, there was a significant increase in the number of DNA breaks in all three styrene exposure groups, but only at the three‐day exposure sampling time; no increases were seen at the 20‐day sampling. There were no increases in DNA breaks at either time point for the FPG Comet assay following exposure to SO. As previously stated, Comet assay data are not evidence of mutation but of primary DNA damage, which can be repaired. This comprehensive study adds substantially to the weight‐of‐the‐evidence that styrene/SO is not mutagenic/clastogenic in vivo to rodents.
As supporting information, we have included literature for DNA strand breakage (e.g., Comet assay), DNA adducts, protein adducts, and SCEs; all of these are markers of exposure and not evidence of mutation induction.
For our critical review, we applied current recommendations for assay conduct, assay acceptability, and interpretation of data. Based on this approach, we found a large number of the studies to be uninterpretable, but that there were enough reliable/interpretable studies to reach a number of conclusions: (1) Unmetabolized styrene is not mutagenic/clastogenic. For the in vitro test systems, when styrene is metabolized to SO (and the SO is not further metabolized to nongenotoxicants), positive results are obtained. (2) When SO is the test material, it is clearly mutagenic in the bacterial Ames test. (3) The majority of publications evaluating styrene/SO for gene mutation in cultured mammalian cells were uninterpretable because of major technical deficiencies based on current OECD TGs. A single study using the MLA was, in spite of some technical deficiencies, interpreted to be evidence that SO can induce mutation in cultured mammalian cells. (4) No in vivo studies for the mutagenicity of styrene or SO were identified and, therefore, no conclusions can be made concerning the ability of in vivo styrene/SO exposure to induce gene mutations in the somatic cells of rodents. (5) SO was found to be clastogenic in in vitro mammalian cell assays. (6) Most of the rodent in vivo CA and MN studies could not be interpreted. However, from the few studies that could be interpreted, we conclude that there was no evidence that styrene is clastogenic in vivo and the Gate et al. (2012) study provides strong evidence that neither styrene or SO are clastogenic. (7) Regarding the exposure endpoints, there is evidence that styrene exposure to rodents can result in an increased level of SCEs. SO exposure can result in both chemical‐specific and nonchemical‐specific DNA adducts in vitro and in vivo in rodents. SO‐specific protein adducts have been observed in styrene‐exposed rodents. DNA strand breaks are induced in mammalian cells following in vitro exposure to SO.
In conclusion, while SO is clearly mutagenic and clastogenic in vitro, we find no evidence that styrene or SO can induce chromosomal damage in rodents in vivo. We note that there are no in vivo gene mutation studies in rodents.
Author Contributions
Drs. Martha Moore, Lynn H. Pottenger, and Tamara House‐Knight critically reviewed the relevant styrene literature, contributed to the interpretation of that information, and drafted the manuscript. Drs. Pottenger and House‐Knight focused on the adduct literature, and Dr. Moore reviewed and interpreted all the other endpoints. All authors read and approved the final version of the manuscript.
Acknowledgment
The authors wish to gratefully acknowledge the administrative support of Sara Oglesbee and Sarah Herrington in obtaining the literature used for this review and in preparing the manuscript. We also appreciate comments on the manuscript by William Fuller and Robinan Gentry. In addition, we acknowledge input from Bhaskar Gollapudi in identifying the literature and developing the strategy for conducing the critical review. This work was supported by the Styrene Information and Research Center (SIRC). SIRC was provided an opportunity to review the manuscript prior to submission and their comments regarding clarity of presentation were considered. The scientific opinions and critical evaluation of the literature are those exclusively of the authors.
REFERENCES
- Amacher DE, Turner GN. 1982. Mutagenic evaluation of carcinogens and non‐carcinogens in the L5178Y/TK assay utilizing postmitochondrial fractions (S9) from normal rat liver. Mutat Res 97(1):49–65. [DOI] [PubMed] [Google Scholar]
- Ames BN, McCann J, Yamasaki E. 1975. Methods for detecting carcinogens and mutagens with the salmonella/mammalian‐microsome mutagenicity test. Mutat Res 31(6):347–363. [DOI] [PubMed] [Google Scholar]
- Barale R. 1991. The genetic toxicology of styrene and styrene oxide. Mutat Res 257(2):107–126. [DOI] [PubMed] [Google Scholar]
- Bartsch H, Malaveille C, Camus M, Martel‐Planche G, Brun G, Hautefeuille A, Sabadie N, Barbin A, Kuroki T, Drevon C, et al. 1980. Validation and comparative studies on 180 chemicals using S. Typhimurium strains and v79 Chinese hamster cells in the presence of various metabolizing systems, in The predictive value of short‐term screening tests in carcinogenicity evaluation. Appl Methods Oncology 3:269–291. [Google Scholar]
- Basile A, Ferranti P, Mamone G, Manco I, Pocsfalvi G, Malorni A, Acampora A, Sannolo N. 2002. Structural analysis of styrene oxide/haemoglobin adducts by mass spectrometry: Identification of suitable biomarkers for human exposure evaluation. Rapid Commun Mass Spectrom 16(9):871–878. [DOI] [PubMed] [Google Scholar]
- Bastlová T, Vodicka P, Peterková K, Hemminki K, Lambert B. 1995. Styrene oxide‐induced HPRT mutations, DNA adducts and DNA strand breaks in cultured human lymphocytes. Carcinogenesis 16(10):2357–2362. [DOI] [PubMed] [Google Scholar]
- Bausinger J, Speit G. 2014. Induction and repair of DNA damage measured by the comet assay in human T lymphocytes separated by immunomagnetic cell sorting. Mutat Res 769:42–48. [DOI] [PubMed] [Google Scholar]
- Beije B, Jenssen D. 1982. Investigation of styrene in the liver perfusion/cell culture system. No indication of styrene‐7,8‐oxide as the principal mutagenic metabolite produced by the intact rat liver. Chem Biol Interact 39(1):57–76. [DOI] [PubMed] [Google Scholar]
- Bergmark E, Belew M, Osterman‐Golkar S. 1990. Separation and enrichment of alkylated globin chains as a means of improving the sensitivity of hemoglobin adduct measurements. Acta Chem Scand 44:630–635. [DOI] [PubMed] [Google Scholar]
- Bjørge C, Brunborg G, Wiger R, Holme JA, Scholz T, Dybing E, Søderlund EJ. 1996. A comparative study of chemically induced DNA damage in isolated human and rat testicular cells. Reprod Toxicol 10(6):509–519. [DOI] [PubMed] [Google Scholar]
- Bonatti S, Abbondandolo A, Corti G, Fiorio R, Mazzaccaro A. 1978. The expression curve of mutants induced by styrene oxide at the HGPRT locus in V79 cells. Mutat Res 52(2):295–300. [DOI] [PubMed] [Google Scholar]
- Bond JA, Bolt H. 1989. Review of the toxicology of styrene. Crit Rev Toxicol 19(3):227–249. [DOI] [PubMed] [Google Scholar]
- Boogaard PJ, de Kloe KP, Wong BA, Sumner SC, Watson WP, van Sittert NJ. 2000. Quantification of DNA adducts formed in liver, lungs, and isolated lung cells of rats and mice exposed to 14C‐styrene by nose‐only inhalation. Toxicol Sci 57(2):203–216. [DOI] [PubMed] [Google Scholar]
- Brams A, Buchet JP, Crutzen‐Fayt MC, de Meester C, Lauwerys R, Léonard A. 1987. A comparative study, with 40 chemicals, of the efficiency of the salmonella assay and the SOS chromotest (kit procedure). Toxicol Lett 38(1):123–133. [DOI] [PubMed] [Google Scholar]
- Brouns RE, Poot M, De Vrind R, Hoek‐Kon T, Henderson P, Kuyper CM. 1979. Measurement of DNA‐excision repair in suspensions of freshly isolated rat hepatocytes after exposure to some carcinogenic compounds: Its possible use in carcinogenicity screening. Mutat Res 64(6):425–432. [DOI] [PubMed] [Google Scholar]
- Busk L. 1979. Mutagenic effects of styrene and styrene oxide. Mutat Res 67(3):201–208. [PubMed] [Google Scholar]
- Cemeli E, Mirkova E, Chiuchiarelli G, Alexandrova E, Anderson D. 2009. Investigation on the mechanisms of genotoxicity of butadiene, styrene and their combination in human lymphocytes using the comet assay. Mutat Res 664(1–2):69–76. [DOI] [PubMed] [Google Scholar]
- Cheh AM. 1986. Mutagen production by chlorination of methylated alpha, beta‐unsaturated ketones. Mutat Res 169:1–9. [DOI] [PubMed] [Google Scholar]
- Claxton LD, Houk VS, Monteith LG, Myers LE, Hughes TJ. 1991. Assessing the use of known mutagens to calibrate the Salmonella typhimurium mutagenicity assay: I. Without exogenous activation. Mutat Res 253(2):137–147. [DOI] [PubMed] [Google Scholar]
- Clay P. 2004. Styrene monomer does not induce unscheduled DNA synthesis in the mouse liver following inhalation exposure. Mutagenesis 19(6):489–492. [DOI] [PubMed] [Google Scholar]
- Cohen JT, Carlson G, Charnley G, Coggon D, Delzell E, Graham JD, Greim H, Krewski D, Medinsky M, Monson R, et al. 2002. A comprehensive evaluation of the potential health risks associated with occupational and environmental exposure to styrene. J Toxicol Environ Health Part B 5:1–263. [DOI] [PubMed] [Google Scholar]
- Conner MK, Alarie Y, Dombroske RL. 1979. Sister chromatid exchange in regenerating liver and bone marrow cells of mice exposed to styrene. Toxicol Appl Pharmacol 50(2):365–367. [DOI] [PubMed] [Google Scholar]
- Conner MK, Alarie Y, Dombroske RL. 1980. Sister chromatid exchange in murine alveolar macrophages, bone marrow, and regenerating liver cells induced by styrene inhalation. Toxicol Appl Pharmacol 55(1):37–42. [DOI] [PubMed] [Google Scholar]
- Conner MK, Alarie Y, Dombroske RL. 1982. Multiple tissue comparisons of sister chromatid exchanges induced by inhaled styrene Genotoxic Effects of Airborne Agents, Vol. 25 Boston, MA: Springer; pp. 433–441. [Google Scholar]
- De Flora S, Zanacchi P, Camoirano A, Bennicelli C, Badolati GS. 1984. Genotoxic activity and potency of 135 compounds in the Ames reversion test and in a bacterial DNA‐repair test. Mutat Res 133:161–198. [DOI] [PubMed] [Google Scholar]
- de Meester C, Poncelet F, Roberfroid M, Rondelet J, Mercier M. 1977. Mutagenicity of styrene and styrene oxide. Mutat Res 56(2):147–152. [Google Scholar]
- de Meester C, Duverger‐van Bogaert M, Lambotte‐Vandepaer M, Mercier M, Poncelet F. 1981. Mutagenicity of styrene in the Salmonella typhimurium test system. Mutat Res 90(4):443–450. [DOI] [PubMed] [Google Scholar]
- Dunkel VC, Simmon VF. 1980. Mutagenic activity of chemicals previously tested for carcinogenicity in the national cancer institute bioassay program. IARC Sci. Publ. 27:283–302. [PubMed] [Google Scholar]
- Dunkel VC, Zeiger E, Brusick D, McCoy E, McGregor D, Mortelmans K, Rosenkranz HS, Simmon VF. 1985. Reproducibility of microbial mutagenicity assays. 2. Testing of carcinogens and noncarcinogens in Salmonella typhimurium and Escherichia coli . Environ Mutagen 7(suppl 5):1–248. [DOI] [PubMed] [Google Scholar]
- Einisto P, Hooberman BH, Sinsheimer JE. 1993. Base‐pair mutations caused by six aliphatic epoxides in Salmonella typhimurium TA100, TA104, TA4001, and TA4006. Environ Mol Mutagen 21(3):253–257. [DOI] [PubMed] [Google Scholar]
- El‐Tantawy MA, Hammock BD. 1980. The effect of hepatic microsomal and cytosolic subcellular fractions on the mutagenic activity of epoxide‐containing compounds in the Salmonella assay. Mutat Res 79(1):59–71. [DOI] [PubMed] [Google Scholar]
- Engelhardt G, Gamer A, Vodicka P, Bárta I, Hoffmann HD, Veenstra G. 2003. A re‐assessment of styrene‐induced clastogenicity in mice in a subacute inhalation study. Arch Toxicol 77(1):56–61. [DOI] [PubMed] [Google Scholar]
- Fabiani R, Rosignoli P, De Bartolomeo A, Fuccelli R, Morozzi G. 2012. Genotoxicity of alkene epoxides in human peripheral blood mononuclear cells and HL60 leukaemia cells evaluated with the comet assay. Mutat Res 747(1):1–6. [DOI] [PubMed] [Google Scholar]
- Fabry L, Leonard A, Roberfroid M. 1978. Mutagenicity tests with styrene oxide in mammals. Mutat Res 51(3):377–381. [DOI] [PubMed] [Google Scholar]
- Gate L, Micillino JC, Sebillaud S, Langlais C, Cosnier F, Nunge H, Darne C, Guichard Y, Binet S. 2012. Genotoxicity of styrene‐7,8‐oxide and styrene in fisher 344 rats: A 4‐week inhalation study. Toxicol Lett 211(3):211–219. [DOI] [PubMed] [Google Scholar]
- Glatt HR, Oesch F, Frigerio A, Garattini S. 1975. Epoxides metabolically produced from some known carcinogens and from some clinically used drugs. 1. Differences in mutagenicity. Int J Cancer 16(5):787–797. [DOI] [PubMed] [Google Scholar]
- Glatt HR, Jung R, Oesch F. 1983. Bacterial mutagenicity investigation of epoxides: Drugs, drug metabolites, steroids and pesticides. Mutat Res 111(2):99–118. [DOI] [PubMed] [Google Scholar]
- Godderis L, Aka P, Mateuca R, Kirsch‐Volders M, Lison D, Veulemans H. 2006. Dose‐dependent influence of genetic polymorphisms on DNA damage induced by styrene oxide, ethylene oxide and gamma‐radiation. Toxicology 219(1–3):220–229. [DOI] [PubMed] [Google Scholar]
- Helal SF, Elshafy WS. 2012. Health hazards among workers in plastic industry. Toxicol Ind Health 29(9):812–819. [DOI] [PubMed] [Google Scholar]
- Hemminki K. 1986. Covalent binding of styrene oxide to amino‐acids human serum proteins and hemoglobin In: Sorsa M, Norppa H, editors. Progress in Clinical and Biological Research, Vol. 207. Monitoring of Occupational Genotoxicants. New York: Alan R. Liss, Inc; pp. 159–168. [PubMed] [Google Scholar]
- Hemminki K, Falck K. 1979. Correlation of mutagenicity and 4‐(p‐nitrobenzyl)‐pyridine alkylation by epoxides. Toxicol Lett 4:103–106. [Google Scholar]
- Herrero ME, Arand M, Hengstler JG, Oesch F. 1997. Recombinant expression of human microsomal epoxide hydrolase protects V79 Chinese hamster cells from styrene oxide‐ but not from ethylene oxide‐induced DNA strand breaks. Environ Mol Mutagen 30(4):429–439. [DOI] [PubMed] [Google Scholar]
- Hughes TJ, Simmons DM, Monteith LG, Claxton LD. 1987. Vaporization technique to measure mutagenic activity of volatile organic chemicals in the Ames/Salmonella assay. Environ Mutagen 9(4):421–441. [DOI] [PubMed] [Google Scholar]
- Hynes DE, DeNicola DB, Carlson GP. 1999. Metabolism of styrene by mouse and rat isolated lung cells. Toxicol Sci 51:195–201. [DOI] [PubMed] [Google Scholar]
- IARC . 1979. IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans: Some monomers, plastics and synthetic elastomers, and acrolein, Vol. 19 Lyon, France: World Health Organization, International Agency for Research on Cancer. [PubMed] [Google Scholar]
- IARC . 1985. Styrene Oxide. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 36 Lyon, France: International Agency for Research on Cancer. [Google Scholar]
- IARC . 1994. Styrene. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 60 Lyon, France: International Agency for Research on Cancer. [Google Scholar]
- IARC . 2002. IARC Monographs on the evaluation of carcinogenic risks to humans. Volume 82. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. Lyon France: World Health Organization, International Agency for Research on Cancer. [PMC free article] [PubMed] [Google Scholar]
- IARC Monographs Vol 121 Group . 2018. Carcinogenicity of quinoline, styrene, and styrene‐7,8‐oxide. Lancet Oncol 19(6):728–729. [DOI] [PubMed] [Google Scholar]
- Ishidate M, Yoshikawa K. 1980. Chromosome aberration tests with Chinese hamster cells in vitro with and without metabolic activation—A comparative study on mutagens and carcinogens. Arch Toxicol 4:41–44. [DOI] [PubMed] [Google Scholar]
- Ishidate M Jr, Sofuni T, Yoshikawa K. 1981. Chromosomal aberration tests in vitro as a primary screening tool for environmental mutagens and/or carcinogens. Gann monograph. Cancer Res 27:95–108. [Google Scholar]
- Jantunen K, Maki‐Paakkanen J, Hannu N. 1986. Induction of chromosome aberrations by styrene and vinylacetate in cultured human lymphocytes: Dependence on erythrocytes. Mutat Res 159(1–2):109–116. [DOI] [PubMed] [Google Scholar]
- Jarabek AM, Pottenger LH, Andrews LS, Casciano D, Embry MR, Kim JH, Preston RJ, Reddy MV, Schoeny R, Shuker D, et al. 2009. Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Crit Rev Toxicol 39:659–678. [DOI] [PubMed] [Google Scholar]
- Kerklaan PRM, Zoetemelk CEM, Mohn GR. 1985. Mutagenic activity of various chemicals in Salmonella strain TA100 and glutathione‐ deficient derivatives. On the role of glutathione in the detoxification or activation of mutagens inside bacterial cells. Biochem Pharmacol 34(12):2151–2156. [DOI] [PubMed] [Google Scholar]
- Khudoley VV, Mizgireuv I, Pliss GB. 1987. The study of mutagenic activity of carcinogens and other chemical agents with Salmonella typhimurium assays: Testing of 126 compounds. Arch Geschwulstforsch 57(6):453–462. [PubMed] [Google Scholar]
- Kim HY, Finneman JI, Harris CM, Harris TM. 2000. Studies of the mechanisms of adduction of 2′‐deoxyadenosine with styrene oxide and polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol 13:625–637. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Allen JW, Bryant MF, Campbell JA, Collins BW, Doerr CL, Erexson GL, Kwanyuen P, Morgan DL. 1992. Cytogenetic studies of mice exposed to styrene by inhalation. Mutat Res 280(1):35–43. [DOI] [PubMed] [Google Scholar]
- Kligerman AD, Allen JW, Erexson GL, Morgan DL. 1993. Cytogenetic studies of rodents exposed to styrene by inhalation. IARC Sci Publ 127:217–224. [PubMed] [Google Scholar]
- Köhlerová R, Stetina R. 2003. The repair of DNA damage induced in human peripheral lymphocytes with styrene oxide. Acta Med Austriaca 46(3):95–100. [PubMed] [Google Scholar]
- Koskinen M, Hemminki K. 1999. Separate deamination mechanisms for isomeric styrene oxide induced N1‐adenine adducts. Org Lett 1:1233–1235. [DOI] [PubMed] [Google Scholar]
- Koskinen M, Plná K. 2000. Specific DNA adducts induced by some mono‐substitued epoxides in vitro and in vivo . Chem Biol Interact 129(3):209–229. [DOI] [PubMed] [Google Scholar]
- Koskinen M, Vodickova L, Vodicka P, Warner SC, Hemminki K. 2001a. Kinetics of formation of specific styrene oxide adducts in double‐stranded DNA. Chem Biol Interact 138(2):111–124. [DOI] [PubMed] [Google Scholar]
- Koskinen M, Vodička P, Vodičkova L, Hemminki K. 2001b. (32)P‐postlabelling/HPLC analysis of various styrene‐induced DNA adducts in mice. Biomarkers 6(3):175–189. [DOI] [PubMed] [Google Scholar]
- Kumar R, Vodicka P, Peltonen K, Hemminki K. 1997. 32P‐postlabelling analysis of isomeric 7‐alkylguanine adducts of styrene oxide. Carcinogenesis 18:407–414. [DOI] [PubMed] [Google Scholar]
- Laffon B, Pásaro E, Méndez J. 2001. Genotoxic effects of styrene‐7,8‐oxide in human white blood cells: Comet assay in relation to the induction of sister chromatid exchanges and micronuclei. Mutat Res 491:163–172. [DOI] [PubMed] [Google Scholar]
- Latriano L, Wazneh L, Dong Z, Lu SJ, Snyder C, Jeffrey AM. 1991. Exposure to styrene: Comparison of DNA and haemoglobin adducts as biomarkers In: Garner RC, Farmer PB, Steel GT, Wright AS, editors. Human Carcinogen Exposure: Biomonitoring and Risk Assessment. Oxford, MI: Oxford University Press. [Google Scholar]
- Latt SA, Allen J, Bloom SE, Carrano A, Falke E, Kram D, Schneider E, Schreck R, Tice R, Whitfield B, et al. 1981. Sister‐chromatid exchanges: A report of the GENE‐TOX program. Mutat Res 87:17–62. [DOI] [PubMed] [Google Scholar]
- Leavens TL, Farris GM, James RA, Shah R, Wong VA, Marshall MW, Bond JA. 1997. Genotoxicity and cytotoxicity in male B6C3F1 mice following exposure to mixtures of 1,3‐butadiene and styrene. Environ Mol Mutagen 29(4):335–345. [DOI] [PubMed] [Google Scholar]
- Linnainmaa K, Meretoja T, Sorsa M, Vainio H. 1978a. Cytogenetic effects of styrene and styrene oxide on human lymphocytes and Allium cepa. Scand J Work Environ Health 4(suppl 2):156–162. [PubMed] [Google Scholar]
- Linnainmaa K, Meretoja T, Sorsa M, Vainio H. 1978b. Cytogenetic effects of styrene and styrene oxide. Mutat Res 58(2):277–286. [DOI] [PubMed] [Google Scholar]
- Liu SF, Fang QM, Jin ZL, Rappaport MS. 2001. Investigation of protein‐styrene oxide adducts as a molecular biomarker of human exposed to styrene. J Environ Sci 13(4):391–397. [PubMed] [Google Scholar]
- Loprieno N, Abbondandolo A, Barale R, Baroncelli S, Bonatti S, Bronzetti G, Cammellini A, Corsi C, Corti G, Frezza D, et al. 1976. Mutagenicity of industrial compounds: Styrene and its possible metabolite styrene oxide. Mutat Res 40(4):317–324. [DOI] [PubMed] [Google Scholar]
- Lorge E, Hayashi M, Albertini S, Kirkland D. 2008. Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test: I. Theoretical aspects. Mutat Res 655:1): 1–1): 3. [DOI] [PubMed] [Google Scholar]
- Matsuoka A, Hayashi M, Ishidate M. 1979. Chromosomal aberration tests on 29 chemicals combined with S9 mix in vitro . Mutat Res 66(3):277–290. [DOI] [PubMed] [Google Scholar]
- McCann J, Ames BN. 1976. Detection of carcinogens as mutagens in the salmonella/microsome test: Assay of 300 chemicals: Discussion. Proc Natl Acad Sci USA 73:950–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann J, Choi E, Yamasaki E, Ames BN. 1975. Detection of carcinogens as mutagens in the salmonella/microsome test: Assay of 300 chemicals. Proc Natl Acad Sci USA 72(12):5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meretoja T, Vainio H, Järventaus H. 1978. Clastogenic effects of styrene exposure on bone marrow cells of rat. Toxicol Lett 1(5):315–318. [Google Scholar]
- Mikeš P, Kořínek M, Linhart I, Krouželka J, Frantik E, Vodickov L, Neufussov L. 2009. Excretion of urinary N7 guanine and N3 adenine DNA adducts in mice after inhalation of styrene. Toxicol Lett 184:33–37. [DOI] [PubMed] [Google Scholar]
- Milvy P, Garro AJ. 1976. Mutagenic activity of styrene oxide (1,2‐epoxyethylbenzene), a presumed styrene metabolite. Mutat Res 40(1):15–18. [DOI] [PubMed] [Google Scholar]
- Morita T, Asano N, Awogi T, Sasaki YF, Sato S, Shimada H, Sutou S, Suzuki T, Wakata A, Sofuni T, et al. 1997. Evaluation of the rodent micronucleus assay in the screening of IARC carcinogens (groups 1, 2A and 2B) the summary report of the 6th collaborative study by CSGMT/JEMS MMS. Collaborative study of the micronucleus group test. Mammalian Mutagenicity Study Group [published erratum appears in mutation research 391(3):259‐67]. Mutat Res 389(1):3–122. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Mutlu E, Sharma V, Collins L, Bodnar W, Yu R, Lai Y, Moeller B, Lu K, Swenberg J. 2014. The endogenous exposome. DNA Repair 19:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestmann ER, Lynch BS, Ratpan F. 2005. Perspectives on the genotoxic risk of styrene. J Toxicol Environ Health Part B 8(2):95–107. [DOI] [PubMed] [Google Scholar]
- Nishi Y, Hasegawa MM, Taketomi M, Ohkawa Y, Inui N. 1984a. Comparison of 6‐thioguanine‐resistant mutation and sister chromatid exchanges in Chinese hamster V79 cells with forty chemical and physical agents. Cancer Res 44(8):3270–3279. [PubMed] [Google Scholar]
- Nishi Y, Hasegawa MM, Taketomi M, Ohkawa Y, Inui N. 1984b. Interrelationships of SCEs, mutation at the hgprt locus, and toxicity in Chinese hamster v79 cells. Basic Life Sci. 29(Pt A):361–384. [DOI] [PubMed] [Google Scholar]
- Norppa H. 1981. Styrene and vinyltoluene induce micronuclei in mouse bone marrow. Toxicol Lett 8(4–5):247–251. [DOI] [PubMed] [Google Scholar]
- Norppa H, Vainio H. 1983. Genetic toxicity of styrene and some of its derivatives. Scand J Work Environ Health 9(2):108–114. [DOI] [PubMed] [Google Scholar]
- Norppa H, Elovaara E, Husgafvel‐Pursiainen K, Sorsa M, Vainio H. 1979. Effects of styrene oxide on chromosome aberrations, sister chromatid exchange and hepatic drug biotrans‐formation in Chinese hamsters in vivo . Chem Biol Interact 26(3):305–315. [DOI] [PubMed] [Google Scholar]
- Norppa H, Sorsa M, Vainio H. 1980. Chromosomal aberrations in bone marrow of Chinese hamsters exposed to styrene and ethanol. Toxicol Lett 5(3–4):241–244. [DOI] [PubMed] [Google Scholar]
- Norppa H, Hemminki K, Sorsa M, Vainio H. 1981. Effect of monosubstituted epoxides on chromosome aberrations and SCE in cultured human lymphocytes. Mutat Res 91(3):243–250. [DOI] [PubMed] [Google Scholar]
- Norppa H, Maki‐Paakkanen J, Jantunen K, Einisto P, Raty R. 1988. Mutagenicity studies on styrene and vinyl acetate. Ann N Y Acad Sci 534:671–678. [DOI] [PubMed] [Google Scholar]
- NRC . 2014. Review of the Styrene Assessment in the National Toxicology Program 12th Report on Carcinogens. Washington, DC: The National Academies Press. [PubMed] [Google Scholar]
- NTP . 2008. Report on Carcinogens Background Document for Styrene. United States Department of Health and Human Services, Public Health Services, National Toxicology Program, Research Triangle Park, N.C. [Google Scholar]
- OECD, authors. OECD Guideline for the Testing of Chemicals. Test 471: Bacterial Reverse Mutation Test. Paris: The Organisation for Economic Co‐operation and Development. 1997a
- OECD . 1997b. OECD guideline for the testing of chemicals. Test 486: Unscheduled DNA synthesis (UDS) test with mammalian liver cells in vivo. Paris: The Organisation for Economic Co‐operation and Development.
- OECD, authors. OECD Guideline for the Testing of Chemicals: Mammalian Bone Marrow Chromosomal Aberration Test. Test 475. Paris: The Organisation for Economic Co‐operation and Development. 2016a
- OECD, authors. OECD Guideline for the Testing of Chemicals: In vitro Mammalian Cell Gene Mutation Tests Using the Thymidine Kinase Gene. Test 490. Paris: The Organisation for Economic Co‐operation and Development. 2016b
- OECD, authors. OECD Guideline for the Testing of Chemicals: In Vitro Mammalian Cell Gene Mutation Tests Using the Hprt and Xprt Genes. Test 476. Paris: The Organisation for Economic Co‐operation and Development. 2016c
- OECD, authors. OECD Guideline for the Testing of Chemicals: In vitro Mammalian Chromosomal Aberration Test. Test 473. Paris: The Organisation for Economic Co‐operation and Development. 2016d
- OECD, authors. OECD Guideline for the Testing of Chemicals: In vitro Mammalian Cell Micronucleus Test. Test 487. Paris: The Organisation for Economic Co‐operation and Development. 2016e
- OECD, authors. OECD Guideline for the Testing of Chemicals: Mammalian Erythrocyte Micronucleus Test. Test 474. Paris: The Organisation for Economic Co‐operation and Development. 2016f
- OECD, authors. OECD Guideline for the Testing of Chemicals: In vivo Mammalian Alkaline Comet Assay. Test 489. Paris: The Organisation for Economic Co‐operation and Development. 2016g
- OECD . 2017. Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014‐2015. Series on Testing & Assessment, Report 238. Paris: Organisation for Economic Cooperation and Development. Available at: http://www.oecd.org/officialdocuments/displaydocument/?cote5env/jm/mono(2016)33&doclanguage5en. Accessed September 27, 2016.
- Osterman‐Golkar S, Christakopoulos A, Zorcec V, Svensson K. 1995. Dosimetry of styrene 7,8‐oxide in styrene‐ and styrene oxide‐exposed mice and rats by quantification of haemoglobin adducts. Chem Biol Interact 95:79–87. [DOI] [PubMed] [Google Scholar]
- Otteneder M, Eder E, Lutz WK, Wu D. 1999. Analysis of DNA adducts of styrene 7, 8‐oxide at the O 6 ‐position of guanine. Chem Res Toxicol 12(1):93–99. [DOI] [PubMed] [Google Scholar]
- Otteneder M, Lutz U, Lutz WK. 2002. DNA adducts of styrene‐7,8‐oxide in target and non‐target organs for tumor induction in rat and mouse after repeated inhalation exposure to styrene. Mutat Res 500(1–2):111–116. [DOI] [PubMed] [Google Scholar]
- Pagano DA, Yagen B, Hernandez O, Bend JR, Zeiger E. 1982. Mutagenicity of (R) and (S) styrene 7,8‐oxide and the intermediary mercapturic acid metabolites formed from styrene 7,8‐oxide. Environ Mutagen 4(5):575–584. [DOI] [PubMed] [Google Scholar]
- Pauwels W, Vodiceka P, Severi M, Plna K, Veulemans H, Hemminki K. 1996. Adduct formation on DNA and haemoglobin in mice intraperitoneally administered with styrene. Carcinogenesis 17(12):2673–2680. [DOI] [PubMed] [Google Scholar]
- Penttila M, Sorsa M, Vainio H. 1980. Inability of styrene to induce nondisjunction in drosophila or a positive micronucleus test in the Chinese hamster. Toxicol Lett 6:119–123. [DOI] [PubMed] [Google Scholar]
- Pero RW, Bryngelsson T, Hogstedt B, Akesson B. 1982. Occupational and in vitro exposure to styrene assessed by unscheduled DNA synthesis in resting human lymphocytes. Carcinogenesis 3(6):681–685. [DOI] [PubMed] [Google Scholar]
- Philippin G, Cadet J, Gasparutto D, Mazon G, Fuchs RP. 2014. Ethylene oxide and propylene oxide derived N7‐alkylguanine adducts are bypassed accurately in vivo. DNA Repair 22:133–136. [DOI] [PubMed] [Google Scholar]
- Phillips DH, Farmer PB. 1994. Evidence for DNA and protein binding by styrene and styrene oxide. Crit Rev Toxicol 24(Suppl):S35–S46. [DOI] [PubMed] [Google Scholar]
- Pohlová H, Rössner P, Srám RJ. 1984. Cytogenetic analysis of human peripheral blood lymphocytes in culture exposed in vitro to styrene and styrene oxide. J Hyg Epidemiol Microbiol Immunol 29(3):269–274. [PubMed] [Google Scholar]
- Pongracz K, Kaur S, Burlingame AL, Bodell WJ. 1989. O6‐substituted‐2′‐deoxyguanosine‐3′‐phosphate adducts detected by 32P post‐labeling of styrene oxide treated DNA. Carcinogenesis 10(6):1009–1013. [DOI] [PubMed] [Google Scholar]
- Pongracz K, Kaur S, Burlingame AL, Bodell WJ. 1992. Identification of N2‐substituted 2′‐deoxyguanosine‐3′‐phosphate adducts detected by 32P‐postlabeling of styrene‐oxide‐treated DNA. Carcinogenesis 13(3):315–319. [DOI] [PubMed] [Google Scholar]
- Pottenger LH, Gollapudi BB. 2010. Genotoxicity testing: Moving beyond qualitative “screen and bin” approach towards characterization of dose‐response and thresholds. Environ Mol Mutagen 51:792–799. [DOI] [PubMed] [Google Scholar]
- Pottenger LH, Andrews LS, Bachman AN, Boogaard PJ, Cadet J, Embry MR, Farmer PB, Himmelstein MW, Jarabek AM, Martin EA, et al. 2014. An organizational approach for the assessment of DNA adduct data in risk assessment: Case studies for aflatoxin B1, tamoxifen and vinyl chloride. Crit Rev Toxicol 44(4):348–391. [DOI] [PubMed] [Google Scholar]
- Pottenger LH, Boysen G, Brown K, Cadet J, Fuchs RP, Johnson GE, Swenberg JA. 2018. Understanding the importance of low‐molecular weight (ethylene oxide‐ and propylene oxide‐induced) DNA adducts and mutations in risk assessment: Insights from 15 years of research and collaborative discussions. Environ Mol Mutagen 60(Dec 10):100–121. 10.1002/em.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston RJ, Abernethy DJ. 1993. Studies of the induction of chromosomal aberration and sister chromatid exchange in rats exposed to styrene by inhalation. IARC Sci Publ 127:225–233. [PubMed] [Google Scholar]
- Qian C, Dipple A. 1995. Different mechanisms of aralkylation of adenosine at the 1 and N6‐positions. Chem Res Toxicol 8(3):389–395. [DOI] [PubMed] [Google Scholar]
- Rappaport SM, Ting D, Jin Z, Yeowell‐O'Connell K, Waidyanatha S, McDonald T. 1993. Application of Raney nickel to measure adducts of styrene oxide with hemoglobin and albumin. Chem Res Toxicol 6:238–244. [DOI] [PubMed] [Google Scholar]
- Rappaport SM, Yeowell‐O'Connell K. 1999. Protein adducts as dosimeters of human exposure to styrene, styrene‐7,8‐oxide, and benzene. Toxicol Lett 108:117–126. [DOI] [PubMed] [Google Scholar]
- Rios‐Blanco MN, Faller TH, Nakamura J, Kessler W, Kreuzer PE, Ranasinghe A, Filser JG, Swenberg JA. 2000. Quantitation of DNA and hemoglobin adducts and apurinic/apyrimidinic sites in tissues of F344 rats exposed to propylene oxide by inhalation. Carcinogenesis 21:2011–2018. [DOI] [PubMed] [Google Scholar]
- Rosman LB, Beylin VG, Gaddamidi V, Hooberman BH, Sinsheimer JE. 1986. Mutagenicity of para‐substituted alpha‐methylstyrene oxide derivatives with salmonella. Mutat Res 171(2–3):63–70. [DOI] [PubMed] [Google Scholar]
- Rusyn I, Asakura S, Li Y, Kosyk O, Koc H, Nakamura J, Upton PB, Swenberg JA. 2005. Effects of ethylene oxide and ethylene inhalation on DNA adducts, apurinic/apyrimidinic sites and expression of base excision DNA repair genes in rat brain, spleen, and liver. DNA Repair 4(10):1099–1110. [DOI] [PubMed] [Google Scholar]
- Savela K, Hesso A, Hemminki K. 1986. Characterization of reaction products between styrene oxide and deoxynucleosides and DNA. Chem Biol Interact 60(3):235–246. [DOI] [PubMed] [Google Scholar]
- Sbrana I, Lascialfari D, Rossi AM, Loprieno N, Bianchi M, Tortoreto M, Pantarotto C. 1983. Bone marrow cell chromosomal aberrations and styrene biotransformation in mice given styrene on a repeated oral schedule. Chem Biol Interact 45(3):349–357. [DOI] [PubMed] [Google Scholar]
- Scott D, Preston RJ. 1994a. A critical review of the cytogenetic effects of styrene with an emphasis on human population monitoring: A synopsis. Crit Rev Toxicol, 24(suppl): S47‐S48. [DOI] [PubMed] [Google Scholar]
- Scott D, Preston RJ. 1994b. A re‐evaluation of the cytogenetic effects of styrene. Mutat Res 318(3):175–203. [DOI] [PubMed] [Google Scholar]
- Seiler JP. 1990. Chirality‐dependent DNA reactivity as the possible cause of the differential mutagenicity of the two components in an enantiomeric pair of epoxides. Mutat Res 245(3):165–169. [DOI] [PubMed] [Google Scholar]
- Sepai O, Anderson S, Street B, Bird I, Farmer PB, Bailey E. 1993. Monitoring of exposure to styrene oxide by GC‐MS analysis of phenylhydroxyethyl esters in hemoglobin. Arch Toxicol 67(1):28–33. [DOI] [PubMed] [Google Scholar]
- Sharief Y, Brown AM, Backer LC, Campbell JA, Westbrook‐Collins B, Stead AG, Allen JW. 1986. Sister chromatid exchange and chromosome aberration analyses in mice after in vivo exposure to acrylonitrile, styrene, or butadiene monoxide. Environ Mutagen 8(3):439–448. [DOI] [PubMed] [Google Scholar]
- Shield AJ, Sanderson BJ. 2001. Role of glutathione S‐transferase Mu (GSTM1) in styrene‐7,8‐oxide toxicity and mutagenicity. Environ Mol Mutagen 37(4):285–289. [DOI] [PubMed] [Google Scholar]
- Shield AJ, Sanderson BJ. 2004. A recombinant model for assessing the role of GSTM1 in styrene‐7, 8‐oxide toxicity and mutagenicity. Toxicology 195(1):61–68. [DOI] [PubMed] [Google Scholar]
- Siethoff C, Feldmann I, Jakubowski N, Linscheid M. 1999. Quantitative determination of DNA adducts using liquid chromatography/electrospray ionization mass spectrometry and liquid chromatography/high‐resolution inductively coupled plasma mass spectrometry. J Mass Spectrom 34(4):421–426. [DOI] [PubMed] [Google Scholar]
- Simmon VF, Kauhanen K, Tardiff RG. 1977. Mutagenic activity of chemicals identified in drinking water. Dev Toxicol Environ Sci 2:249–258. [Google Scholar]
- Simula AP, Priestly BG. 1992. Species differences in the genotoxicity of cyclophosphamide and styrene in three in vivo assays. Mutat Res 271(1):49–58. [DOI] [PubMed] [Google Scholar]
- Sina JF, Bean CL, Dysart GR, Taylor VI, Bradley MO. 1983. Evaluation of the alkaline elution/rat hepatocyte assay as a predictor of carcinogenic/mutagenic potential. Mutat Res 113(5):357–391. [DOI] [PubMed] [Google Scholar]
- Sinha AK, Jersey GC, Linscome VA, Adams RJ, Mueller AM, MClintock ML. 1983. Cytogenetic evaluation of bone marrow cells from rats exposed to styrene vapor for one year. Fundam Appl Toxicol 3:95–98. [DOI] [PubMed] [Google Scholar]
- Sinsheimer JE, Chen R, Das SK, Hooberman BH, Osorio S, You Z. 1993. The genotoxicity of enantiomeric aliphatic epoxides. Mutat Res 298(3):197–206. [DOI] [PubMed] [Google Scholar]
- Speit G, Henderson L. 2005. Review of the in vivo genotoxicity tests performed with styrene. Mutat Res 589(1):67–79. [DOI] [PubMed] [Google Scholar]
- Speit G, Linsenmeyer R, Schütz P, Kuehner S. 2012. Insensitivity of the in vitro cytokinesis‐block micronucleus assay with human lymphocytes for the detection of DNA damage present at the start of the cell culture. Mutagenesis 27(6):743–747. [DOI] [PubMed] [Google Scholar]
- Stoltz DR, Whitey RJ. 1977. Mutagenicity testing of styrene and styrene epoxide in Salmonella typhimurium . Bull Environ Contam Toxicol 17(6):739–742. [DOI] [PubMed] [Google Scholar]
- Sugiura K, Goto M. 1981. Mutagenicities of styrene oxide derivatives on bacterial test systems: Relationship between mutagenic potencies and chemical reactivity. Chem Biol Interact 35(1):71–91. [DOI] [PubMed] [Google Scholar]
- Sugiura K, Kimura T, Goto M. 1978. Mutagenicities of styrene oxide derivatives on Salmonella typhimurium (TA 100): Relationship between mutagenic potencies and chemical reactivity. Mutat Res 58(2):159–165. [DOI] [PubMed] [Google Scholar]
- Sugiura K, Maeda A, Goto M. 1979. Substitutional effects of styrene oxides on survival and mutation induction in cultured Chinese hamster cells (v‐79). Chemosphere 6:369–372. [Google Scholar]
- Swenberg JA, Fryar‐Tita E, Jeong Y‐C, Boysen G, Starr T, Walker VE, Albertini RJ. 2008. Biomarkers in toxicology and risk assessment: Informing critical dose–response relationships. Chem Res Toxicol 21:253–265. [DOI] [PubMed] [Google Scholar]
- Thybaud V, Lorge E, Levy DD, Benthem J, Douglas GR, Marchetti M, Moore MM, Schoeny R. 2017. Main issues addressed in the 2014–2015 revisions to the OECD genetic toxicology test guidelines. Environ Mol Mutagen 58(5):284–295. [DOI] [PubMed] [Google Scholar]
- Ting D, Smith MT, Doane‐Setzer P, Rappaport SM. 1990. Analysis of styrene oxide‐globin adducts based upon reaction with Raney nickel. Carcinogenesis 11:755–760. [DOI] [PubMed] [Google Scholar]
- Tucker JD, Auletta A, Cimino MC, Dearfield KL, Jacobson‐Kram D, Tice RR, Carrano AV. 1993. Sister‐chromatid exchange: Second report of the gene‐Tox program. Mutat Res 297(2):101–180. [DOI] [PubMed] [Google Scholar]
- Turchi G, Bonatti S, Citti L, Gervasi PG, Abbondandolo A, Presciuttini S. 1981. Alkylating properties and genetic activity of 4‐vinylcyclohexene metabolites and structurally related epoxides. Mutat Res 83(3):419–430. [DOI] [PubMed] [Google Scholar]
- Vaghef H, Hellman B. 1998. Detection of styrene and styrene oxide‐induced DNA damage in various organs of mice using the comet assay. Pharmacol Toxicol 83(2):69–74. [DOI] [PubMed] [Google Scholar]
- Vainio H, Paakkonen R, Ronnholm K, Raunio V, Pelkonen O. 1976. A study on the mutagenic activity of styrene and styrene oxide. Scand J Work Environ Health 2(3):147–151. [DOI] [PubMed] [Google Scholar]
- Vainio H, Norppa H, Hemminki K, Sorsa M. 1981. Metabolism and genotoxicity of styrene. Adv Exp Med Biol 136(Pt A):257–274. [DOI] [PubMed] [Google Scholar]
- Vainio H, Norppa H, Belvedere G. 1984. Metabolism and mutagenicity of styrene and styrene oxide. Prog Clin Biol Res 141:215–225. [PubMed] [Google Scholar]
- Vodicka P, Hemminki K. 1988a. Depurination and imidazole ring‐opening in nucleosides and DNA alkylated by styrene oxide. Chem Biol Interact 68(1–2):117–126. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Hemminki K. 1988b. Identification of alkylation products of styrene oxide in single‐ and double‐stranded DNA. Carcinogenesis 9(9):1657–1660. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Hemminki K. 1991a. 32P‐postlabeling of N‐7, N2 and O6 2′‐deoxyguanosine 3′‐monophosphate adducts of styrene oxide. Chem Biol Interact 77(1):39–50. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Hemminki K. 1991b. The prospects of the development of the method for monitoring of occupational exposure to some alkylating agents. Sci Total Environ 101(1–2):121–130. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Koskinen M, Vodickova L, Stetina R, Smerak P, Barta I, Hemminki K. 2001. DNA adducts, strand breaks and micronuclei in mice exposed to styrene by inhalation. Chem Biol Interact 137(3):213–227. [DOI] [PubMed] [Google Scholar]
- Vodicka P, Koskinen M, Arand M, Oesch F, Hemminki K. 2002. Spectrum of styrene‐induced DNA adducts: The relationship to other biomarkers and prospects in human biomonitoring. Mutat Res 511(3):239–254. [DOI] [PubMed] [Google Scholar]
- Vodicka PE, Linhart I, Novak J, Koskinen M, Vodickova L, Hemminki K. 2006. 7‐Alkylguanine adduct levels in urine, lungs and liver of mice exposed to styrene by inhalation. Toxicol Appl Pharmacol 210:1–2): 1–8. [DOI] [PubMed] [Google Scholar]
- Wade DR, Airy SC, Sinsheimer JE. 1978. Mutagenicity of aliphatic epoxides. Mutat Res 58(2):217–223. [DOI] [PubMed] [Google Scholar]
- Watabe T, Isobe M, Sawahata T, Yoshikawa K, Yamada S, Takabatake E. 1978. Metabolism and mutagenticity of styrene. Scand J Work Environ Health 4(suppl 2):142–155. [PubMed] [Google Scholar]
- Watabe T, Hiratsuka A, Isobe M, Ozawa N. 1980. Metabolism of D‐limonene by hepatic microsomes to non‐mutagenic epoxides toward salmonella typhimurium. Biochem Pharmacol 29(1):1068–1071. [DOI] [PubMed] [Google Scholar]
- Williams GM, Mori H, McQueen CA. 1989. Structure‐activity relationships in the rat hepatocyte DNA‐repair test for 300 chemicals. Mutat Res 221(3):263–286. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Thompson LH. 2007. Molecular mechanisms of sister‐chromatid exchange. Mutat Res 616:11–23. [DOI] [PubMed] [Google Scholar]
- Yang J, Wang B, Rusling JF. 2005. Genotoxicity sensor response correlated with DNA nucleobase damage rates measured by LC‐MS. Mol Biosyst, 1: 251–259. Royal Society of Chemistry {foreign} [DOI] [PubMed] [Google Scholar]
- Yoshikawa K, Isobe M, Watabe I, Takabatake E. 1980. Studies on metabolism and toxicity of styrene: III. The effect of metabolic inactivation by rat‐liver S9 on the mutagenicity of phenyloxirane toward salmonella typhimurium. Mutat Res 78(3):219–226. [DOI] [PubMed] [Google Scholar]
- Zeiger E. 1987. Carcinogenicity of mutagens: Predictive capability of the salmonella mutagenesis assay for rodent carcinogenicity. Cancer Res 47(5):1287–1296. [PubMed] [Google Scholar]
- Zeiger E, Anderson B, Haworth S, Lawlor T, Mortelmans K. 1988. Salmonella mutagenicity tests. 4. Results from the testing of 300 chemicals. Environ Mol Mutagen 11(suppl 12):1–158. [PubMed] [Google Scholar]
- Zeiger E, Anderson B, Haworth S, Lawlor T, Mortelmans K. 1992. Salmonella mutagenicity tests: V. Results from the testing of 311 chemicals. Environ Mol Mutagen 19, 2–141. [DOI] [PubMed] [Google Scholar]