

Abstract
Regulation of gene transcription via the cAMP-mediated second messenger pathway has been implicated in the actions of antidepressant drugs, but studies to date have not demonstrated such an effectin vivo. To directly study the regulation of cAMP response element (CRE)-mediated gene transcription by antidepressants, transgenic mice with a CRE–LacZ reporter gene construct were administered one of three different classes of antidepressants: a norepinephrine selective reuptake inhibitor (desipramine), a serotonin selective reuptake inhibitor (fluoxetine), or a monoamine oxidase inhibitor (tranylcypromine). Chronic, but not acute, administration of these antidepressants significantly increased CRE-mediated gene transcription, as well as the phosphorylation of CRE binding protein (CREB), in several limbic brain regions thought to mediate the action of antidepressants, including the cerebral cortex, hippocampus, amygdala, and hypothalamus. These results demonstrate that chronic antidepressant treatment induces CRE-mediated gene expression in a neuroanatomically differentiated pattern and further elucidate the molecular mechanisms underlying the actions of these widely used therapeutic agents.
Keywords: CRE enhancer, β-galactosidase, gene transcription, phosphorylation, fluoxetine, desipramine, tranylcypromine
Although the acute action of antidepressant treatment is known to be mediated by blockade of the reuptake or breakdown of serotonin (5-HT) and norepinephrine (NE), the molecular adaptations underlying the therapeutic action of these agents has not been determined. One signal transduction pathway that has been implicated in the action of antidepressant treatment is the cAMP second messenger cascade. Recent studies have demonstrated that different classes of antidepressants upregulate the cAMP system at several sites, including increased Gs activation of adenylyl cyclase (Ozawa and Rasenick, 1991) and upregulation of cAMP-dependent protein kinase (PKA) (Nestler et al., 1989; Perez et al., 1989). In addition, inhibition of cAMP metabolism produces antidepressant-like effects in behavioral models of depression (Wachtel, 1983; Griebel et al., 1991;O'Donnell, 1993) as well as therapeutic responses in depressed patients (Horowski and Sastre-Y-Hernandez, 1985; Bobon et al., 1988; Fleischhacker et al., 1992; Malison et al., 1997).
One potential target of the cAMP system that could mediate the action of antidepressants is the cAMP response element binding protein (CREB) (Montminy, 1990; Meyer and Habener, 1993). CREB is a transcription factor that mediates the actions of cAMP on gene expression and could thereby underlie some of the long-term effects of antidepressant treatment. A role for CREB in the action of antidepressants is supported by studies demonstrating that chronic antidepressant treatment increases the expression of CREB in limbic regions of rat brain (Nibuya et al., 1996). CREB regulates gene transcription by binding to a cAMP response element (CRE), acis-acting enhancer element in the regulatory region of various genes. The function of CREB is regulated largely by its state of phosphorylation at Ser133, which results in activation of gene transcription (Montminy, 1990;Meyer and Habener, 1993). Phosphorylation of CREB at Ser133 can occur via activation of the cAMP cascade and PKA, but also via activation of calcium-dependent protein kinases (i.e., protein kinase C and calcium/calmodulin-dependent protein kinase) (Duman et al., 1997,1999). This raises the possibility that CREB could act as a common downstream target of different classes of antidepressants that influence 5-HT and/or NE (Duman et al., 1997, 1999).
The focus of the present study is to determine the influence of antidepressant treatment on the function of CREB. This is a critical issue because although CREB expression is upregulated by antidepressant treatment, the function of this transcription factor could be unchanged without a corresponding increase in phosphorylation. To address this issue, the influence of antidepressant administration on CRE-mediated gene expression and CREB phosphorylation were examined in CRE–LacZ transgenic mice. The transgene in these mice is a CRE–LacZ reporter gene construct, and stimulation of the CRE site leads to increased expression of the LacZ gene product, β-galactosidase. These mice have been used to establish the role of CRE-mediated gene expression in cellular and behavioral models of learning and memory and in circadian rhythm (Impey et al., 1996, 1998; Obrietan et al., 1998; Pham et al., 1999).
MATERIALS AND METHODS
CRE–LacZ transgenic mice. All experiments were conducted in mice heterozygous for the CRE–LacZ reporter gene construct. This construct consists of six tandem CREs upstream of a minimal HSV promoter driving the expression of β-galactosidase (Meinkoth et al., 1990). The mice were generated using C57BL6/SJL F2 blastocytes for microinjection; founders were bred to C57BL6 mice (Impey et al., 1996). The animals were bred and maintained under standard conditions (12 hr light/dark cycle, food and water ad libitum). PCR was used to determine the genotype of individual mice. All animal use procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Yale Animal Care and Use Committee.
Drug administration paradigms. For chronic paradigms, fluoxetine (10 mg/kg, i.p.), desipramine (15 mg/kg, i.p.), or tranylcypromine (10 mg/kg, i.p.) was administered daily for 14 d. For acute paradigms, mice received 0.9% saline (i.p.) daily for 13 d and a single injection of tranylcypromine, desipramine, or fluoxetine (i.p.) on day 14 (same dose as above). Control animals were administered 0.9% saline (i.p.) for 14 d. Haloperidol (1 mg/kg, i.p.) or cocaine (10 mg/kg, i.p.) were administered daily for 14 d.
β-Galactosidase staining. Mice were killed 6 hr after the last injection. After perfusion with 0.9% NaCl solution and 4% paraformaldehyde, the brains were post-fixed overnight and cryoprotected in 20% glycerol. Fluorescence immunohistochemistry was performed using standard protocols. Briefly, 40 μm sections were incubated with rabbit anti β-galactosidase antibody at 1:1000 dilution (ICN Biochemicals, Costa Mesa, CA) and Alexa 594 goat anti-rabbit IgG at 3 μg/ml (Molecular Probes, Eugene, OR). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (see below). Images were captured on a Zeiss Axioskop fluorescence microscope.
Phosphorylation of CREB. After blocking with BSA and goat serum, free-floating sections were incubated overnight with anti-phosphorylated CREB (phospho-CREB) antibody from rabbit (New England Biolabs, Beverly, MA) at 1:250 dilution. A second overnight incubation was performed with 2.5 μg/ml fluorescin-labeled anti-rabbit IgG antibody (Jackson ImmunoResearch, West Grove, PA), and a third was performed with 4 μg/ml Alexa 488-antifluorescin antibody (Molecular Probes). This method involving three antibodies results in a considerable enhancement of the fluorescence signal. Between each incubation step with antibody, stringent washing steps with PBS containing NaF and Triton X-100 were performed.
Counterstaining. Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA) was used for counterstaining. The DAPI fluorophore binds to cellular DNA, allowing for fluorescence visualization of the cells. DAPI produces a blue fluorescence with excitation at 360 nm and emission at 460 nm when bound to DNA. There is no emission overlap with fluorescin, rhodamine, Texas Red, or the fluorophores used for β-galactosidase or phospho-CREB immunolabeling.
Data analysis. Images were evaluated using a four point score system (0 = minimal, 1 = weak, 2 = intermediate, 3 = strong immunoreactivity) by two independent investigators blinded to the treatment condition. The Kruskal–Wallis test was performed for statistical analysis. The level of significance wasp < 0.05. A trend or tendency was assumed byp < 0.10. Similar results were obtained when quantified by densitometry of digitalized images. For this approach, the images were converted into gray-scale pictures. The appropriate areas were outlined, and staining intensity was determined using IPLab Spectrum–Scientific Image Processing, Version 3.1.2 (Scanalytics, Inc.).
RESULTS
Chronic antidepressant administration increases CRE-mediated gene expression
To measure CRE-mediated gene expression in the CRE–LacZ transgenic mice, levels of the LacZ gene product, β-galactosidase, were determined by fluorescence immunohistochemistry. Relatively low levels of β-galactosidase were observed in most brain regions in saline-treated mice, particularly the dentate gyrus of hippocampus and cerebral cortex (Figs. 1,2). Relatively higher levels of immunoreactivity were observed in amygdala and hypothalamus (Figs. 1,2).
Fig. 1.
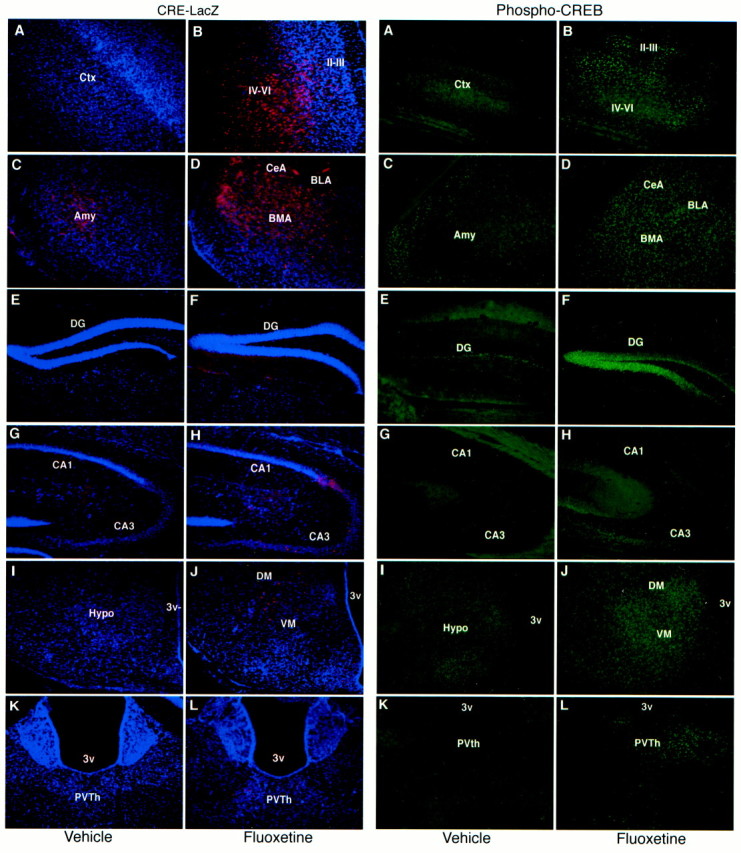
Chronic fluoxetine administration increases CRE-mediated gene expression and CREB phosphorylation in brain. Mice were administered saline or fluoxetine once daily for 14 d, and the brains were processed for β-galactosidase (CRE–LacZ) or phospho-CREB immunohistochemistry 6 hr after the last treatment. Representative images of β-galactosidase (left/red,A–L) or phospho-CREB (right/green,A–L) are shown. The brain regions that were examined include cerebral cortex (A,B), amygdala (C, D), dentate gyrus granule cell layer (E,F), CA3 pyramidal cell layer (G,H), hypothalamus (I,J), and thalamus (K,L). Images from saline-treated (A,C, E, G, I,K) and fluoxetine-treated (B,D, F, H, J,L) animals are shown. The subregions that are labeled include layers II–III and IV–VI of cerebral cortex; central (CeA), basolateral (BLA), and basomedial (BMA) nuclei of the amygdala; dentate gyrus (DG) granule cell layer, and CA3 and CA1 pyramidal cell layers of hippocampus; dorsomedial (DM) and ventromedial (VM) nuclei of the hypothalamus; paraventricular nucleus of the thalamus (PVTh); and third ventricle (3v).
Fig. 2.
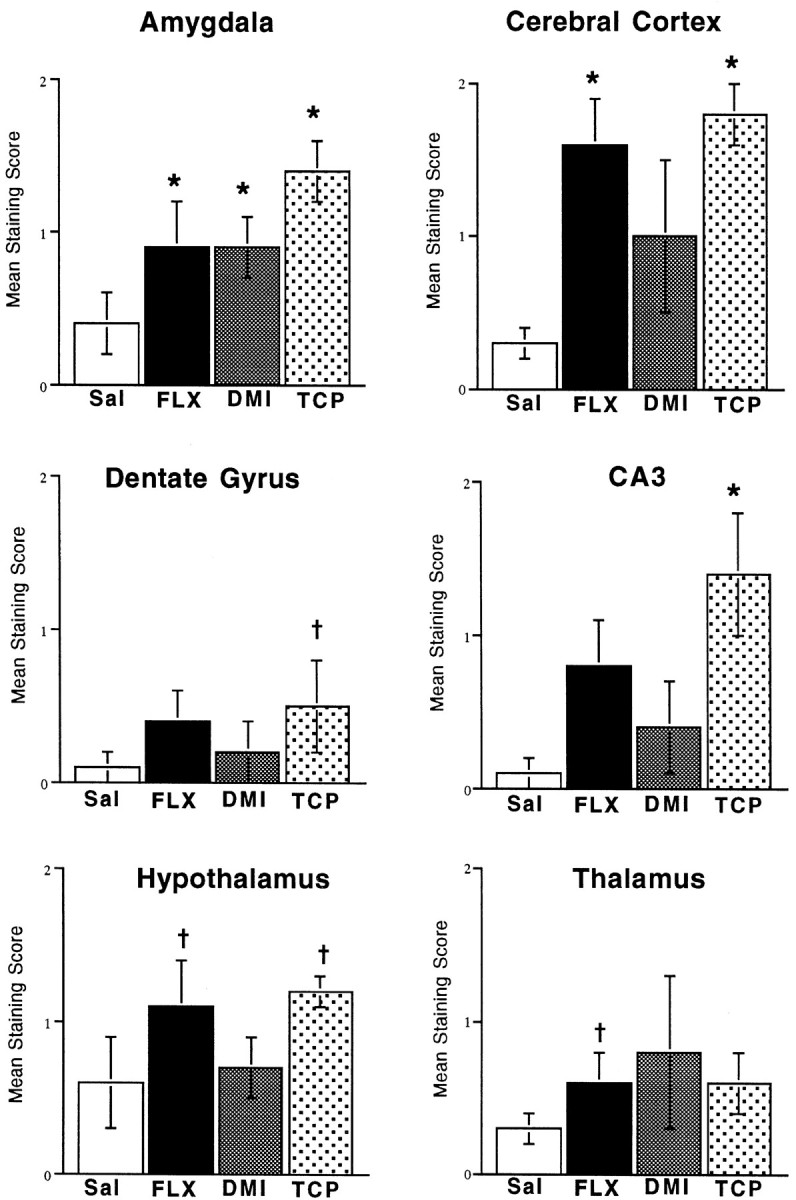
Chronic antidepressant administration increases CRE-mediated gene expression in brain. Mice were administered saline, fluoxetine, desipramine, or tranylcypromine once daily for 14 d, and 6 hr after the last treatment β-galactosidase immunohistochemistry was conducted as described in Materials and Methods. Levels of β-galactosidase were scored as described in Materials and Methods. The results are presented as the mean ± SEM of four to six separate determinations. *p < 0.05, † p < 0.10 compared with saline (Kruskal–Wallis).
The influence of chronic administration of several different classes of antidepressants, including a 5-HT selective reuptake inhibitor (fluoxetine), a norepinephrine selective reuptake inhibitor (desipramine), and a monoamine oxidase inhibitor (tranylcypromine), on levels of CRE-induced LacZ expression were tested. Mice were administered saline or antidepressants for 14 d, and the brains were processed for levels of β-galactosidase immunoreactivity 6 hr after the last treatment. The results demonstrate that chronic antidepressant administration significantly increases levels of CRE-mediated gene expression in several brain regions, although differences were observed in the regional pattern of expression between the different classes of antidepressants. Chronic administration of each of the antidepressants significantly increased levels of β-galactosidase immunoreactivity in the amygdala. The subregions of amygdala that were influenced include the central, basolateral, and basomedial nuclei. Chronic administration of tranylcypromine or fluoxetine, but not desipramine, also induced CRE-mediated gene transcription in the cerebral cortex, including superficial and deep layers. Chronic administration of tranylcypromine additionally increased levels of β-galactosidase immunoreactivity in the hippocampus (CA3 pyramidal cell layer), and there was a tendency for an increase in the hypothalamus (dorsomedial and ventromedial nuclei). A tendency for an increase was also observed in the dentate gyrus granule cell layer, but this effect was small and did not achieve significance. Chronic fluoxetine administration also resulted in a tendency for induction of β-galactosidase immunoreactivity in the thalamus, and there were as well tendencies toward elevated levels in the CA3 pyramidal cell layer and hypothalamus. No alterations were observed in the CA1 pyramidal cell layer of the hippocampus. For most experiments, the immunohistochemical results were quantified using a subjective scoring scale (see Materials and Methods). However, similar results were obtained for chronic fluoxetine when the digitalized images were quantified by densitometry (Table 1).
Table 1.
Influence of chronic fluoxetine administration on CRE-mediated gene expression
| β-Galactosidase expression | ||
|---|---|---|
| SAL | FLU | |
| Amygdala | 173 ± 15 | 217 ± 11* |
| Cortex | 196 ± 17 | 241 ± 9* |
| CA3 | 138 ± 4 | 184 ± 17* |
| Dentate gyrus | 135 ± 7 | 141 ± 12 |
| Hypothalamus | 125 ± 8 | 167 ± 8* |
| Thalamus | 120 ± 4 | 152 ± 14* |
Mice received a chronic (14 d) administration of vehicle (SAL) or fluoxetine (FLU) and were killed 6 h after the last injection. Levels of β-galactosidase were determined by immunohistochemistry as described in Materials and Methods. Levels of β-galactosidase labeling were quantified by densitometry of digitalized images as described in Materials and Methods. The data are the mean ± SEM;n = 6 separate determinations. *p < 0.05 compared with saline (Kruskal–Wallis).
In contrast to the chronic paradigms, acute antidepressant administration induced CRE-mediated gene expression to a much lower level. For the acute studies, mice received saline injections for 13 d, and on day 14 received either saline or an antidepressant at the same dose used for the chronic paradigm. The repeated saline injections were conducted to acclimate the mice to the stress associated with the handling and injections. The brains were then processed for levels of β-galactosidase immunohistochemistry 6 hr after saline or drug treatment. No significant effects were observed in any of the brain regions examined after acute administration of fluoxetine or desipramine (Table 2). Acute administration of tranylcypromine significantly increased β-galactosidase immunoreactivity in hypothalamus, and there was a similar trend in the thalamus and dentate gyrus. However, there was no significant effect of acute tranylcypromine administration in any of the other brain regions examined.
Table 2.
Influence of acute antidepressant administration on CRE-mediated gene expression
| β-Galactosidase expression | ||||
|---|---|---|---|---|
| SAL | FLU | DMI | TCP | |
| Amygdala | 0.4 ± 0.2 | 0.5 ± 0.4 | 0.2 ± 0.2 | 1.0 ± 0.6 |
| Cortex | 0.3 ± 0.1 | 0.2 ± 0.2 | 0.3 ± 0.2 | 1.8 ± 0.7 |
| CA1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.4 ± 0.4 |
| CA3 | 0.3 ± 0.1 | 0.1 ± 0.1 | 0.4 ± 0.3 | 1.1 ± 0.6 |
| Dentate gyrus | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.8 ± 0.4 |
| Hypothalamus | 0.6 ± 0.3 | 0.3 ± 0.3 | 0.3 ± 0.2 | 1.6 ± 0.6* |
| Thalamus | 0.3 ± 0.1 | 0.2 ± 0.2 | 0.2 ± 0.1 | 1.4 ± 0.4* |
Mice received a single dose of vehicle (SAL), fluoxetine (FLU), desipramine (DMI), or tranylcypromine (TCP) and were killed 6 h later. Levels of β-galactosidase were determined by immunohistochemistry as described in Materials and Methods. The data are the mean ± SEM; n = 4–5 separate determinations. *p < 0.05 compared with saline (Kruskal–Wallis).
To determine whether the induction of CRE-mediated gene expression is specific to antidepressants, two other classes of psychotropic drugs were examined. This included an antipsychotic (haloperidol) and a psychostimulant (cocaine). However, chronic administration of these nonantidepressant drugs did not significantly influence levels of β-galactosidase immunoreactivity in any of the brain regions examined (Table 3).
Table 3.
Influence of chronic cocaine or haloperidol administration on CRE-mediated gene expression
| Phospho-CREB | |||
|---|---|---|---|
| Saline | Cocaine | Haloperidol | |
| Amygdala | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.4 ± 0.4 |
| Cortex | 0.3 ± 0.2 | 0.3 ± 0.3 | 0.4 ± 0.4 |
| CA1 | 0.1 ± 0.4 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| CA3 | 0.1 ± 0.4 | 0.0 ± 0.0 | 0.2 ± 0.2 |
| DG | 0.2 ± 0.3 | 0.0 ± 0.0 | 0.2 ± 0.2 |
| Hypothalamus | 0.4 ± 0.2 | 0.3 ± 0.3 | 0.4 ± 0.4 |
| Thalamus | 0.3 ± 0.2 | 0.3 ± 0.3 | 0.8 ± 0.6 |
Vehicle, cocaine, or haloperidol was administered chronically for 14 d, and mice were killed 6 h after the last injection. Levels of β-galactosidase were determined by immunohistochemistry as described in Materials and Methods. The data are the mean ± SEM;n = 7 separate determinations.
Chronic antidepressant administration increases the phosphorylation of CREB
To study the possible mechanisms underlying the induction of CRE-mediated gene expression, the influence of antidepressants on the phosphorylation of CREB was examined. Levels of phospho-CREB were determined by fluorescence immunohistochemistry using an antibody specific for the phosphorylated form of CREB. Alternate sections from the same brains used for analysis of β-galactosidase immunohistochemistry were used for most experiments. Phospho-CREB immunoreactivity was observed in most brain regions of saline-treated mice, including the dentate gyrus granule cell layer and the cerebral cortex. Relatively high levels of immunoreactivity were found in amygdala and hypothalamus.
Chronic administration of fluoxetine significantly increased levels of phospho-CREB immunoreactivity in several brain regions, including amygdala, cerebral cortex, dentate gyrus granule cell layer, thalamus, and hypothalamus (Figs. 1, 3). The subregions that were influenced within each of these areas were similar to those observed for β-galactosidase immunohistochemistry. Chronic administration of desipramine also increased phospho-CREB immunoreactivity in the dentate gyrus granule cell layer, although to a lower level than observed with fluoxetine. No significant effects were observed in response to chronic desipramine administration in any of the other brain regions examined. The influence of tranylcypromine administration on levels of phospho-CREB immunoreactivity was not determined.
Fig. 3.
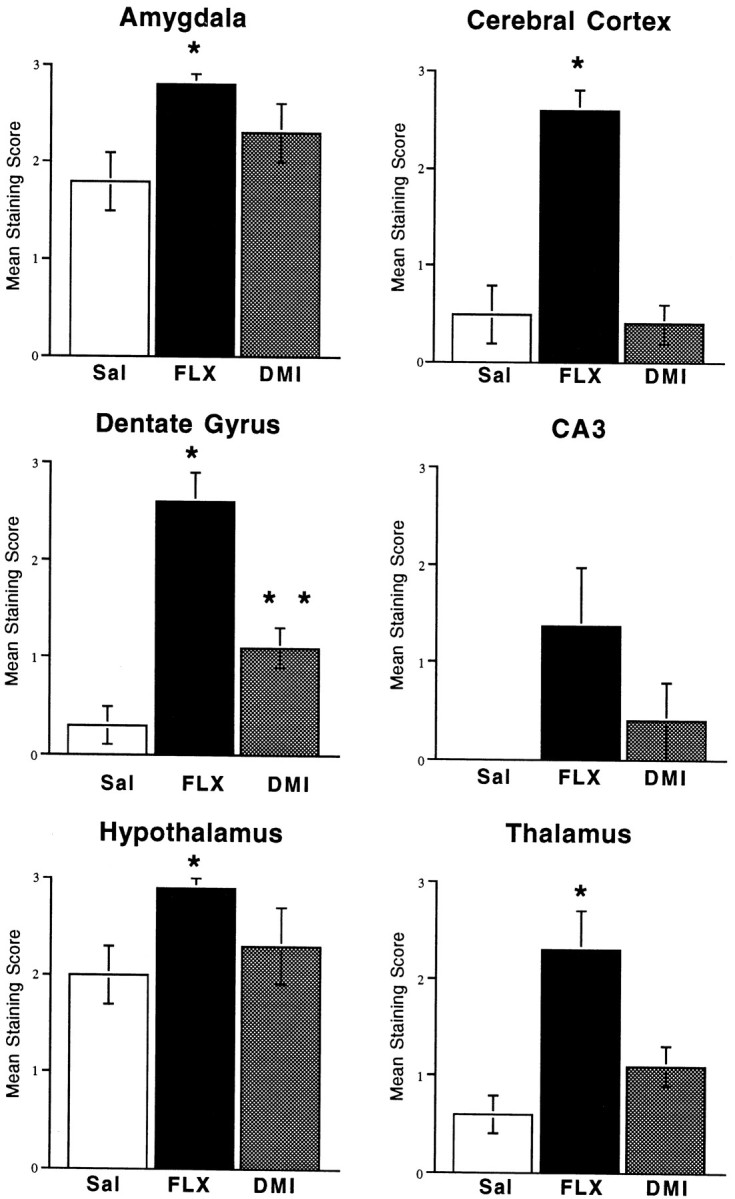
Chronic antidepressant administration increases CREB phosphorylation in brain. Mice were administered saline, fluoxetine, or desipramine once daily for 14 d, and 6 hr after the last treatment phospho-CREB immunohistochemistry was conducted as described in Materials and Methods. Levels of phospho-CREB staining were scored, and results are presented as the mean ± SEM of four to six separate determinations. *p < 0.05 compared with saline (Kruskal–Wallis).
The influence of acute antidepressant administration on levels of phospho-CREB was also examined. Alternate sections from the mouse brains used for β-galactosidase immunohistochemistry were used for these experiments. Acute administration of fluoxetine or desipramine did not significantly influence levels of phospho-CREB immunostaining in any of the brain regions examined (Table 4). However, there was a tendency for acute administration of these antidepressants to increase levels of phospho-CREB in the dentate gyrus granule cell layer of hippocampus.
Table 4.
Influence of acute antidepressant administration on levels of phospho-CREB immunohistochemistry
| Phospho-CREB | |||
|---|---|---|---|
| SAL | FLU | DMI | |
| Amygdala | 1.8 ± 0.3 | 1.9 ± 0.1 | 1.1 ± 0.3 |
| Cortex | 0.5 ± 0.3 | 0.3 ± 0.2 | 0.0 ± 0.0 |
| CA1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| CA3 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Dentate gyrus | 0.3 ± 0.2 | 1.0 ± 0.3 | 1.1 ± 0.2 |
| Hypothalamus | 2.0 ± 0.3 | 1.9 ± 0.1 | 2.1 ± 0.1 |
| Thalamus | 0.6 ± 0.2 | 1.1 ± 0.1 | 0.8 ± 0.2 |
Mice received a single dose of vehicle (SAL), fluoxetine (FLU), or desipramine (DMI) and were killed 6 h later. Levels of phospho-CREB were determined by immunohistochemistry as described in Materials and Methods. The data are the mean ± SEM;n = 4–5 separate determinations. *p < 0.05 compared with saline (Kruskal–Wallis).
Several other classes of psychotropic drugs were also examined to determine whether the induction of phospho-CREB was specific to antidepressants. Chronic administration of haloperidol or cocaine did not significantly influence levels of phospho-CREB immunoreactivity in any of the brain regions examined (data not shown).
DISCUSSION
The results of this study demonstrate that chronic antidepressant administration increases CRE-mediated gene expression and CREB phosphorylation in a region- and drug-specific manner. The most consistent effects observed between the different antidepressants tested were in the amygdala and cerebral cortex. In addition, significant effects were observed in several other limbic brain regions, including the hippocampus, hypothalamus, and thalamus. Induction of CRE-mediated gene expression and CREB phosphorylation were observed in response to chronic, but not acute, antidepressant administration, consistent with the time course for the therapeutic action of these agents. Induction of CRE-mediated gene expression in these brain regions appears to be relatively specific to antidepressants in that administration of nonantidepressant psychotropic drugs does not result in similar effects. These results demonstrate that induction of CRE-mediated gene expression in specific limbic brain structures is an intracellular target of different classes of antidepressants.
The time lag for induction of CRE-mediated gene expression could result from the time required for upregulation of one or more of the upstream components of the cAMP cascade. Previous studies have demonstrated that upregulation of PKA enzyme activity and induction of CREB expression is dependent on chronic antidepressant treatment (Duman et al., 1999). There is also a corresponding delay in the upregulation of CRE binding in response to antidepressant administration (Nibuya et al., 1996;Frechilla et al., 1998). In addition, it is possible that adaptations of 5-HT and NE autoreceptors and monoamine neurotransmission contribute to a stable upregulation of the cAMP cascade and consequently of CRE-mediated gene expression (Blier and de Montigny, 1994).
Both the NE and 5-HT selective reuptake inhibitors increase CRE-mediated gene expression and CREB phosphorylation in amygdala. In contrast, fluoxetine, but not desipramine, significantly increases β-galactosidase and CREB phosphorylation in cerebral cortex. Chronic fluoxetine administration also increases phospho-CREB in several other limbic structures, including the dentate gyrus, hypothalamus, and thalamus. The reasons for these regional differences between fluoxetine and desipramine are not clear because these structures receive diffuse projections from both the NE and 5-HT systems. However, there is evidence for a more rapid adaptation of autoreceptor inhibition of the 5-HT than the NE neurotransmitter system (Blier and de Montigny, 1994). Alternatively, it is possible that higher brain levels of fluoxetine are achieved because of its relatively long half-life. It is notable that there is a tendency for desipramine to increase phospho-CREB in brain regions in which fluoxetine produces a significant effect (i.e., dentate gyrus and thalamus). It is possible that a higher dose of desipramine would produce a significant response, although we were not able to test this possibility because of the side effects of this antidepressant. It will be interesting to examine more selective NE reuptake inhibitors with fewer side effects, such as reboxetine, to further study the influence of the NE system on CRE-mediated gene expression.
The corresponding induction of phospho-CREB and β-galactosidase in amygdala and cerebral cortex indicates that CREB phosphorylation could mediate the induction of CRE-mediated gene expression in these brain regions. However, it was somewhat surprising to find that there was not a significant induction of CRE-mediated gene expression in some brain regions where there was an induction of phospho-CREB (i.e., dentate gyrus, hypothalamus, and thalamus in response to fluoxetine). It is conceivable that transcription factors other than CREB are also required for induction of CRE-mediated gene expression and that these transcription factors are not regulated by antidepressants in all brain regions. This is consistent with previous reports that phosphorylation of CREB at Ser133 is not always sufficient to induce CRE-mediated gene expression (Enslen et al., 1994; Thompson et al., 1995; Impey et al., 1998). Alternatively, the induction of β-galactosidase may not be as responsive as phospho-CREB. This possibility is supported by the observation that there was a trend for an induction of β-galactosidase in some of the brain regions where an induction of phospho-CREB was observed (i.e., thalamus and hypothalamus).
Antidepressant induction of CREB phosphorylation and CRE-mediated gene expression is observed in brain structures that are thought to play a role in the regulation of emotion and responses to stress. Moreover, clinical studies have reported that there are alterations in blood flow, volume, and neurochemistry of many of these same brain regions in depressed patients. For example, previous studies have demonstrated that amygdala mediates some of the behavioral actions of antidepressants and that neurochemical adaptations to antidepressants are observed in this brain region (Ordway et al., 1991; Beck and Fibiger, 1995; Duncan et al., 1996; Dawes et al., 1998; Morelli et al., 1999). In addition, clinical brain imaging studies report alterations in blood flow and glucose metabolism in amygdala of depressed patients (Drevets et al., 1992). The amygdala plays a significant role in fear conditioning and conditioned avoidance behavior and is thought to encode the emotional component of aversive stimulus conditioning (Fanselow et al., 1999; Holland and Gallagher, 1999). The possibility that CREB influences the function of amygdala is supported by a recent report that overexpression of CREB in this brain regions alters long-term memory of fearful conditions (Josselyn et al., 2000). On the basis of these observations, it is also possible that neurochemical alterations in amygdala could contribute to the displaced emotion, as well as anxiety, that is often observed in depressed patients. The possibility that CREB influences amygdala function can be directly tested by studying the influence of the cAMP-CREB cascade on behavioral models of depression, as well as behaviors that are controlled by amygdala (i.e., fear conditioning and conditioned avoidance behavior).
Chronic antidepressant treatment also results in neurochemical and cellular adaptations in cerebral cortex and hippocampus. A role for CREB is supported by the results of our previous reports and the present study, which demonstrate that chronic antidepressant treatment increases the expression and function of CREB in these brain regions (Nibuya et al., 1995, 1996; Duman et al., 1997, 1999). In addition, we have found that antidepressant administration increases the expression of BDNF in hippocampus. The possibility that induction of BDNF is mediated by CREB is supported by recent reports that the promoter of the BDNF gene contains a CRE (Shieh et al., 1998; Tao et al., 1998). Upregulation of CREB and BDNF could act to oppose the damaging effects of stress on hippocampal neurons (Sapolsky, 1996; McEwen, 1999). Clinical studies also report a reduction in the volume and/or number of neurons in hippocampus and cerebral cortex of depressed patients (Sheline et al., 1996; Drevets et al., 1997; Ongur et al., 1998;Rajkowska et al., 1999). A role for CREB in the pathophysiology of depression is supported by a postmortem study demonstrating that CREB levels are decreased in the cerebral cortex of depressed patients and increased in patients receiving antidepressant medication at the time of death (Dowlatshahi et al., 1998).
Alterations in the function of hippocampus and cerebral cortex could also contribute to the cognitive deficits that are often observed in depressed patients. The cAMP cascade is reported to be integrally involved in the cellular adaptations underlying learning and memory in these brain regions (Abel et al., 1997; Taylor et al., 1999). On the basis of these findings and the results of the present study, it is possible that amelioration of the cognitive deficits in depressed patients could result, at least in part, from upregulation of CRE-mediated gene expression in response to antidepressant treatment.
The actions of antidepressants on the hypothalamus and thalamus are not as well characterized. The dorsomedial and ventromedial regions of the hypothalamus are reported to play a role in vegetative behaviors (e.g., eating and sexual drive) that are also abnormal in depression. It is possible that induction of CRE-mediated gene expression in these hypothalamic nuclei normalizes these abnormalities. There was no effect in the paraventricular nucleus of the hypothalamus, which controls the hypothalamic–pituitary adrenal (HPA) axis in response to stress. Interestingly, the paraventricular nucleus of the thalamus is another stress-responsive brain structure that is reported to exert negative control on the HPA axis (Bhatnagar and Dallman, 1998). Upregulation of CRE-mediated gene expression in this structure could contribute to normalization of HPA function in response to chronic antidepressant treatment.
The results of this study demonstrate that induction of CRE-mediated gene expression is a common action of antidepressant treatment. The challenge now is to directly test the role of CREB on the cellular, behavioral, and endocrine responses that are regulated by specific limbic brain structures. Studies are currently underway to address these questions using viral vectors and transgenic mice to determine the functional responses to overexpression of CREB in specific brain regions. Another challenge is to identify the target genes, in addition to BDNF, that are influenced by CREB and antidepressant treatment. One approach is to use DNA microarray technology to identify gene targets of the cAMP-CREB cascade and antidepressants. The results of the present study, in combination with these future approaches, should provide a more complete characterization of the role of CREB in the action of antidepressants, as well as the gene targets that mediate the therapeutic response to these agents.
Footnotes
This work was supported by United States Public Health Service Grants MH45481, MH53199, and 2 PO1 MH25642, a Veterans Administration (VA) National Center Grant for Post-Traumatic Stress Disorder, VA Medical Center, and German Research Council Grant DFG Th698/1-1.
Correspondence should be addressed to R. S. Duman, 34 Park Street, New Haven, CT 06508. E-mail:ronald.duman@yale.edu.
REFERENCES
- 1.Abel T, Nguyen PV, Marad M, Deuel TAS, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Beck CHM, Fibiger HC. Chronic desipramine alters stress-induced behaviors and regional expression of the immediate early gene, c-fos. Pharmacol Biochem Behav. 1995;51:331–338. doi: 10.1016/0091-3057(94)00391-u. [DOI] [PubMed] [Google Scholar]
- 3.Bhatnagar S, Dallman MF. Neuroanatomical basis for facilitation of hypothalamic-pituitary-adrenal responses to a novel stressor after chronic stress. Neuroscience. 1998;84:1025–1039. doi: 10.1016/s0306-4522(97)00577-0. [DOI] [PubMed] [Google Scholar]
- 4.Blier P, de Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994;15:220–226. doi: 10.1016/0165-6147(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 5.Bobon D, Breulet M, Gerard-Vandenhove MA, Guilot-Goffioul F, Plonteux G, Sastre-y-Hernandez M, Schratzer M, Troisfontaines B, Vov-Frenckell R, Wachel H. Is phosphodiesterase inhibition a new mechanism of antidepressant action? A double blind double-dummy study between rolipram and desipramine in hospitalized major and/or endogenous depressives. Eur Arch Psychiatry Neurol Sci. 1988;238:2–6. doi: 10.1007/BF00381071. [DOI] [PubMed] [Google Scholar]
- 6.Dawes LC, Lopez R, Frazer A. Effects of antidepressant treatment on inhibitory avoidance behavior and amygdaloid β-adrenoceptors in rats. Neuropsychopharmacology. 1998;19:300–313. doi: 10.1016/S0893-133X(98)00016-5. [DOI] [PubMed] [Google Scholar]
- 7.Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet. 1998;352:1754–1755. doi: 10.1016/S0140-6736(05)79827-5. [DOI] [PubMed] [Google Scholar]
- 8.Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A functional anatomical study of unipolar depression. J Neurosci. 1992;12:3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drevets WC, Price JL, Simpson JR. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 10.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 11.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry. 1999;46:1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 12.Duncan GE, Knapp DJ, Johnson KB, Breese GR. Functional classification of antidepressants based on antagonism of swim stress-induced fos-like immunoreactivity. J Pharmacol Exp Ther. 1996;277:1076–1080. [PubMed] [Google Scholar]
- 13.Enslen H, Sun P, Brickey D, Soderling SH, Klamo E. Characterization of Ca2+-calmodulin-dependent protein kinase IV role in transcriptional regulation. J Biol Chem. 1994;269:15520–15527. [PubMed] [Google Scholar]
- 14.Fanselow MS, LeDoux JE. Why we think plasticity underlying pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 15.Fleischhacker WW, Hinterhuber H, Bauer H, Pflug B, Berner P, Simhandl C, Wilf R, Gerlach W, Jaklitsch H, Sastre-y-Hernandez M, Schmeding-Wiegel H, Sperner-Unterweger B, Voet B, Schubert H. A multicenter double-blind study of three different doses of the new cAMP-phosphodiesterase inhibitor rolipram in patients with major depressive disorder. Neuropsychobiology. 1992;26:59–64. doi: 10.1159/000118897. [DOI] [PubMed] [Google Scholar]
- 16.Frechilla D, Otano A, Del Rio J. Effect of chronic antidepressant treatment on transcription factor binding activity in rat hippocampus and frontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:787–802. doi: 10.1016/s0278-5846(98)00040-2. [DOI] [PubMed] [Google Scholar]
- 17.Griebel G, Misslin R, Vogel E, Bourguignon J. Behavioral effects of rolipram and structurally related compounds in mice: behavioral sedation of cAMP phosphodiesterase inhibitors. Pharmacol Biochem Behav. 1991;39:321–323. doi: 10.1016/0091-3057(91)90186-6. [DOI] [PubMed] [Google Scholar]
- 18.Holland PC, Gallagher M. Amygdala circuitry in attentional and representational processes. Trends Cognit Sci. 1999;3:65–73. doi: 10.1016/s1364-6613(98)01271-6. [DOI] [PubMed] [Google Scholar]
- 19.Horowski R, Sastre-Y-Hernandez M. Clinical effects of the neurotrophic selective cAMP phosphodiesterase inhibitor rolipram in depressed patients: global evaluation of the preliminary reports. Curr Ther Res. 1985;38:23–29. [Google Scholar]
- 20.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 21.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 22.Josselyn SA, Carlezon WA, Shi CJ, Neve RL, Nestler EJ, Davis M 2000 Long-term memory is facilitated by CREB overexpression in the amygdala. Pharmacol Biochem Behav, in press. [DOI] [PMC free article] [PubMed]
- 23.Malison R, Price LH, Nestler EJ, Heninger GR, Duman RS. Efficacy of Papaverine addition in treatment-refractory major depression. Am J Psychiatry. 1997;154:579–580. doi: 10.1176/ajp.154.4.579. [DOI] [PubMed] [Google Scholar]
- 24.McEwen BS : Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 25.Meinkoth J, Alberts AS, Feramisco JR. Construction of mammalian cell lines with indicator genes driven by regulated promoters. Ciba Found Symp. 1990;150:47–56. doi: 10.1002/9780470513927.ch4. [DOI] [PubMed] [Google Scholar]
- 26.Meyer TE, Habener JF. Cyclic adenosine 3′,5′-monophosphate response element-binding protein (CREB) and related transcription-activating deoxyribonucleic acid-binding proteins. Endocr Rev. 1993;14:269–290. doi: 10.1210/edrv-14-3-269. [DOI] [PubMed] [Google Scholar]
- 27.Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- 28.Morelli M, Pinna A, Ruiu S, del Zompo M. Induction of Fos-like immunoreactivity in the central extended amygdala by antidepressant drugs. Synapse. 1999;31:1–4. doi: 10.1002/(SICI)1098-2396(199901)31:1<1::AID-SYN1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 29.Nestler EJ, Terwilliger RZ, Duman RS. Chronic antidepressant administration alters the subcellular distribution of cyclic AMP-dependent protein kinase in rat frontal cortex. J Neurochem. 1989;53:1644–1647. doi: 10.1111/j.1471-4159.1989.tb08564.x. [DOI] [PubMed] [Google Scholar]
- 30.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- 33.O'Donnell JM. Antidepressant-like effects of rolipram and other inhibitors of cyclic AMP phosphodiesterase on behavior maintained by differential reinforcement of low response rate. J Pharmacol Exp Ther. 1993;264:1168–1178. [PubMed] [Google Scholar]
- 34.Ongur D, Drevets WC, Price JL : Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ordway GA, Gambarana C, Tejani-Butt SM, Areso P, Hauptmann M, Frazer A. Preferential reduction of binding of 125I-iodopindolol to beta-1-adrenoceptors in the amygdala of rat after antidepressant treatments. J Pharmacol Exp Ther. 1991;257:681–690. [PubMed] [Google Scholar]
- 36.Ozawa H, Rasenick MM. Chronic electroconvulsive treatment augments coupling of the GTP-binding protein Gs to the catalytic moiety of adenylyl cyclase in a manner similar to that seen with chronic antidepressant drugs. J Neurochem. 1991;56:330–338. doi: 10.1111/j.1471-4159.1991.tb02599.x. [DOI] [PubMed] [Google Scholar]
- 37.Perez J, Tinelli D, Brunello N, Racagni G. cAMP-dependent phosphorylation of soluble and crude microtubule fractions of rat cerebral cortex after prolonged desmethylimipramine treatment. Eur J Pharmacol. 1989;172:305–316. doi: 10.1016/0922-4106(89)90060-6. [DOI] [PubMed] [Google Scholar]
- 38.Pham TA, Impey S, Storm DR, Stryker MP. CRE-mediated gene transcription in neocortical plasticity during the developmental critical period. Neuron. 1999;22:63–72. doi: 10.1016/s0896-6273(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 39.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 40.Sapolsky RM. Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress. 1996;1:1–19. doi: 10.3109/10253899609001092. [DOI] [PubMed] [Google Scholar]
- 41.Sheline YI, Wang P, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shieh PB, Hu SC, Bobb K, Timmusk T, Gosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 43.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 44.Taylor JR, Birnbaum S, Ubriani R, Arnsten AFT. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci. 1999;19:1–5. doi: 10.1523/JNEUROSCI.19-18-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson MA, Ginty DD, Bonni A, Greenberg ME. L-type voltage-sensitive Ca2+ channel activation regulates c-fos transcription at multiple levels. J Biol Chem. 1995;270:4224–4235. doi: 10.1074/jbc.270.9.4224. [DOI] [PubMed] [Google Scholar]
- 46.Wachtel H. Potential antidepressant activity of rolipram and other selective cyclic adenosine 3′,5′-monophosphate phosphodiesterase inhibitors. Neuropharmacology. 1983;22:267–272. doi: 10.1016/0028-3908(83)90239-3. [DOI] [PubMed] [Google Scholar]