

Abstract
Genome architecture is well diversified among eukaryotes in terms of size and content, with many being radically shaped by ancient and ongoing genome conflicts with transposable elements (e.g., the large transposon‐rich genomes common among plants). In ciliates, a group of microbial eukaryotes with distinct somatic and germ‐line genomes present in a single cell, the consequences of these genome conflicts are most apparent in their developmentally programmed genome rearrangements. This complicated developmental phenomenon has largely overshadowed and outpaced our understanding of how germ‐line and somatic genome architectures have influenced the evolutionary dynamism and potential in these taxa. In our review, we highlight three central concepts: how the evolution of atypical ciliate germ‐line genome architectures is linked to ancient genome conflicts; how the complex, epigenetically guided transformation of germline to soma during development can generate widespread genetic variation; and how these features, coupled with their unusual life cycle, have increased the rate of molecular evolution linked to genome architecture in these taxa.
Keywords: ciliates, evolution, genome evolution, transposable elements, genome architecture
Introduction
Genomes are highly dynamic, undergoing constant modification by genetic and epigenetic processes, while also maintaining vigil against the spread of transposable elements (TEs).1, 2, 3, 4, 5, 6 While TEs are often described as parasitic or selfish DNA that are assumed to proliferate at the expense of the host genome's fitness (i.e., by increasing genome instability),7, 8 they remain essential and well‐regulated genomic components, for example, possessing roles as centromeres and/or telomeres in Dictyostelium discoideum and Drosophila melanogaster, respectively.9, 10 Maintaining some control over genome instability provides massive fitness benefits to the host genome/organism, and is linked to genome dynamism,11, 12, 13, 14, 15, 16, 17 which is exaggerated and well studied in pathogenic lineages (Phytophthora infestans and Entamoeba histolytica).11, 12, 13 A dramatic example is the separation of germ‐line and somatic genomes, which provides the means to protect the heritable genome while reaping the benefits of a highly dynamic and responsive soma.
Distinct somatic and germ‐line genomes are found in diverse lineages across the eukaryotic tree of life and are best understood in multicellular eukaryotes, where they are partitioned into separate tissues (e.g., pollen in plants, eggs in animals, and spores in fungi). However, in single‐cell ciliates, these two genomes are found in dimorphic nuclei: a diploid, transcriptionally silent germ‐line micronuclear genome (MIC), which becomes transcriptionally active only during sex, and a highly polyploid and transcriptionally active somatic macronuclear genome (MAC) that supports the cell. Complex, epigenetically guided processing underlies the development of new somatic nuclei from a zygotic nucleus.18, 19, 20, 21, 22, 23, 24 This involves the elimination of germline‐limited DNA (e.g., TEs, centromeres, germline‐limited genes, and internally eliminated sequences (IESs)) and the assembly of functional somatic regions (i.e., macronuclear‐destined sequences (MDS)).18, 25, 26, 27, 28, 29, 30 Although details differ across ciliate lineages, the delineation during development between somatic MDSs and the germline‐limited IESs, which separate MDSs, involves RNA‐guided mechanisms that resemble epigenetic responses to TE invasion/control in other eukaryotes.17, 19, 20, 21, 22, 23, 24
Here, we describe how the atypical genome architectures in ciliates, coupled with a predominantly asexual life cycle punctuated by rare sexual events (similar to yeasts and other protists), provide them with an immense evolutionary potential and the means for rapid adaptation. This is largely due to the evolutionary impacts of ancient genome conflict with TEs, which are well known to provide the basis for evolutionary innovation in other eukaryotes.17 The general exploration of the interrelations between ciliate genome architecture, programmed genome rearrangements, and their life history (i.e., asexual growth with infrequent sexual events) will draw more attention to the role of genome architecture in evolution.
TEs and germ‐line genome architecture in ciliates
Due to mechanistic similarities between the developmentally regulated genome rearrangements in ciliates and transposon regulation in other taxa, Klobutcher and Herrick31 proposed that evolution of nuclear dualism in ciliates was an evolutionary response to TE invasion, providing the means to purge them from the somatic genome. While the somatic genomes of most ciliates studied to date are effectively free of TEs, the germ‐line genomes are enriched with repetitive regions/sequences that interrupt gene‐coding sequences (an exception being the model ciliate Tetrahymena thermophila, where these regions are predominantly intergenic or intronic) and need to be excised during development of a new somatic genome.25, 26, 27, 28, 29, 30, 31, 32, 33 While the evolutionary origin of these repetitive regions is unclear given their vast diversity within a single germ‐line genome, many harbor signatures of once functional TEs (i.e., repetitive boundary sequences or small terminal inverted repeats, such as TA; Fig. 1C).25, 27, 28, 31, 32, 33 For example, Tec elements (transposons from the Tc1/Mariner family) in Euplotes are present in great abundance, at >10,000 copies and ∼20–25% of the estimated germ‐line genome size.32 These highly abundant transposons are flanked by direct TA repeats, as are the majority of Euplotes IESs.31, 32, 33 This observation, coupled with the presence of several documented TEs interrupting coding sequences (which are accurately eliminated during development), implies a common excision mechanism targeting both TEs and IESs. As Klobutcher and Herrick31 describe, one explanation for the evolution and widespread distribution of IESs in ciliate germlines may derive from a period of replicative transposon transposition (bloom; Fig. 1B), followed by their inactivation and subsequent degeneration into IESs, where only the terminal sequences necessary for excision (pointer sequences) have remained (decay; Fig. 1C). More recently, a survey of IESs from the complete T. thermophila germ‐line genome has found that ∼42% of the IESs (comprising 10.9 Mbp of the 150 Mbp germ‐line genome) are putative TEs and their decayed remnants.27 While all IESs identified among diverse ciliate lineages, spanning ∼1 GYA,34 are demarcated by direct repeats at their MDS–IES boundaries,25, 26, 27, 28, 29, 30, 33 these data suggest that TEs have played a role in the origin of some of the IESs found in ciliate germ‐line genomes.
Figure 1.
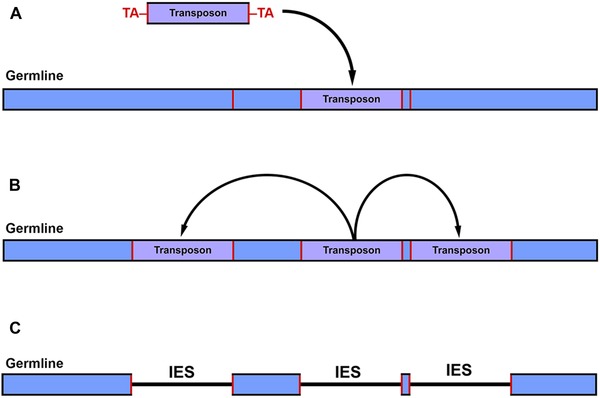
Origin of germ‐line genome architecture in ciliates. (A) Invasion of a transposable element into the germ‐line genome at a target insertion site (red boundaries, germline). (B) Over time, it proliferates (bloom) throughout the germ‐line genome. (C) These TEs decay over time into internally eliminated sequences (IESs), ultimately generating the traditional nonscrambled genome architecture. The original insertion target sequences (red) remain as pointer sequences that guide MDS organization in many ciliates (e.g., Paramecium and Euplotes).
While TEs are tightly linked with the evolution of ciliate germ‐line genome architecture, they have also become an indispensable player in programmed DNA elimination. In Paramecium tetraurelia and T. thermophila, domesticated PiggyBac transposases (e.g., PiggyMAC or TPB encoded in the Paramecium and Tetrahymena somatic genomes, respectively) perform the bulk excision of germline‐limited DNA, including TEs, from the developing somatic genome.35, 36, 37, 38, 39 Silencing P. tetraurelia’s PiggyMac transposase during development ultimately results in the retention of most of the ∼45,000 IESs, resulting in a nonfunctional somatic genome.25 Although taming transposons appears to be required for the massive DNA elimination observed in ciliates, the degree of domestication (i.e., recruitment into the somatic genome versus limited to the germline) is variable. For example, the transposases of thousands of presumably active germline‐limited telomere‐bearing elements (TBEs, part of the Tc1/Mariner family) facilitate the developmentally regulated genome rearrangements during Oxytricha trifallax development.40 Silencing these germline‐limited TBEs in O. trifallax during development hampers the genome rearrangement process, resulting in the accumulation of quasi‐germline chromosomes (misarranged, atypically large, harboring IESs) in the new somatic nucleus.40 Their absence from the somatic genome suggests that these germline‐limited transposases are not domesticated to the same degree as in Paramecium and Tetrahymena. Interestingly, a germline‐limited PiggyBac transposase has been identified in Tetrahymena and is required for precise excision of germline‐limited DNA, whereas the somatic PiggyBac, which is responsible for the bulk of IES excision, does so at variable boundaries.41 These data from ciliates are yet another example of how TE proteins, regardless of their domestication status, have often been co‐opted into numerous pathways as adaptations to a variety of evolutionary conflicts spanning the tree of life.37, 41, 42, 43
Origins of ciliate scrambled germ‐line genome architecture
Descriptions of ciliate germ‐line genome architecture fall into two categories: a nonscrambled organization, with consecutive somatic MDSs separated by germline‐limited IESs in the same orientation (the most obvious result of the TE invasion–bloom–decay hypothesis; Fig. 1),31 and scrambled, where some MDSs are in nonconsecutive order and/or encoded on opposing DNA strands (Fig. 2).26, 29, 30, 44, 45, 46, 47, 48, 49 While nonscrambled germ‐line organization is common across the ciliate phylogeny, emerging evidence from poorly sampled lineages suggests that scrambled germ‐line loci may be more common than previously expected.30
Figure 2.
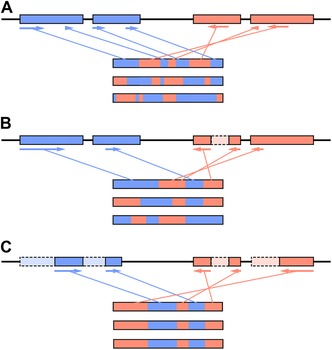
Example of the origin of a scrambled germ‐line locus. (A) Following the duplication of a germ‐line locus (top), functional proteins/chromosomes (three examples below) can be assembled from a myriad of combinations of the duplicate MDSs (blue and red). Arrows indicate the 5′‐3′ orientation in the germline and represent the portions of the MDSs that are assembled into the top‐most chromosome/gene. (B) Eventually some portions of MDSs decay (red, dashed box), forcing the alternative processing of the germ‐line loci to still produce functional chromosomes. (C) Eventually, the decay becomes complete, resulting in a single combination of the remaining functional MDS portions in a scrambled orientation.
A proposed model for the origin of scrambled germ‐line loci involves an initial duplication event that generates long stretches of identical DNA, irrespective of orientation.48 From these duplicated loci, combinatorial rearrangements can take place during development (guided by the large pool of redundant pointer sequences), generating identical somatic sequences (Fig. 2A).43, 44, 45, 46, 47, 48, 49 Over time, decay/divergence of redundant pointers and/or identical coding regions could become fixed, with negligible impacts on fitness (Fig. 2B and C). Alternative processing has been suggested to be the intermediate stage between duplication and fixation of a single orientation of MDSs,45, 46, 47, 48, 49 where numerous paralogous genes/chromosomes can be formed from duplicated germ‐line loci. However, given enough time, these highly diverged regions can be targeted for elimination if absent from the parental genome. For example, in Oxytricha piRNAs, two types of RNAs are involved in forming a faithful reproduction of the old parental genome. Small piRNAs protect MDSs from elimination, which presumably then use RNA copies of whole chromosomes that guide accurate rearrangement of these protected MDSs, regardless their orientation in the germline.19, 21, 23, 50
Generation of diversity through genome rearrangements
Despite the critical importance of accurate complex genome reorganization in the development of a new somatic genome in ciliates, the process itself remains susceptible to heritable changes (errors) linked with epigenetic processes and environmental conditions. For example, in P. tetraurelia, mating‐type determination involves the retention of a single 195 bp IES at the mating‐type locus, where the IES is retained in MT‐E (IES+) and absent in MT‐O (IES–).51, 52, 53, 54, 55 When growing under optimal conditions, spontaneous switches in mating type from MT‐E to MT‐O occur in ∼1/3000 cells, whereas the opposite is much rarer, <1/50,000 cells.55 Environmental conditions (e.g., temperature differences) can strongly alter the above patterns of mating‐type inheritance. Interestingly, when exposed to reduced temperatures (13 °C), MT‐O individuals predominantly maintain their mating type; however, the frequency of spontaneous switches from MT‐O to MT‐E, the difference being the retention of a single specific IES in MT‐E (IES+), increases dramatically with increasing temperature.53, 54 More generally in Paramecium, the protein machinery involved in the developmentally regulated genome rearrangements are themselves error prone, as the accidental elimination of portions of MDSs has been noted to occur and nearly 1/10th of IESs are inaccurately excised or incompletely excised.56 However, the severity of the impact of IES retention in Paramecium is likely offset by its rather great ploidy (∼800N).57
Although the frequency of aberrant structural variation events during development in Paramecium may be relatively common, they rarely appear to be fixed in the population of chromosomes. For example, for a Paramecium to express the surface antigens found only in the MT‐E (IES+) rather than MT‐O (IES−) cells, enough copies of the mating‐type locus retaining the IES must be present; otherwise, the cell remains MT‐O.53, 54 This suggests that aberrant excision events and/or retained IESs, even when present at low abundance, can impact a cell's fitness by either reducing the number of functional copies of a gene (i.e., through frameshifting or the introduction of premature stop codons), or through the insertion of novel sequences that alter the protein structure itself or its regulatory regions.
Although IESs can alter pre‐existing protein structure/expression, genes from scrambled germ‐line loci are a particular source of genomic diversity/novelty (Fig. 3). For example, in Chilodonella uncinata there are at least four divergent β‐tubulin genes that are generated from the alternative processing of three scrambled germ‐line loci.45, 46 Genes from scrambled germ‐line loci comprise surprisingly great proportions of C. uncinata’s largest gene families; however, these large gene families are also fairly new (lacking homologs in other taxa).29 The expansion of novel (i.e., lineage‐specific) gene families through gene scrambling is common among taxa with substantial germ‐line scrambling (e.g., O. trifallax and its relatives; Figs. 2 and 3B).48 In these taxa, small RNAs from the parental genome aid in demarcating portions of the germ‐line genome that ought to be retained,21, 23 as in Paramecium and Tetrahymena, but full chromosomes (i.e., long template RNAs) are transcribed to ensure the correct rearrangement order.19, 20, 50
Figure 3.
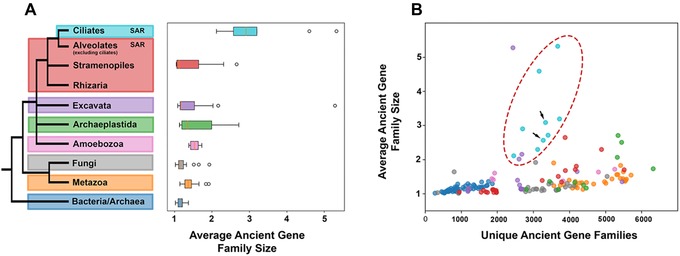
(A) Ciliate genomes harbor fewer, but larger gene families than other eukaryotes. This trend holds true across most ciliate taxa (B, red ellipse), including parasitic ciliates (e.g., Ichthyophthirius multifilis and Pseudocohnilembus persalinus), which possess a substantially reduced proteome. (B) The ciliates with scrambled germ‐line genomes (black arrows, Stylonychia lemnae and Oxytricha trifallax; lower left and upper right, respectively), possess comparable paralog diversity in ancient gene families to ciliates lacking genome scrambling, despite evidence for scrambling‐associated gene family expansion. These gene families are more likely to be lineage‐specific or are too divergent for gene family binning and as such do not show up on this plot.
Potential mistakes that occur during unscrambling (resembling alternative processing) ultimately diversify the population of chromosomes. Once present, these alternatively arranged chromosomes can undergo functional divergence (i.e., subfunctionalization or neofunctionalization). Analyses of alternatively processed paralogs from both Chilodonella and Oxytricha show strong purifying selection acting upon shared MDSs, whereas those paralog‐specific sequences can be incredibly divergent.45, 46, 47, 48, 49 While some of these paralogs may be nonfunctional, RNA‐seq analyses of different time points show little overlap of expression between alternatively processed paralogs.48 These data clearly demonstrate how genome unscrambling can generate novel genetic diversity upon which selection can act. So long as these alternatively processed chromosomes do not negatively impact the cell's fitness, these rearranged chromosomes provide the template for unscrambling the respective germ‐line loci during the next sexual event.19, 20, 50
Although not all IESs require small RNAs to identify and guide their excision (i.e., nonmaternally controlled IESs), the parental genome's influence through RNA over the developing somatic genome appears common across diverse ciliates. For example, experimental deletions in the parental somatic genome in Paramecium correspond to changes in the pool of scanning RNAs that aid in delineating germline from soma, ultimately resulting in the inheritance of the same deletion in the developing somatic genome.58 Similarly, experiments hindering the production of these small RNAs in Paramecium and Tetrahymena result in the retention of large portions of the germline‐limited DNA in the developing genome.59, 60 This transnuclear crosstalk through RNA intermediates helps the parental soma to further shape the next generation's somatic genome.
Accelerated protein evolution and amitosis
Asexuality dominates the majority of a ciliate's life cycle, while sexual events (and the mutagenic potential of genome rearrangements) are brief points. During many rounds of asexual division between sexual events, the diploid germline divides through conventional mitosis, steadily accumulating mutations.61 Although most of the mutations that arise in the germline are likely deleterious, they remain hidden from selection, providing the time needed for compensatory mutations to arise with minimal impact on fitness.61 Eventually, these mutations will be exposed to selection in the somatic genome after sex. However, during these abundant asexual divisions, the polyploid somatic genome undergoes amitosis (ciliates in the class Karyorelictea being the exception),62, 63, 64, 65 which separates masses of chromosomes in the absence of mitotic spindles, resulting in the unequal partitioning of DNA (i.e., aneuploidy).18, 62
This absence of controlled segregation (i.e., a metaphase plate) results in unique daughter nuclei that are not only initially distinct in ploidy, but potentially in genomic content as well. For example, during the development of a new somatic genome, two alleles (i.e., an exposed germ‐line mutation now exposed in the soma, retained IES, or a unique alternatively processed gene or chromosome) arise at roughly equal copy numbers, one of which is deleterious, in the somatic genome (Fig. 4A). Even after the first amitotic division, the proportion of the deleterious allele would be different between daughter cells, due to the random segregation of bulk DNA (Fig. 4B). The daughter nucleus and cell with the lower proportion of the deleterious allele would be favored by selection. Over time and many amitotic divisions, the random assortment of alleles will result in asexual cell lineages with increasingly fewer copies of this disadvantageous allele. These cells will outgrow most other cells, ultimately comprising greater proportions of the population (Fig. 4C).
Figure 4.
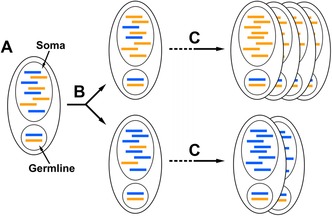
Phenotypic assortment through amitosis enhances the impact of selection. (A) Two alleles, one slightly deleterious (blue), are present in roughly equal copy number in the somatic nucleus. (B) After the first amitotic division, the alleles are separated randomly, resulting in nonidentical daughter cells/nuclei. (C) Over many amitotic divisions, an incredibly fit lineage emerges (top), dominating the population of cells, whereas cells predominantly possessing the slightly deleterious allele divide more slowly, becoming an increasingly smaller proportion of the population of cells (bottom).
The efficiency of phenotypic assortment through amitosis (and the efficacy of selection) is strongly tied to the structure of the somatic genome. For instance, in Chilodonella and Oxytricha, the somatic nuclei harbor thousands of unique gene‐sized chromosomes, with each chromosome at independent copy numbers ranging from several hundred to nearly a million copies.66, 67, 68, 69 The fate of every gene in these genomes is independent of the others. Selection can favor those nuclei harboring fewer deleterious mutations/arrangements, which could be lost over time without impacting other genes/chromosomes. By contrast, in Paramecium and Tetrahymena, most genes share the large (>50 kbp) chromosomes with ∼100–400 other genes, and are at much lower ploidy (∼800N in P. tetraurelia and ∼45N in T. thermophila).57, 70 Complete chromosome loss would be catastrophic and deleterious mutations may be more likely to be retained, albeit at minimal ploidy (e.g., retention of an IES in a subpopulation of chromosomes from MT‐E Paramecia). The combination of ciliate genome architecture and amitosis are tied to the observed elevated rates of protein evolution.46, 48, 71, 72
The protein‐coding genes in those ciliates with extremely processed somatic genomes (i.e., composed of millions of gene‐sized chromosomes) exist nearly free of any gene linkage. This organization greatly enhances the rate and efficacy of phenotypic assortment since the evolutionary fates of alleles and chromosomes are unique. Coupled with the ability to alternatively process germ‐line loci, these taxa are able to generate highly divergent proteins in relatively short periods of time.29, 44, 45, 46, 47, 48, 49 Even though this trend is strongest in ciliates possessing gene‐sized chromosomes, this general trend holds true for most of the ciliate clade, even among highly conserved proteins.71, 72 The exception to this pattern is members of the ciliate class Karyorelictea, whose MACs are unable to divide and must be generated from a diploid MIC with every cell division. In these taxa, there is evidence for substantially greater purifying selection acting on orthologous and paralogous genes compared to ciliates able to divide their macronuclei through amitosis (unpublished data). This appears to occur despite germ‐line genome architectures similar to other ciliate taxa30 and indicates the ability that amitosis has to enhance patterns of protein evolution.
Conclusion
Genome conflict and the epigenetic process have greatly contributed to the great diversity of observed genome architectures across the tree of life. In ciliates, this has led to a dynamic developmental system, where epigenetic processes have been co‐opted into dramatic genome remodeling, including RNA‐guided DNA elimination and chromosome copy number control. Errors in these processes provide the means for the rapid development of new genes and alterations in regulatory networks within a few sexual generations. While ciliate genome architectures are the source of novelty, the ability to amitotically divide their somatic genomes facilitates their adaptability through the proliferation or loss of novel mutations. Unfortunately, without more data (both genomic and experimental) from a greater diversity of ciliates and other eukaryotes, it remains difficult to disentangle the roles of amitosis and genome architecture (both somatic and germline) in the context of adaptability and molecular evolution.
Competing interests
The authors declare no competing interests.
References
- 1. Wassenegger, M. , Heimes S., Riedel L. & Sanger H.L.. 1994. RNA‐directed de novo methylation of genomic sequences in plants. Cell 76: 567–576. [DOI] [PubMed] [Google Scholar]
- 2. Matzke, M.A. , Mette M.F. & Matzke A.J.M.. 2000. Transgene silencing by the host genome defense: implications for the evolution of epigenetic control mechanisms in plants and vertebrates. Plant Mol. Biol. 43: 401–415. [DOI] [PubMed] [Google Scholar]
- 3. Slotkin, K.R. & Martienssen R.. 2007. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8: 272. [DOI] [PubMed] [Google Scholar]
- 4. Shabalina, S.A. & Koonin E.V.. 2008. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 23: 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hollister, J.D. & Gaut B.S.. 2009. Epigenetic silencing of transposable elements: a trade‐off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 19: 1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matzke, M.A. & Mosher R.A.. 2014. RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15: 394. [DOI] [PubMed] [Google Scholar]
- 7. Charlesworth, B. , Sniegowski P. & Stephan W.. 1994. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371: 215. [DOI] [PubMed] [Google Scholar]
- 8. Orgel, L.E. & Crick F.H.C.. 1980. Selfish DNA: the ultimate parasite. Nature 284: 604. [DOI] [PubMed] [Google Scholar]
- 9. Levis, R.W. et al 1993. Transposons in place of telomeric repeats at a Drosophila telomere . Cell 75: 1083–1093. [DOI] [PubMed] [Google Scholar]
- 10. Glöckner, G. & Heidel A.J.. 2009. Centromere sequence and dynamics in Dictyostelium discoideum . Nucleic Acids Res. 37: 1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haas, B.J. et al 2009 Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans . Nature 461: 393–398. [DOI] [PubMed] [Google Scholar]
- 12. Kumari, V. et al 2011. Differential distribution of a SINE element in the Entamoeba histolytica and Entamoeba dispar genomes: role of the LINE‐encoded endonuclease. BMC Genomics 12: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vetukuri, R.R. et al 2013. Phenotypic diversification by gene silencing in Phytophthora plant pathogens. Commun. Integr. Biol. 6: e25890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Molinier, J. , Ries G., Zipfel C. & Hohn B.. 2006. Transgeneration memory of stress in plants. Nature 442: 1046–1049. [DOI] [PubMed] [Google Scholar]
- 15. Oliver, K.R. & Greene W.K.. 2009. Transposable elements: powerful facilitators of evolution. Bioessays 31: 703–714. [DOI] [PubMed] [Google Scholar]
- 16. Kathiria, P. et al 2010. Tobacco mosaic virus infection results in an increase in recombination frequency and resistance to viral, bacterial, and fungal pathogens in the progeny of infected tobacco plants. Plant Physiol. 153: 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maurer‐Alcalá, X.X. & Katz L.A.. 2015. An epigenetic toolkit allows for diverse genome architectures in eukaryotes. Curr. Opin. Genet. Dev. 35: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prescott, D.M. 1994. The DNA of ciliated protozoa. Microbiol. Rev. 58: 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nowacki, M. et al 2008. RNA‐mediated epigenetic programming of a genome‐rearrangement pathway. Nature 451: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jönsson, F. , Postberg J. & Lipps H.J.. 2009. The unusual way to make a genetically active nucleus. DNA Cell Biol. 28: 71–78. [DOI] [PubMed] [Google Scholar]
- 21. Fang, W. et al 2012. Piwi‐interacting RNAs protect DNA against loss during Oxytricha genome rearrangement. Cell 151: 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bracht, J.R. et al 2013. Genomes on the edge: programmed genome instability in ciliates. Cell 152: 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zahler, A.M. et al 2012. Mating of the stichotrichous ciliate Oxytricha trifallax induces production of a class of 27 nt small RNAs derived from the parental macronucleus. PLoS One 7: e42371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Allen, S.E. & Nowacki M.. 2017. Necessity is the mother of invention: ciliates, transposons, and transgenerational inheritance. Trends Genet. 33: 197–207. [DOI] [PubMed] [Google Scholar]
- 25. Arnaiz, O. et al 2012.. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 8: e1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen, X. et al 2014. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158: 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamilton, E.P. et al 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife 5: e19090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guérin, F. et al 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maurer‐Alcalá, X.X. , Knight R. & Katz L.A.. 2018. Exploration of the germline genome of the ciliate Chilodonella uncinata through Single‐cell omics (transcriptomics and genomics). MBio 9: e01836‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maurer‐Alcalá, X.X. et al 2018. Twisted tales: insights into genome diversity of ciliates using single‐cell omics. Genome Biol. Evol. 10: 1927–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klobutcher, L.A. & Herrick G.. 1997. Developmental genome reorganization in ciliated protozoa: the transposon link. Prog. Nucleic Acid Res. Mol. Biol. 5656: 1–62. [DOI] [PubMed] [Google Scholar]
- 32. Jaraczewski, J.W. , Frels J.S. & Jahn C.L.. 1994. Developmentally regulated, low abundance Tec element transcripts in Euplotes crassus—implications for DNA elimination and transposition. Nucleic Acids Res. 22: 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klobutcher, L.A. & Herrick G.. 1995. Consensus inverted terminal repeat sequence of Paramecium lESs: resemblance to termini of Tc1‐related and Euplotes Tec transposons. Nucleic Acids Res. 23: 2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parfrey, L.W. et al 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. USA 108: 13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baudry, C. et al 2009. PiggyMac, a domesticated PiggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia . Genes Dev. 23: 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheng, C.Y. et al 2010. A domesticated PiggyBac transposase plays key roles in heterochromatin dynamics and DNA cleavage during programmed DNA deletion in Tetrahymena thermophila . Mol. Biol. Cell 21: 1753–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubois, E. 2012. Transposon invasion of the Paramecium germline genome countered by a domesticated PiggyBac transposase and the NHEJ pathway. Int. J. Evol. Biol. 2012: 436196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng, C.Y. et al 2016. The PiggyBac transposon‐derived genes TPB1 and TPB6 mediate essential transposon‐like excision during the developmental rearrangement of key genes in Tetrahymena thermophila . Genes Dev. 30: 2724–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bischerour, J. et al 2018. Six domesticated PiggyBac transposases together carry out programmed DNA elimination in Paramecium. Elife 7: e37927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nowacki, M. et al 2009. A functional role for transposases in a large eukaryotic genome. Science 324: 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng, L. et al 2017.. A germline‐limited PiggyBac transposase gene is required for precise excision in Tetrahymena genome rearrangement. Nucleic Acids Res. 45: 9481–9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krupovic, M. et al Casposons: a new superfamily of self‐synthesizing DNA transposons at the origin of prokaryotic CRISPR‐Cas immunity. BMC Biol. 12: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jangam, D. , Feschotte C. & Betrán E.. 2017. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 33: 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prescott, D.M. & Greslin A.F.. 1992. Scrambled actin I gene in the micronucleus of Oxytricha nova . Dev. Genet. 13: 66–74. [DOI] [PubMed] [Google Scholar]
- 45. Zufall, R.A. & Katz L.A.. 2007. Micronuclear and macronuclear forms of β‐tubulin genes in the ciliate Chilodonella uncinata reveal insights into genome processing and protein evolution. J. Eukaryot. Microbiol. 54: 275–282. [DOI] [PubMed] [Google Scholar]
- 46. Katz, L.A. & Kovner A.M.. 2010. Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata . J. Exp. Zool. B Mol. Dev. Evol. 314: 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gao, F. , Song W. & Katz L.A.. 2014. Genome structure drives patterns of gene family evolution in ciliates, a case study using Chilodonella uncinata (Protista, Ciliophora, Phyllopharyngea). Evolution 68: 2287–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen, X. et al 2015. Combinatorial DNA rearrangement facilitates the origin of new genes in ciliates. Genome Biol. Evol. 7: 2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao, F. , Roy S.W. & Katz L.A.. 2015. Analyses of alternatively processed genes in ciliates provide insights into the origins of scrambled genomes and may provide a mechanism for speciation. MBio 6: e01998‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lindblad, K.A. et al 2017. Thousands of RNA‐cached copies of whole chromosomes are present in the ciliate Oxytricha during development. RNA 23: 1200–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sonneborn, T.M. 1975. The Paramecium aurelia complex of fourteen sibling species. Trans. Am. Microsc. Soc. 94: 155–178. [Google Scholar]
- 52. Brygoo, Y. 1977. Genetic analysis of mating‐type differentiation in Paramecium tetraurelia . Genetics 87: 633–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meyer, E. & Keller A.M.. 1996. A mendelian mutation affecting mating‐type determination also affects developmental genomic rearrangements in Paramecium tetraurelia . Genetics 143: 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Singh, D.P. et al 2014. Genome‐defence small RNAs exapted for epigenetic mating‐type inheritance. Nature 509: 447–452. [DOI] [PubMed] [Google Scholar]
- 55. Orias, E. , Singh D.P. & Meyer E.. 2017. Genetics and epigenetics of mating type determination in Paramecium and Tetrahymena. Annu. Rev. Microbiol. 71: 133–156. [DOI] [PubMed] [Google Scholar]
- 56. Swart, E.C. et al 2014. Genome‐wide analysis of genetic and epigenetic control of programmed DNA deletion. Nucleic Acids Res. 42: 8970–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Duret, L. et al 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res. 18: 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garnier, O. et al 2004. RNA‐mediated programming of developmental genome rearrangements in Paramecium tetraurelia . Mol. Cell. Biol. 24: 7370–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mochizuki, K. & Gorovsky M.A.. 2004. Conjugation‐specific small RNAs in Tetrahymena have predicted properties of scan (scn) RNAs involved in genome rearrangement. Genes Dev. 18: 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Malone, C.D. et al 2005. Germ Line transcripts are processed by a dicer‐like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila . Mol. Cell. Biol. 25: 9151–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Long, H. et al 2013. Accumulation of spontaneous mutations in the ciliate Tetrhaymena thermophila . Genetics 195: 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raikov, I.B. 1982. The Protozoan Nucleus—Morphology and Evolution Springer. [Google Scholar]
- 63. Orias, E. 1991. Evolution of amitosis of the ciliate macronucleus: gain of the capacity to divide. J. Protozool. 38: 217–221. [DOI] [PubMed] [Google Scholar]
- 64. Katz, L.A. 2001. Evolution of nuclear dualism in ciliates: a reanalysis in light of recent molecular data. Int. J. Syst. Evol. Microbiol. 51: 1587–1592. [DOI] [PubMed] [Google Scholar]
- 65. Yan, Y. et al 2017. Unusual features of non‐dividing somatic macronuclei in the ciliate class Karyorelictea. Eur. J. Protistol. 61: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nowacki, M. et al 2010. RNA‐mediated epigenetic regulation of DNA copy number. Proc. Natl. Acad. Sci. USA 107: 22140–22144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bellec, L. & Katz L.A.. 2012. Analyses of chromosome copy number and expression level of four genes in the ciliate Chilodonella uncinata reveal a complex pattern that suggests epigenetic regulation. Gene 504: 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu, K. et al 2012. Copy number variations of 11 macronuclear chromosomes and their gene expression in Oxytricha trifallax . Gene 505: 75–80. [DOI] [PubMed] [Google Scholar]
- 69. Huang, J. & Katz L.A.. 2014. Nanochromosome copy number does not correlate with RNA levels though patterns are conserved between strains of the ciliate morphospecies Chilodonella uncinata . Protist 165: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Eisen, J.A. et al 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 4: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Katz, L.A. et al 2004. Dramatic diversity of ciliate histone H4 genes revealed by comparisons of patterns of substitutions and paralog divergences among eukaryotes. Mol. Biol. Evol. 21: 555–562. [DOI] [PubMed] [Google Scholar]
- 72. Zufall, R.A. et al Genome architecture drives protein evolution in ciliates. Mol. Biol. Evol. 23: 1681–1687. [DOI] [PubMed] [Google Scholar]