

Abstract
Regulatory guidelines describe the use of estimands in designing and conducting clinical trials. Estimands ensure alignment of the objectives with the design, conduct and analysis of a trial. An estimand is defined by four inter‐related attributes: the population of interest, the variable (endpoint) of interest, the way intercurrent events are handled and the population level summary. A trial may employ multiple estimands to evaluate treatment effects from different perspectives in order to address different scientific questions. As estimands may be an unfamiliar concept for many clinicians treating diabetes, this paper reviews the estimand concept and uses the PIONEER 1 phase 3a clinical trial, which investigated the efficacy and safety of oral semaglutide vs placebo, as an example of the way in which estimands can be implemented and interpreted. In the PIONEER 1 trial, two estimands were employed for each efficacy endpoint and were labelled as: (a) the treatment policy estimand, used to assess the treatment effect regardless of use of rescue medication or discontinuation of trial product, and provides a broad perspective of the treatment effect in the population of patients with type 2 diabetes in clinical practice; and (b) the trial product estimand, used to assess the treatment effect if all patients had continued to use trial product for the planned duration of the trial without rescue medication, thereby providing information on the anticipated treatment effect of the medication. Both approaches are complementary to understanding the effect of the studied treatments.
Keywords: Diabetes, estimand, GLP‐1 receptor agonists, oral semaglutide, PIONEER, regulatory guidance
1. INTRODUCTION
A key objective of many randomized clinical trials is quantification of the treatment effect of an intervention, such as a device or medication, compared with a control such as placebo or an active treatment, to inform clinical and regulatory decision making. Various types of treatment effects can be defined, and different stakeholders may have conflicting views on the relevance and applicability of the treatment effects described. Whenever results from clinical trials are published, it is crucial to accompany the results with a precise explanation of the way in which data that may have been impacted by intercurrent events, such as use of rescue medication or trial product discontinuation, are accounted for, and thus the way in which treatment effects have been estimated.
In 2008, the US Food and Drug Administration (FDA) issued a draft guideline that recommended the use of a last‐observation carried forward (LOCF) approach, in which missing data are replaced with the last observed value for a patient who discontinued the trial or treatment.1 The LOCF approach implicitly assumes that a patient with good short‐term disease control who prematurely discontinues the trial or trial product would also have similarly good disease control in the longer term.2 As this assumption is debatable in many settings, and because the LOCF approach may result in bias in favour of the tested therapy,3 a 2010 National Research Council (NRC) report, commissioned by the FDA, subsequently recommended against use of LOCF as a primary approach to handle missing data unless scientifically justified.2 Other methods to handle missing data have since been adopted, including multiple imputation approaches and the mixed model for repeated measures (MMRM), which are employed according to the type of treatment effect to be estimated.4
In the past, the type of treatment effect to be estimated was often insufficiently described in trial protocols or publications, which could have led to ambiguity and difficulties in interpreting data or comparing results from other trials. Because of a need for greater clarity and transparency, the International Council for Harmonisation (ICH) Steering Committee endorsed the ICH E9 (R1) concept paper in 2014 concerning this topic5 and a draft addendum was made available for public consultation in 20176 by various regulatory agencies, including the European Medicines Agency (EMA)7 and the FDA.8 This draft addendum presents a “structured framework to link trial objectives to a suitable trial design and tools for estimation and hypothesis testing and introduces the concept of an estimand, translating the trial objective into a precise definition of the treatment effect that is to be estimated.”6 The estimand concept is not new but, until recently, estimands have not been explicitly defined in clinical trial protocols or in publications.
The PIONEER programme is a global, clinical development programme for a novel oral formulation of the glucagon‐like peptide‐1 (GLP‐1) analog, semaglutide. Trials in the PIONEER programme, including PIONEER 19 and PIONEER 3,10 are some of the first clinical trials in type 2 diabetes to introduce estimands into trial planning, trial conduct, data analysis and interpretation of results. The decision to adopt the estimand concept in the PIONEER programme was made after the concept paper1 was released and following interactions with regulatory authorities, but before the draft ICH E9 addendum6 was available for public consultation. In this review, we will discuss estimands and their incorporation in the PIONEER development programme. The PIONEER 1 trial will be used as an example, to provide an understanding of the estimands implemented in the PIONEER programme and to clarify the considerations for clinical interpretation. Although future trials may involve different scientific questions and, consequently, may use different estimands than those used in the PIONEER 1 trial, an understanding of the estimand concept will help readers in designing and interpreting future trials.
2. THE ESTIMAND CONCEPT: DEFINING THE TARGET OF ESTIMATION
An estimand is a detailed description of the type of treatment effect that is to be estimated in order to address the scientific question of interest. It is important to emphasize that “estimand” is not a statistical term. Together with the structured framework, rigorous definition of the estimands used provides transparency and alignment of the trial objective, design, conduct and analysis, and thereby ensures that the planned treatment effect is actually the treatment effect that is eventually estimated. In a clinical trial, more than one estimand can be included for the same endpoint in the protocol.
An estimand is defined by four inter‐related attributes:
population of interest, that is, the population targeted by the scientific question of interest, for example, adult patients with type 2 diabetes;
variable (endpoint) of interest, for example, change from baseline in glycated haemoglobin (HbA1c) at Week 26;
the way to handle intercurrent events, for example, the way to account for use of rescue medication or premature trial product discontinuation in addressing the scientific question of interest;
population level summary, for example, the mean difference between treatment groups.
Previously, attributes i, ii and iv were clearly defined in clinical trial protocols. However, attribute iii, intercurrent events, described as “Events that occur after treatment initiation and either preclude observation of the variable or affect its interpretation,”6 was often not explicitly defined. Rather, intercurrent events were defined implicitly by specification of the statistical analyses.11 Thus, the choices for collection of data and the statistical analyses indicated the scientific question of interest rather than using the scientific question to guide decisions concerning data collection and statistical analyses. The ICH E9 (R1) draft addendum emphasizes the importance of utilizing the framework in a sequential manner, such that the estimand determines the method of estimation, that is, the analysis.6
Intercurrent events may “include the use of an alternative treatment (e.g., a rescue medication, a medication prohibited by the protocol or a subsequent line of therapy), discontinuation of treatment, treatment switching and terminal events, […] such as death.”6 For example, during a diabetes trial, patients may be allowed to receive another glucose‐lowering agent as rescue medication if they do not maintain acceptable glycaemic control during the course of the trial; however, the use of rescue medication may alter the treatment effect that is ultimately assessed. Considering change in HbA1c as the endpoint in a placebo‐controlled design, the use of rescue medication will probably occur more commonly in participants receiving placebo than in those receiving an active trial product. If measurements collected after initiation of rescue medication contribute to the estimated treatment effect, this would lead to a smaller estimated difference in HbA1c between treatment arms. Hence, the estimated treatment effect reflects a comparison of the effect of the active trial product plus the potential effect of rescue medication vs the effect of placebo plus the potential effect of rescue medication. The relevance of such a treatment effect depends on the scientific question posed.
Another common intercurrent event in diabetes trials is premature discontinuation of trial product, for example, due to adverse events. The relevance of including measurements collected after trial product discontinuation, again, depends on the question of interest. If the question of interest is to estimate the effect of the trial product in a scenario in which patients had continued to use trial product and did not use rescue medication, HbA1c measurements after a patient discontinued trial product are not relevant. However, if it is of interest to compare treatment policies, and to take into account both efficacy and tolerability, measurements after having discontinued trial product are relevant, as discontinuations may occur because of adverse events.
Different strategies can be used to account for intercurrent events, to construct an estimand that allows assessment of the treatment effect specified by the scientific question of interest. Five strategies are discussed in draft ICH E9 (R1). In brief: (a) the “treatment policy strategy", in which measurements of the variable of interest are used regardless of the occurrence of the intercurrent event; (b) the “hypothetical strategy", which envisages a scenario in which an intercurrent event, such as use of rescue medication or discontinuation of medication, would not have occurred; (c) the “composite strategy", in which an intercurrent event may be integrated into one or more other measures of a clinical outcome as the variable of interest; (d) the “principal stratum strategy", in which the target population might be taken as a subset of the broader population in which the intercurrent event would not occur; and (e) the “while‐on‐treatment strategy", in which the treatment effect achieved before occurrence of the intercurrent event is of interest. In addition to the five strategies discussed in the draft ICH E9 (R1) guideline, others may be relevant.6
The choice of estimand for a clinical trial, and the subsequent analyses to be performed, should be made with the scientific question of interest in mind. For example, it may be of interest to understand the treatment effect in patients if they had continued to use treatment for the duration of the trial, without rescue medication. Alternatively, it may be of interest to understand the overall treatment effect, taking into consideration tolerability and/or efficacy issues that may result in medication discontinuation or additional therapy. There are many different scientific questions of interest relevant to clinical trials and, therefore, different ways in which estimands can be utilized to address these questions.12, 13, 14, 15, 16, 17
The ICH E9 (R1) presents a general framework along with the estimand concept and does not recommend the use of specific estimands, as the relevance of an estimand depends on the clinical setting and scientific question of interest. In early 2018, the EMA published a draft guideline18 that specifically discusses the use of estimands in diabetes trials. The draft guideline recommends that the primary estimand for glycated haemoglobin should utilize the hypothetical strategy to account for the intercurrent event,“initiation of rescue medication” and to use the treatment policy strategy for the intercurrent event, “trial product discontinuation.” Hence, a treatment effect that is free from the impact of other glucose‐lowering agents, regardless of trial product discontinuation, is targeted. A supplementary suggestion in the EMA draft guideline is to use the composite strategy to account for the two intercurrent events through definition of the endpoint: the difference in proportion of patients who reached an absolute HbA1c value of <7.0% (<53 mmol/mol) at end‐of‐trial without the use of additional medication and who continued to use trial product for the duration of the trial.
It is important when planning a new trial to consider all relevant intercurrent events and to be able to justify the strategy chosen to account for these. For example, the treatment policy strategy, in which a treatment effect is assessed regardless of intercurrent events, will often tend to minimize the difference in treatment effect between groups. It should also be taken into consideration whether it is justifiable to account for the intercurrent event of premature trial product discontinuation because of drug‐related adverse events according to the hypothetical strategy, in which the estimated treatment effect assumes that patients continued using trial drug even if not tolerated.
A clear, precise description of the type of treatment effect (estimand) to be estimated in the trial will make it easier for clinical trialists, physicians and other stakeholders to understand and interpret results from clinical trials. Clinical trial sponsors are responsible for choosing and defining the estimands that best answer the questions of interest according to the disease and the population, and for working with decision makers and clinical stakeholders to ensure that the estimand(s) applied is(are) appropriate. A collaborative approach across different areas of expertise is required when defining estimands.
3. HOW ESTIMANDS WERE INCORPORATED INTO THE PIONEER 1 TRIAL
PIONEER 1 was a phase 3a, randomized, placebo‐controlled trial comparing once‐daily oral semaglutide 3, 7 and 14 mg with placebo in adult patients with type 2 diabetes who, at trial entry, were being treated only with advice concerning diet and exercise (NCT02906930).9 This trial, as others in the PIONEER programme (eg, PIONEER 3),10 employed two estimands, labelled “treatment policy estimand” and “trial product estimand,” to address two different scientific questions of interest and to provide information relevant to regulatory agencies and/or clinicians concerning oral semaglutide. The trial was designed to evaluate adult patients with type 2 diabetes who would be eligible for oral semaglutide in clinical practice, and to assess variables of interest relevant to a GLP‐1 analog (eg, HbA1c and body weight). For both estimands employed in the PIONEER programme, two intercurrent events were considered: initiation of rescue medication and premature trial product discontinuation. These events were handled differently according to the scientific question of interest (Figure 1).
Figure 1.
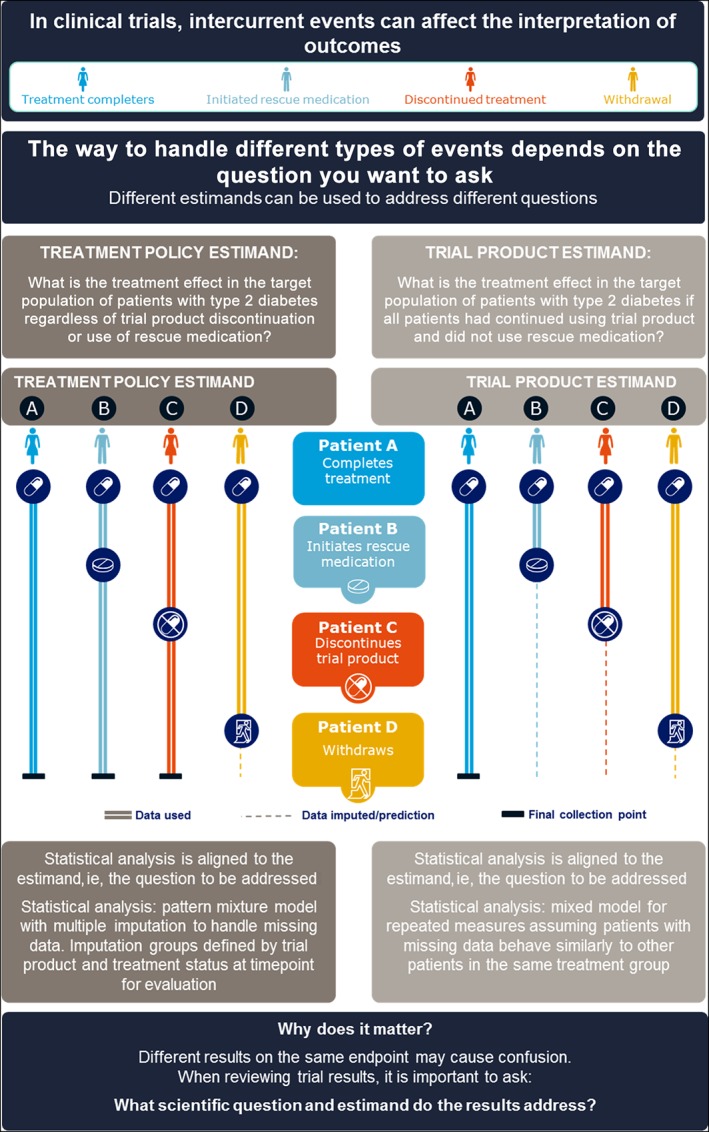
Illustration of estimands used in the PIONEER 1 trial. HbA1c, glycated haemoglobin
The treatment policy estimand aimed to answer the question: What is the treatment effect in the targeted population of patients with type 2 diabetes regardless of trial product discontinuation or use of rescue medication? The treatment policy estimand for the efficacy objectives in the PIONEER 1 trial was the primary estimand, and was defined as the mean difference between oral semaglutide and placebo in change from baseline to Week 26 in HbA1c and body weight in patients with type 2 diabetes, regardless of trial product discontinuation and/or addition of rescue medication or switch to another glucose‐lowering drug. It is noteworthy that, in the PIONEER 1 trial, rescue medication was recommended for participants with persistent and unacceptable hyperglycaemia, that is, confirmed fasting blood glucose greater than 240 mg/dL (13.3 mmol/L) during Weeks 8 to 13, or greater than 200 mg/dL (11.1 mmol/L) from Week 14 onwards.9 The two anticipated intercurrent events were both accounted for by the treatment policy strategy as described in the draft ICH E9 (R1) addendum.6 One implication for trial design and conduct when applying this estimand was that all patients were encouraged to continue participation in the trial and data were collected even after discontinuation of trial product. This contrasts with historical approaches in which data would not have been collected following trial product discontinuation.2 This estimand may, therefore, be of interest to both regulatory authorities and clinicians.
The trial product estimand aimed to answer the question: What is the treatment effect in the targeted population of patients with type 2 diabetes if all patients had continued to use trial product and did not use rescue medication? and this was the secondary estimand in the PIONEER 1 trial. The trial product estimand was defined as the mean difference between oral semaglutide and placebo in change from baseline to Week 26 in HbA1c and body weight in patients with type 2 diabetes if all patients had continued to use trial product for the entire planned duration of the trial and did not use rescue medication. Thus, both intercurrent events were accounted for by the hypothetical strategy as described in the draft ICH E9 (R1).6 The trial product estimand aims at targeting the effect if patients had continued to use treatment with oral semaglutide, compared with the effect if patients had continued to use placebo, without the confounding effects of rescue medication or any other changes in glucose‐lowering medication. The trial product estimand adds clinical value by aiming to provide information concerning the anticipated effect of trial product.
4. STATISTICAL METHODS USED IN THE PIONEER 1 TRIAL
In the PIONEER 1 trial, confirmation of the efficacy of oral semaglutide on change in HbA1c and in body weight from baseline to Week 26 was based on a weighted Bonferroni closed‐testing strategy, to control the overall type 1 error for the hypotheses evaluated by the treatment policy estimand.9 The statistical analysis should be aligned to estimands of interest6 and the following pre‐specified analyses were used to estimate each of the estimands in the PIONEER 1 trial.
The treatment policy estimand was estimated by a pattern mixture model, using multiple imputation to handle missing data from Week 26 for both confirmatory endpoints (Figures 1 and 2). Data collected at Week 26 from all randomized patients, irrespective of premature discontinuation of trial product and/or initiation of rescue medication, were included in the statistical analysis. Imputation was undertaken within groups, defined by trial product and treatment status at Week 26. The assumption is that the behaviour of patients who discontinued trial product or initiated rescue medication, but for whom data were missing at the primary evaluation time point, is best described by patients with the same treatment status for whom data were available at the primary evaluation time point. The treatment policy estimand used in the PIONEER 1 trial may provide a broad perspective of the treatment effect and the statistical approach relies on fewer assumptions than other statistical approaches.6 Both imputation and analysis were based on analysis of covariance models. Results were combined by use of Rubin's rule.19 Recent US prescribing information for the subcutaneous GLP‐1 receptor agonist semaglutide20 reported results with an approach similar to that used in the PIONEER 1 trial to estimate the treatment policy estimand.
Figure 2.
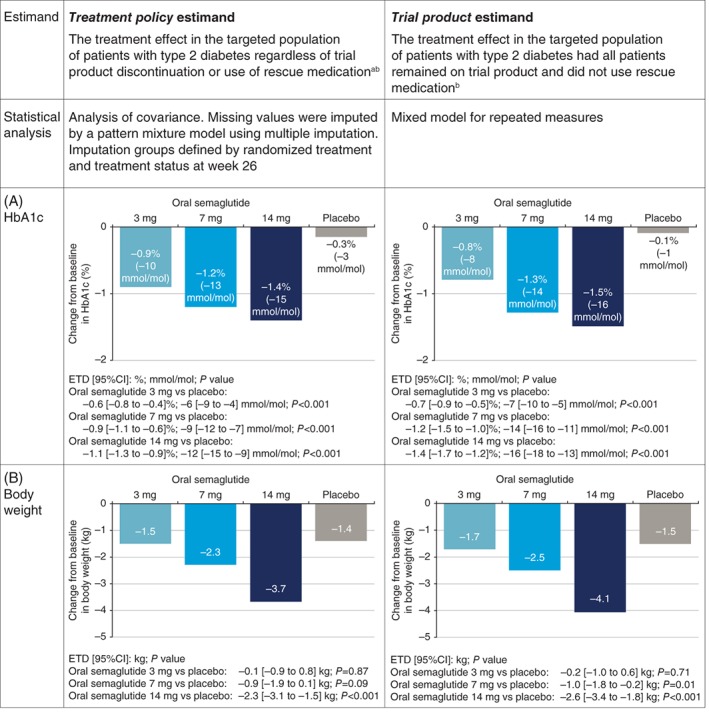
Estimand description and results from the PIONEER 1 trial. Change from baseline in A, HbA1c and B, body weight for the treatment policy estimand and the trial product estimand at Week 26.9 ETDs [95% CI] are shown. American Diabetes Association PIONEER 1: Randomized Clinical Trial Comparing the Efficacy and Safety of Oral Semaglutide Monotherapy with Placebo in Patients with Type 2 Diabetes, American Diabetes Association, 2019. Copyright and all rights reserved. Material from this publication has been used with the permission of American Diabetes Association. CI, confidence interval; ETD, estimated treatment difference; HbA1c, glycated haemoglobin. aRescue medication criteria: confirmed fasting blood glucose greater than 240 mg/dL (13.3 mmol/L) from week 8 to 13, or greater than 200 mg/dL (11.1 mmol/L) from week 14 onwards. bIn PIONEER 1, trial product discontinuation rates were 2.3% to 7.4% with oral semaglutide and 2.2% with placebo
The trial product estimand was estimated using an MMRM that incorporated data from all randomized patients, collected prior to premature trial product discontinuation or initiation of rescue medication (Figures 1 and 2). The independent effects included in the model were treatment and region as categorical fixed effects and baseline value as a covariate, all nested within visit. An unstructured covariance matrix for endpoint measurements within the same patient was employed.9 The MMRM is based on the assumption that data are missing at random, meaning that patients for whom data were missing would be considered to behave similarly to other patients in the same treatment group. The trial product estimand aims to provide information concerning the anticipated effect of trial product, but should not be considered equivalent to the per‐protocol or complete case analysis. The MMRM analysis used to estimate the trial product estimand differs from a per‐protocol or complete‐case analysis because it includes data from all randomized patients, rather than a subset of the randomized patients. Recent EU prescribing information for the subcutaneous GLP‐1 receptor agonist semaglutide21 reported results with an approach similar to that used in the PIONEER 1 trial to estimate the trial product estimand.
5. INTERPRETING RESULTS FROM ESTIMANDS INCORPORATED INTO THE PIONEER 1 TRIAL
In the PIONEER 1 trial, all dose levels of oral semaglutide were superior to placebo in reducing HbA1c and superior reductions in body weight were observed for the 14 mg dose compared with placebo (Figure 2). In the PIONEER 3 trial, compared with sitagliptin, the 7 mg and 14 mg doses of oral semaglutide resulted in superior reductions in HbA1c and body weight.10 While the two estimands used in the PIONEER 1 and PIONEER 3 trials addressed two different scientific questions of interest, both contribute to the full clinical picture. As outlined earlier, the results for both estimands are dependent on the frequency of intercurrent events and will be similar if these are very low. The PIONEER 1 trial had a very high completion rate (92% to 97%), and 85% to 87% of patients across the four treatment arms completed the trial and continued with trial product without use of rescue medication,9 thus providing a high degree of concordance between the results of the two estimands. The frequency and timing of intercurrent events during the PIONEER 1 trial is illustrated in Figure 3.
Figure 3.
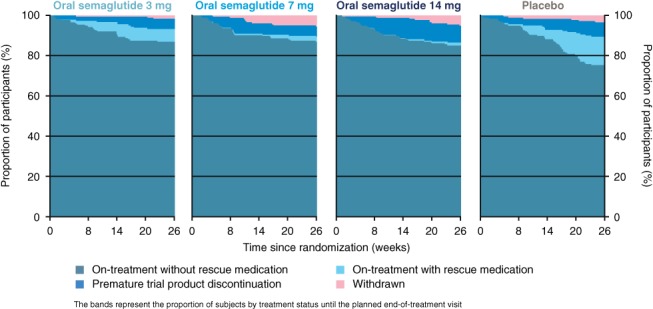
Frequency and timings of intercurrent events in the PIONEER 1 trial9
It is of interest to note the way in which handling of intercurrent events is reflected in the reported results. With the highest dose of oral semaglutide tested (14 mg), the estimated treatment difference in HbA1c, compared with placebo, as assessed by the trial product estimand (that is, the treatment effect if rescue medication had not been initiated and all patients remained on the trial product), was greater by 0.3% points than the estimated treatment effect according to the treatment policy estimand (that is, the treatment effect regardless of discontinuation of trial product or use of rescue medication) (Figure 2). This was primarily the result of the greater reduction in HbA1c with placebo for the treatment policy estimand, which is probably a reflection of the increased use of rescue medication in the placebo group (14%) vs the oral semaglutide 14 mg group (1.1%).9 Discontinuation of trial product occurred more frequently with oral semaglutide 14 mg compared with placebo (Figure 3) and, as the majority did not switch to another glucose‐lowering agent or may have switched to a less effective glucose‐lowering agent, the inclusion of data after trial product discontinuation for the treatment policy estimand could also have contributed to the smaller treatment difference observed between oral semaglutide 14 mg and placebo. Likewise, with oral semaglutide 14 mg, the estimated treatment difference vs placebo for body weight was greater by 0.3 kg for the trial product estimand than for the treatment policy estimand (Figure 2), which may be because semaglutide has been shown to markedly reduce body weight compared with many other glucose‐lowering agents22, 23, 24 and, consequently, patients who discontinued trial product prematurely would not be expected to experience the same weight loss as those continuing the trial product.
6. SUMMARY
The ICH E9 (R1) draft addendum provides a general framework to ensure alignment of trial planning, trial design, trial conduct, data analysis and interpretation of results. Clearly defining the estimand(s) for a trial provides greater clarity with respect to the type of treatment effect being estimated. Defining an estimand is a multidisciplinary task and the relevance of a specific estimand depends on the clinical setting. The ICH E9 (R1) draft addendum does not recommend the use of specific estimands, but rather, introduces the general framework and the concept of estimands.
Including more than one estimand allows evaluation of the treatment effect from different perspectives. Clinicians may find the treatment effect determined by the trial product estimand to be of interest when assessing and comparing effects of different therapeutic choices, as this targets the expected treatment effect assuming that a patient continues to use trial product without the need for rescue medication. Complementing the understanding of the trial product treatment effect is the overall effect, that is, the efficacy and tolerability of a therapeutic choice or pathway. Hence, the treatment policy estimand, which accounts for the addition of rescue medication, as well as discontinuation and/or switch of medication because of tolerability concerns, may provide a broader perspective concerning treatment effect. The approaches are complementary in understanding the full treatment effect of medication within different scenarios.
As seen in the PIONEER trial programme, estimands are now being incorporated in type 2 diabetes clinical trials. Greater familiarity with the concept of estimands and the reasons why estimands have been introduced, along with an appreciation of the clarity provided by use of estimands, will help clinicians who treat diabetes in interpreting and comparing trial results and in making informed clinical decisions.
CONFLICT OF INTEREST
V. R. A. has served as a consultant for Adocia, AstraZeneca, BD, Novo Nordisk, Sanofi and Zafgen; has received research grant support to her employing institution from AstraZeneca/BMS, Calibra, Eisai, Janssen, Novo Nordisk, Sanofi and Theracos; and declares that her spouse is an employee of Merck.
J. B. has received contracted consulting fees that are paid to the University of North Carolina from Adocia, AstraZeneca, Dance Biopharm, Dexcom, Elcelyx Therapeutics, Eli Lilly, Fractyl, GI Dynamics, Intarcia Therapeutics, Lexicon, MannKind, Metavention, NovaTarg, Novo Nordisk, Orexigen, PhaseBio, Sanofi, Senseonics, Shenzhen HighTide, Takeda, vTv Therapeutics and Zafgen; has received grant support from AstraZeneca, Eli Lilly, GI Dynamics, GlaxoSmithKline, Intarcia Therapeutics, Johnson & Johnson, Lexicon, Medtronic, Novo Nordisk, Orexigen, Sanofi, Scion NeuroStim, Takeda, Theracos and vTv Therapeutics; has served as a consultant to Neurimmune AG; holds stock options in Mellitus Health, PhaseBio and Stability Health; and is supported by a grant from the National Institutes of Health (UL1TR002489).
M. J. D. has served as a consultant, advisory board member and speaker for Novo Nordisk, Sanofi‐Aventis and Lilly; has served as an advisory board member for Servier and Janssen; has served as a speaker for Boehringer Ingelheim and Takeda Pharmaceuticals International Inc; has received grants in support of the investigator and investigator‐initiated trials from Novo Nordisk, Sanofi‐Aventis, Lilly, Boehringer Ingelheim and Janssen.
T. S., M. D. and J. Z. are employees of Novo Nordisk A/S. T. S. and M. D. also hold shares in Novo Nordisk A/S.
AUTHOR CONTRIBUTIONS
The article was conceived by V. R. A., T. S. and M. D., V. R. A and T. S. drafted the article with J. B. B., M. D., J. Z. and M. J. D. critically revising the article. All authors approved the final version.
ACKNOWLEDGMENTS
The authors would like to thank Helle Lynggaard (Novo Nordisk A/S) for her contribution in developing the manuscript and Christin Løth Hertz and Helle Lynggaard (Novo Nordisk A/S) for their contribution to the development of Figure 1. The authors would also like to thank Brian Bekker Hansen (Novo Nordisk A/S) for reviewing the manuscript and Debbie Day (Spirit Medical Communications Group) for medical writing and editorial assistance.
Aroda VR, Saugstrup T, Buse JB, Donsmark M, Zacho J, Davies MJ. Incorporating and interpreting regulatory guidance on estimands in diabetes clinical trials: The PIONEER 1 randomized clinical trial as an example. Diabetes Obes Metab. 2019;21:2203–2210. 10.1111/dom.13804
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/dom.13804.
Funding information The PIONEER 1 trial and the medical writing/editorial support for this manuscript were funded by Novo Nordisk A/S
REFERENCES
- 1. US Food and Drug Administration . Guidance for industry. Diabetes mellitus: developing drugs and therapeutic biologics for treatment and prevention, 2008. https://wwwfdagov/downloads/Drugs//Guidances/ucm071624pdf. Accessed April 2, 2019.
- 2. National Research Council . The Prevention and Treatment of Missing Data in Clinical Trials. Panel on Handling Missing Data in Clinical Trials. Committee on National Statistics, Division of Behavioral and Social Sciences and Education. Washington, DC: The National Academies Press; 2010. https://www.ncbi.nlm.nih.gov/books/NBK209904/. Accessed June 13, 2019. [Google Scholar]
- 3. Holzhauer B, Akacha M, Bermann G. Choice of estimands and analysis methods in diabetes trials with rescue medication. Pharm Stat. 2015;14:433‐447. [DOI] [PubMed] [Google Scholar]
- 4. Mehrotra DV, Liu F, Permutt T. Missing data in clinical trials: control‐based mean imputation and sensitivity analysis. Pharm Stat. 2017;16:378‐392. [DOI] [PubMed] [Google Scholar]
- 5. International Council for Harmonisation (ICH) of Technical Requirements for Pharmaceuticals for Human Use . ICH Concept Paper E9 (R1): Addendum to Statistical Principles for Clinical Trials on Choosing Appropriate Estimands and Defining Sensitivity Analyses in Clinical Trials, 2014. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/E9__R1__Final_Concept_Paper_October_23_2014.pdf. Accessed December 17, 2018.
- 6. International Council for Harmonisation (ICH) of Technical Requirements for Pharmaceuticals for Human Use . ICH Harmonised Guideline E9 (R1): Estimands and Sensitivity Analysis in Clinical Trials, 2017. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/E9-R1EWG_Step2_Guideline_2017_0616.pdf. Accessed December 17, 2018.
- 7. European Medicines Agency . ICH E9 (R1) draft addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials, 2017. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/08/WC500233916.pdf. Accessed December 17, 2018.
- 8. US Food and Drug Administration . ICH E9 (R1) statistical principles for clinical trials: addendum: estimands and sensitivity analysis in clinical trials, 2017. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM582738.pdf. Accessed December 17, 2018.
- 9. Aroda VR, Rosenstock J, Terauchi Y, et al. PIONEER 1: randomized clinical trial comparing the efficacy and safety of oral semaglutide monotherapy with placebo in patients with type 2 diabetes. Diabetes Care. 2019. 10.2337/dc19-0749. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 10. Rosenstock J, Allison D, Birkenfeld AL, et al. Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: the PIONEER 3 randomized clinical trial. JAMA. 2019;321:1466‐1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akacha M, Bretz F, Ohlssen D, Rosenkranz G, Schmidli H. Estimands and their role in clinical trials. Stat Biopharm Res. 2017;9:268‐271. [Google Scholar]
- 12. Akacha M, Kothny W. Estimands: a more strategic approach to study design and analysis. Clin Pharmacol Ther. 2017;102:894‐896. [DOI] [PubMed] [Google Scholar]
- 13. LaVange LM, Permutt T. A regulatory perspective on missing data in the aftermath of the NRC report. Stat Med. 2016;35:2853‐2864. [DOI] [PubMed] [Google Scholar]
- 14. Mallinckrodt CH, Lin Q, Lipkovich I, Molenberghs G. A structured approach to choosing estimands and estimators in longitudinal clinical trials. Pharm Stat. 2012;11:456‐461. [DOI] [PubMed] [Google Scholar]
- 15. Mallinckrodt C, Molenberghs G, Rathmann S. Choosing estimands in clinical trials with missing data. Pharm Stat. 2017;16:29‐36. [DOI] [PubMed] [Google Scholar]
- 16. Permutt T. A taxonomy of estimands for regulatory clinical trials with discontinuations. Stat Med. 2016;35:2865‐2875. [DOI] [PubMed] [Google Scholar]
- 17. Mehrotra DV, Hemmings RJ, Russek‐Cohen E. ICH E9/R1 Expert Working Group. Seeking harmony: estimands and sensitivity analyses for confirmatory clinical trials. Clin Trials. 2016;13:456‐458. [DOI] [PubMed] [Google Scholar]
- 18. European Medicines Agency . Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus, 2018. https://www.ema.europa.eu/documents/scientific-guideline/draft-guideline-clinical-investigation-medicinal-products-treatment-prevention-diabetes-mellitus_en.pdf. Accessed March 25, 2019.
- 19. Little RJA, Rubin DB. Statistical Analysis with Missing Data. New York, NY: John Wiley & Sons; 1987. [Google Scholar]
- 20. Novo Nordisk . OZEMPIC® (semaglutide injection) Prescribing Information. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209637lbl.pdf. Accessed December 17, 2018. [Google Scholar]
- 21. Novo Nordisk . OZEMPIC (semaglutide injection) SmPC. https://www.ema.europa.eu/en/documents/product-information/ozempic-epar-product-information_en.pdf. Accessed April 18, 2019.
- 22. Aroda VR, Bain SC, Cariou B, et al. Efficacy and safety of once‐weekly semaglutide versus once‐daily insulin glargine as add‐on to metformin (with or without sulfonylureas) in insulin‐naïve patients with type 2 diabetes (SUSTAIN 4): a randomised, open‐label, parallel‐group, multicentre, multinational, phase 3a trial. Lancet Diabetes Endocrinol. 2017;5:355‐366. [DOI] [PubMed] [Google Scholar]
- 23. Aroda VR, Ahmann A, Cariou B, et al. Comparative efficacy, safety, and cardiovascular outcomes with once‐weekly subcutaneous semaglutide in the treatment of type 2 diabetes: insights from the SUSTAIN 1‐7 trials. Diabetes Metab. 2019, pii: S1262‐3636(18)30222‐2. 10.1016/j.diabet.2018.12.001. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 24. Mishriky BM, Cummings DM, Powell JR, et al. Comparing once‐weekly semaglutide to incretin‐based therapies with type 2 diabetes: a systematic review and meta‐analysis. Diabetes Metab. 2018;45:102‐109. [DOI] [PubMed] [Google Scholar]