

Abstract
We have generated previously transgenic rats that overexpress peripheral myelin protein 22 (PMP22) in Schwann cells. In the nerves of these animals, Schwann cells have segregated with axons to the normal 1:1 ratio but remain arrested at the promyelinating stage, apparently unable to elaborate myelin sheaths. We have examined gene expression of these dysmyelinating Schwann cells using semiquantitative reverse transcription-PCR and immunofluorescence analysis. Unexpectedly, Schwann cell differentiation appears to proceed normally at the molecular level when monitored by the expression of mRNAs encoding major structural proteins of myelin. Furthermore, an aberrant coexpression of early and late Schwann cell markers was observed. PMP22 itself acquires complex glycosylation, suggesting that trafficking of the myelin protein through the endoplasmic reticulum is not significantly impaired. We suggest that PMP22, when overexpressed, accumulates in a late Golgi–cell membrane compartment and uncouples myelin assembly from the underlying program of Schwann cell differentiation.
Keywords: transgenic disease model, Charcot-Marie-Tooth neuropathy, myelin disease, axon-Schwann cell interaction, abnormal gene dosage, myelin protein function
Rapid saltatory impulse conduction in the peripheral nervous system depends on the myelination of axons by Schwann cells (SCs) and the coordinated expression of a set of myelin-specific proteins and lipids (for review, see Nave, 1995; Snipes and Suter, 1995; Martini and Schachner, 1997; Stoffel and Bosio, 1997;Coetzee et al., 1998). Peripheral myelin protein 22 (PMP22) is a minor constituent of the peripheral myelin sheath but is essential for peripheral nerve function (Spreyer et al., 1991; Welcher et al., 1991;Snipes et al., 1992; Adlkofer et al., 1995, 1997a). The PMP22 gene is tightly regulated in SC development with a time course similar to those of other myelin proteins. PMP22 expression is not restricted to the nervous system, however, but is also widespread in the vertebrate embryo (Baechner et al., 1995; Parmantier et al., 1995; de Wulf et al., 1999; Hagedorn et al., 1999). Consistent with these findings, PMP22 has been suggested to play not only a role in the development and maintenance of myelin and axons but also to regulate common cellular functions, including cell proliferation, cell shape, and apoptosis (Manfioletti et al., 1990; Fabbretti et al., 1995; Zoidl et al., 1995;Hanemann and Müller, 1998; Brancolini et al., 1999).
The human hereditary neuropathies Charcot-Marie-Tooth disease (CMT) (Dyck et al., 1993) are autosomal-dominant in the majority of families and most commonly linked to chromosome 17 (CMT1A). CMT1A has been associated with an intrachromosomal duplication of 1.5 Mb genomic DNA, which includes the intact PMP22 gene (Lupski et al., 1991; Patel et al., 1992). Point mutations of the PMP22 gene have been found in the dysmyelinated Trembler and Trembler-J mice (Suter et al., 1992a,b) and in patients with CMT1A and Dejerine-Sottas disease (Valentijn et al., 1992; Roa et al., 1993; Naef and Suter, 1998). The mechanisms by which both point mutations and overexpression of the PMP22 gene cause dysmyelination and demyelination are not fully understood, but toxic gain-of-function, possibly because of impaired intracellular trafficking, has been implicated (Adlkofer et al., 1997b; Naef et al., 1997; D'Urso et al., 1998; Naef and Suter, 1999; Tobler et al., 1999).
To obtain an animal model of CMT1A and address its pathology at the cellular level, we have generated a line of transgenic rats that express additional copies of the wild-type PMP22 gene (Sereda et al., 1996). Hemizygous transgenics (termed “CMT rats”) display a peripheral hypomyelinating neuropathy associated with muscle weakness, reduced nerve conduction velocities, and onion bulb formation, similar to CMT1A patients. Homozygous PMP22 transgenic rats of the same line show a much more dramatic phenotype, and peripheral nerves remain unmyelinated. SCs of homozygous rats appear developmentally arrested at the promyelining stage (Sereda et al., 1996), comparable with transgenic mice with multiple copies of a PMP22 transgene (Huxley et al., 1996, 1998; Magyar et al., 1996).
In normal development, the promyelinating stage is a critical point in the transition of proliferative, premyelinating SCs (which subdivide axon bundles) to the nonproliferative, myelin-forming SCs, which enwrap single axons and assemble myelin sheaths (Webster, 1993; Mirsky and Jessen, 1996; Zorick and Lemke, 1996). A failure of SC differentiation with a developmental arrest at the promyelinating stage has been observed in mouse mutants lacking the essential transcription factors Krox-20 and SCIP/Tst-1/Oct-6 (Topilko et al., 1994;Bermingham et al., 1996; Jaegle et al., 1996; Zorick et al., 1996). Furthermore, point mutations in the human EGR-2/Krox-20 gene have been identified in patients with congenital hypomyelination (Warner et al., 1998).
Thus, the promyelinating stage appears to mark a threshold in development at which different mutations arrest SC differentiation along a genetically determined program. We reasoned that understanding this abnormal SC arrest in homozygous PMP22 transgenic rats would help to define the role of PMP22 in CMT1A disease and in normal SC development. Here, we report the unexpected finding that SCs are arrested at the promyelinating stage in homozygous PMP22 transgenic rats but continue to differentiate at the molecular level. Myelination and terminal SC differentiation seem to be “uncoupled,” suggesting that PMP22, when sufficiently overexpressed, blocks late steps of myelin assembly.
MATERIALS AND METHODS
Transgenic animals and histology. The generation of PMP22 transgenic rats (in which three copies of a genomic DNA fragment constitute one PMP22 transgene locus) and genotyping of homozygous and hemizygous animals have been described previously (Sereda et al., 1996). Because homozygous animals are born to clinically affected parents (CMT rats), the poor performance of male rats limits the number of available homozygous rats. Animals were anesthetized with pentobarbital (240 mg/kg body weight) and transcardially perfused with saline, followed by 5% glutaraldehyde–4% paraformaldehyde in cacodylate buffer, pH 7.2. Animals were stored in the same fixative before further processing. Tissue was removed from the sciatic nerves (midthigh level), brachial plexus, and lumbar nerve roots, osmicated, and processed routinely for resin embedding. Sections were stained with methylene blue–azure II for light microscopy, and grids (60 μm) were double-stained with uranyl acetate and lead citrate for electron microscopy.
Teased nerve fibers. Single nerve fibers were teased from unfixed sciatic nerves in a drop of PBS using fine forceps under microscopic control. Teased fibers were transferred onto poly-l-lysine-coated slides, allowed to air dry, and then processed for immunocytochemistry as described below.
RNA analysis. Total RNA was isolated from sciatic nerves of 5-week-old rats using the guanidine thiocyanate–water-saturated phenol procedure (Chomczynski and Sacchi, 1987). For reverse transcription (RT)-PCR analysis, cDNA was synthesized from total RNA using random hexamer primers and murine leukemia virus reverse transcriptase (Life Technologies, Gaithersburg, MD) according to the manufacturer's recommendations. All PCR amplifications were semiquantitative, performed in 4 × 20 μl reaction buffer, containing 2 ng of cDNA, 400 nm of each primer, 200 μm of each deoxynucleotide triphosphate, 3 μm MgCl, and 1 U of Taq polymerase (Amersham Pharmacia Biotech, Uppsala, Sweden) from the same master mix. To amplify SCIP cDNA, PCR was performed in the presence of 6 μm MgCl and 10% DMSO. All reactions were trace-labeled with 32P-dCTP (8 μCi). Standard PCR parameters were denaturation at 94°C (30 sec), annealing at 56°C (30 sec), and extension at 72°C (60 sec). PCR parameters for SCIP cDNA were denaturation at 94°C (60 sec), annealing at 50°C (60 sec), and extension at 72°C (90 sec). After increasing cycle numbers, 20 μl samples were analyzed to determine the linear range of the assay; 10 μl aliquots of the radiolabeled PCR product were size separated on 5% polyacrylamide gels, and the radioactivity of a fragment was quantified with a phosphoimager (BAS 1000; Fuji, Tokyo, Japan). To obtain an internal standard, a cDNA fragment of the ubiquitously expressed glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA was coamplified with primers 5′-CTACATGGTCTACATGTTCCAGTA-3′ (sense) and 5′-TGATGGCATGGACTGTGGTCAT-3′ (antisense) from the published cDNA sequence (Tso et al., 1985). To amplify endogenous plus transgene-derived PMP22 cDNA, primers were used corresponding to exon 3 (5′-CTGTACCACATCCGCCTTGG-3′) and exon 5 (5′-TCAACACGAGGCTGACGGTC-3′) of the mouse PMP22 gene. Protein zero (P0) cDNA was amplified with primers 5′-GGTGGTGCTGTTGCTGCTG-3′ (sense) from exon 4 and 5′-TTGGTGCTTCGGCTGTGGTC-3′ (antisense) from exon 6 of the published cDNA (Lemke and Axel, 1985). Exons 1 to 5 of the myelin basic protein (MBP) cDNA were amplified using primers 5′-TTCTTTAGCGGTGACAG-3′ (sense) and 5′-CTGTCTCTTCCTCCCCA-3′ (antisense) from the published sequence of Roach et al. (1983). Krox-20 cDNA was amplified with primer 5′-CTTTGACCAGATGAACGGAGT-3′ (sense) of exon 2 and primer 5′-ATGCCCGCACTCACAATATT-3′ (antisense) of exon 3 from the cDNA sequence of Chavrier et al. (1988). SCIP cDNA was amplified with primers 5′-ACGGACACGCACACGGACAT-3′ (sense) and 5′-GAGGGCTTGGG GCACTTGAG-3′ (antisense) from the sequence of Monuki et al. (1990). The low-affinity nerve growth factor receptor (LNGFR/p75) was amplified with primers 5′-GTGAACCCTGCCT-GGACAATG-3′ (sense) and 5′-ACGACCACAGCAGCCAAGATG-3′ (antisense) from the cDNA sequence of Radeke et al. (1987). PCR products that had the predicted size of 422 bp (GAPDH), 219 bp (Krox-20), 601 bp (LNGFR), 230 and 306 bp (MBP), 636 bp (PMP22), 190 bp (P0), and 463 bp (SCIP), were quantified by phosphoimager analysis and compared within the linear range of this assay.
Immunofluorescence. Sciatic nerves were dissected from transgenic rats and age-matched littermates at various postnatal time points [from postnatal day 0 to 8 months], snap-frozen into O.C.T. medium (Sakura), and sectioned at −20°C (8 and 10 μm). Cross-sections and longitudinal sections were thaw-mounted onto poly-l-lysine-coated slides. For the detection of LNGFR/p75 and SCIP/Oct-6, fixation was in 4% paraformaldehyde (room temperature for 5 min). For labeling of P0, PMP22, and bromodeoxyuridine (BrdU), cryosections were delipidated for 2 min in methanol/acetone (95:5 at −20°C) and washed in PBS. Nonspecific binding was blocked for 1 hr at room temperature using 10% fetal calf serum and 0.1% Triton X-100 in PBS (PBS/T). Primary antibodies (specified below) were added in the same blocking solution (4°C, overnight). After three washes in PBS/T (room temperature for 10 min), the sections were incubated with the appropriate secondary antibody in blocking solution (room temperature for 1–2 hr). Slides were washed three times in PBS/T (room temperature for 10 min) and briefly rinsed in H2O, and sections were mounted in Aqua PolyMount (Polysciences, Warrington, PA). Between washing steps, some sections were stained for 10 min with 50 ng/ml 4′,6′-diamidino-2-phenylindole (DAPI). Slides were examined under epifluorescence and photographed (Axiophot; Zeiss, Oberkochen, Germany) before the processing of digitalized images (Adobe Photoshop; Adobe Systems, Mountain View, CA). An alternative fixation protocol of sections used acetone (2 min at −20°C) and methanol (30 min at room temperature). Blocking, antibody incubation, and washing were in the presence of 0.2% Tween 20.
Antibodies. A rabbit polyclonal anti-PMP22 (peptide-2) antiserum was diluted 1:500 (Snipes et al., 1992). A mouse monoclonal anti-P0 antibody (a gift of Dr. J. Archelos, University of Graz, Graz, Austria) was diluted 1:2000 (Archelos et al., 1993). A rabbit polyclonal anti-p75/LNGFR antiserum (Chemicon, Temecula, CA) was diluted 1:200. A rabbit polyclonal anti-SCIP/Oct-6 antiserum (a gift of Dr. G. Lemke, Salk Institute, La Jolla, CA) was diluted 1:40 (Zorick et al., 1996). A mouse monoclonal anti-BrdU antibody was obtained from GenQuest BioScience Inc. (San Jose, CA) and used at 1:10. Secondary antibodies were goat anti-mouse IgG Cyc3 conjugate (Jackson ImmunoResearch, West Grove, PA), diluted 1:2000, and goat anti-rabbit DTAF conjugate (Jackson ImmunoResearch), diluted 1:50.
BrdU-labeling. To detect DNA synthesizing cells, rats were injected intraperitoneally with BrdU (50 mg/kg body weight) and killed 1 hr later. Sciatic nerves were cryosectioned and fixed as described for immunofluorescence. Sections were incubated in 2 N HCl for 30 min at room temperature, followed by 0.1 msodium borate, pH 8.5, for 10 min at room temperature before immunocytochemical detection of BrdU.
Deglycosylation assay. Protein extracts from single sciatic nerves of homozygous, hemizygous, and wild-type rats (aged 5 weeks) were made in 2 ml of PBS, pH 7.4, and 1% SDS using a polytron homogenizer at the highest setting. The homogenate was immediately boiled for 3 min, and insoluble material was pelleted by centrifugation. Protein concentrations were determined using BCA reagents (Pierce, Rockford, IL). Endoglycosidase digestions with N-glycosidase F (PNGase F) and endoglycosidase H (EndoH) (both from Boehringer Mannheim, Mannheim, Germany) were performed according to the manufacturer's suggestions in 80 μl for 24–36 hr at 37°C. PNGase F buffer was 50 mm sodium phosphate, pH 7.2, 25 mm EDTA, 1% Triton X-100, and 1% β-mercaptoethanol. EndoH buffer was 50 mmsodium phosphate, pH 5.5, 25 mm EDTA, 1% Triton X-100, and 1% β-mercaptoethanol.
Western blot analysis. Equal amounts of solubilized protein (lanes for the PNGase treatment contained 2.5 μg and lanes for the EndoH treatment contained 10 μg of protein) were separated on denaturating 12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (BA-S 85; Schleicher and Schüll, Dassel, Germany) by semidry electroblotting. Membranes were blocked with 5% nonfat dry milk and 1% BSA (in TBS and 0.02% Tween) for 1 hr at room temperature. Incubation with primary antibodies in blocking buffer was at 4°C overnight, followed by washing in TBS containing 0.1% Tween (TBS/T) and a 1 hr incubation at room temperature with horseradish peroxidase-conjugated secondary antibodies (Amersham Pharmacia Biotech). After washing in TBS/T, immunoreactive protein was detected using the enhanced chemiluminescence kit (ECL; Amersham Pharmacia Biotech). Western blots were stripped and reprobed with different antibodies. Anti-PMP22 antibodies were diluted 1:1000 and anti-P0 antibodies were diluted 1:2000. For total protein analysis, 4 μg of nerve extract was separated on a 12% SDS-polyacrylamide gel and visualized by staining with Coomassie Brilliant blue.
RESULTS
Absence of peripheral myelin in homozygous PMP22-overexpressing rats
Homozygous PMP22 transgenic rats can be easily distinguished from the hemizygous CMT rat by a more severe and early-onset phenotype, as described in our initial report (Sereda et al., 1996). The homozygous animals studied here developed marked hindlimb paresis and often became paraplegic by 4 weeks of age. They also exhibited dyspnoea upon exertion, decreased body weight, contractures, and kyphoscoliosis, as well as impaired general ealth and developmental retardation. These symptoms were first apparent at 2 weeks of age, followed by progressive deterioration. Despite this condition, some homozygous animals survived for more than 8 months.
Light microscopy of sciatic nerves obtained from homozygous rats at 5 weeks of age revealed severe dysmyelination with virtually all axons never myelinated (Sereda et al., 1996). This is confirmed by the findings in high-copy number PMP22 transgenic mice, which were analyzed as early as postnatal day 10 (Huxley et al., 1996, 1998; Magyar et al., 1996). Also, in 8-month-old homozygous rats, myelin sheaths were lacking (Fig.1A,B), indicating a stable differentiation arrest rather than a temporal delay of myelination. Furthermore, axonal calibers appeared reduced as observed previously in PMP22 mutant mice (Sancho et al., 1999). Electron microscopy analysis revealed that SCs were arrested at the promyelinating stage (Fig. 1C). SCs associated with large-caliber axons had segregated in the normal 1:1 ratio but failed to assemble myelin. Only rarely (i.e., in <1 of 1000 SC–axons units), myelin sheaths were observed (Fig. 1C, A3). The appearance of such myelin sheaths might be explained by somatic PMP22 transgene inactivation allowing single cells to escape from the arrest. Also at the EM level, we never observed onion bulbs or signs of myelin degradation, suggesting that the absence of myelin stems from a failure of myelin formation rather than abnormal demyelination.
Fig. 1.
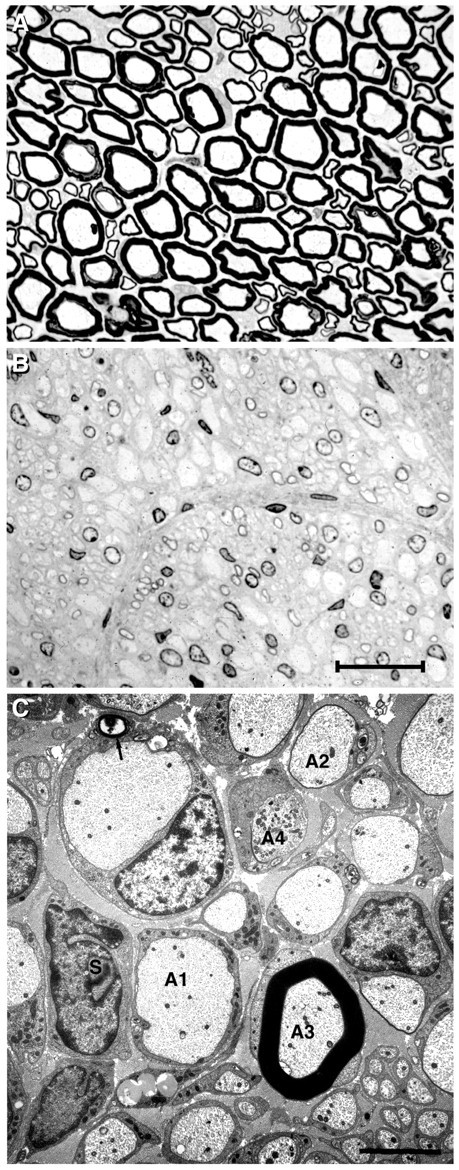
Absence of peripheral myelin is not a developmental delay in rats homozygous for the PMP22 transgene. Resin sections of sciatic nerves from 8-month-old wild-type (A) and PMP22 transgenic (B) animals were stained with methylene blue–azure II and photographed at the same magnification. In the absence of myelin, SC nuclear chromatin and nucleoli are prominently stained. Scale bar, 15 μm.C, PMP22 transgenic rats at 8 months of age. By electron microscopy, putative myelin-forming SCs of the sciatic nerve have segregated in a 1:1 manner with axons (A1) but reveal a block of myelination. Nearly all axons are surrounded by SC cytoplasm but not myelinated. Occasional axons (A2) have a very thin sheath of two or three lamellae. This field has been deliberately selected to also include the rare example of a myelinated axon (A3; frequency <1 in 1000) surrounded with a sheath of normal thickness. Another axon contains a collection of mitochondria (A4). Onion bulbs are absent, although occasional SCs (S) are unassociated with axons. Rare intracellular myelin figures are depicted with anarrow. The amount of endoneurial collagen is considerably increased. Unmyelinated fibers appear normal. Scale bar, 4 μm.
In some cases, arrested SCs contained intracellular myelin figures, some of which were associated with vacuoles of unknown origin (Fig.2A). They could be myelin debris or products of abnormal myelin assembly. Other vesicular structures in the affected Schwann cell appeared to be closely associated with its adaxonal surface (Fig. 2B). Occasionally, cisternae of the endoplasmic reticulum (ER) were also vacuolated or distended by electron dense material (Fig.2C).
Fig. 2.
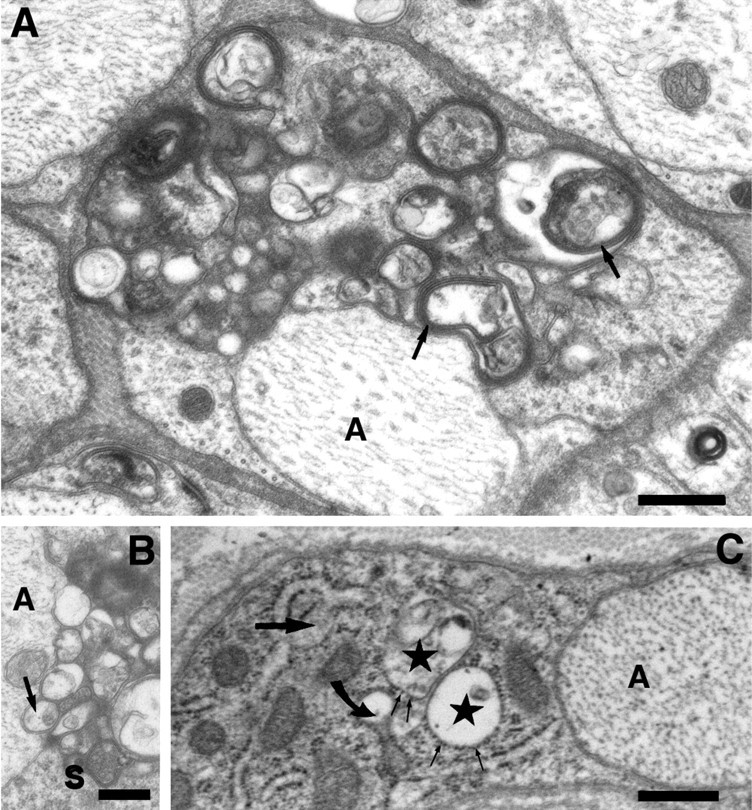
A, At higher magnification, intracellular myelin figures (arrows) are sometimes recognized, which are not associated with the axon (A). Scale bar, 0.5 μm. B, Other vesicular structures (arrow) within the Schwann cell (S) appear to be closely associated with adaxonal surface (A, axon). Scale bar, 0.5 μm.C, Schwann cell abnormalities occasionally include the ER, as suggested by ER cisternae distended by a moderately electron dense material (large straight arrow). Other profiles (stars), designated as rough ER by the presence of ribosomes on their surface, are vacuolated. A transition between the two forms is also evident (curved arrow). Scale bar, 0.5 μm. All electron micrographs were taken from a homozygous PMP22 transgenic rat at 8 months of age.
Molecular SC differentiation
Whereas the ultrastructure of dysmyelinated nerves in homozygous rats revealed an apparent differentiation block of affected SCs, a reinvestigation of the molecular phenotype suggested the opposite. Because tissue samples were small, semiquantitative RT-PCR analysis was used to analyze the steady-state mRNA levels of SC differentiation markers using cyclophilin or GAPDH mRNA as internal standards. The low-affinity nerve growth factor receptor (LNGFR/p75) should only be expressed by nonmyelinating SCs and immature SC precursors (Mirsky and Jessen, 1996). Also, the transcription factors SCIP/Oct-6/Tst-1 and Krox-20/EGR-2 are expressed in SC before myelin formation (Monuki et al., 1989; Topilko et al., 1994; Zorick et al., 1996). The myelin-associated proteins PMP22, MBP, and P0 are highly upregulated during terminal SC differentiation and myelination (Mirsky and Jessen, 1996; Sommer and Suter, 1998).
Surprisingly, we found abundant myelin gene expression in the sciatic nerves of homozygous rats at the age of 5 weeks, despite the absence of myelin profiles. Both P0 mRNA and MBP mRNA levels were as high in homozygous as in wild-type sciatic nerves, and in some animals, even higher than in the age-matched controls (Fig.3A). As predicted, PMP22 mRNA itself was always higher in PMP22 transgenic animals than in age-matched wild-type. Furthermore, the mRNAs encoding SCIP and LNGFR (but not Krox-20) were more abundant in PMP22 homozygous nerves than in wild-type nerves (Fig. 3B).
Fig. 3.
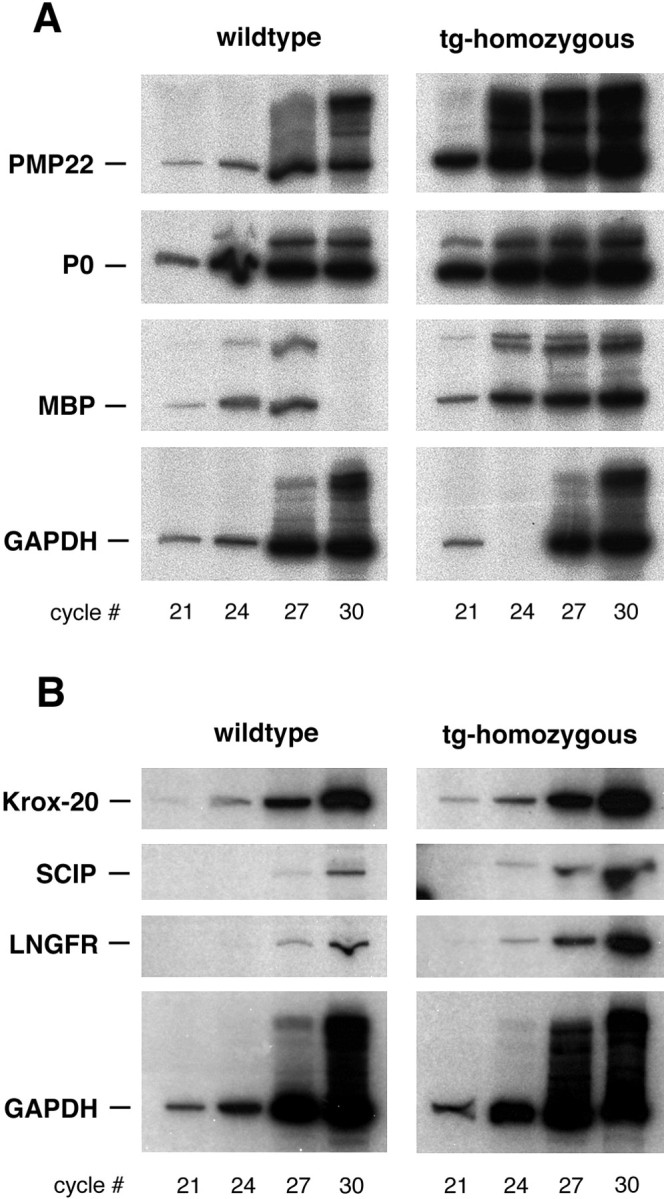
Normal transcription of myelin protein genes in sciatic nerves lacking myelin. Semiquantitative RT-PCR analysis was used to compare the steady-state levels of mRNAs encoding (A) the myelin proteins PMP22, P0 and MBP, (B) the glial transcription factors Krox-20, SCIP/Tst-1/Oct-6, and the receptor p75/LNGFR between homozygous PMP22 transgenic rats and wild-type controls (5 weeks of age). For quantitation, we used GAPDH mRNA as an internal standard. Within the linear range of this assay, PCR products for PMP22, P0, and MBP were at least twofold more abundant in homozygous transgenics. In the data set shown, two samples were lost before loading. Also, the expression of LNGFR and SCIP (but not of Krox-20) is higher in the unmyelinated nerves of homozygous transgenics (tg) than in wild-type controls.
Coexpression of early and late SC markers
The expression of both myelin protein genes and early SC markers in single nerves was unexpected but could reflect cellular heterogeneity. To confirm the differentiation status of affected SCs at the protein level and to address a possible cellular coexpression of markers, we analyzed frozen sections by immunofluorescence. In the complete absence of myelin, the cross-sectioned sciatic nerve had a spongiform appearance and poor tissue preservation after cryosectioning. Improved quality of immunofluorescence was achieved by longitudinal sectioning or teasing of the nerves.
In wild-type animals, P0 and PMP22 were distributed uniformly in compacted myelin as expected (data not shown). Adult homozygous nerves showed also high levels of P0 and PMP22 immunoreactivity, but, in the absence of myelin as a receiving compartment, this signal was primarily localized to intracellular regions (Fig.4B,C). The widespread signal that appears to include the cell surface of permeabilized nerves strongly suggests, but does not prove, that PMP22 reaches the Schwann cell membrane. This point can be decided when antibodies against extracellular epitopes of PMP22 become available.
Fig. 4.
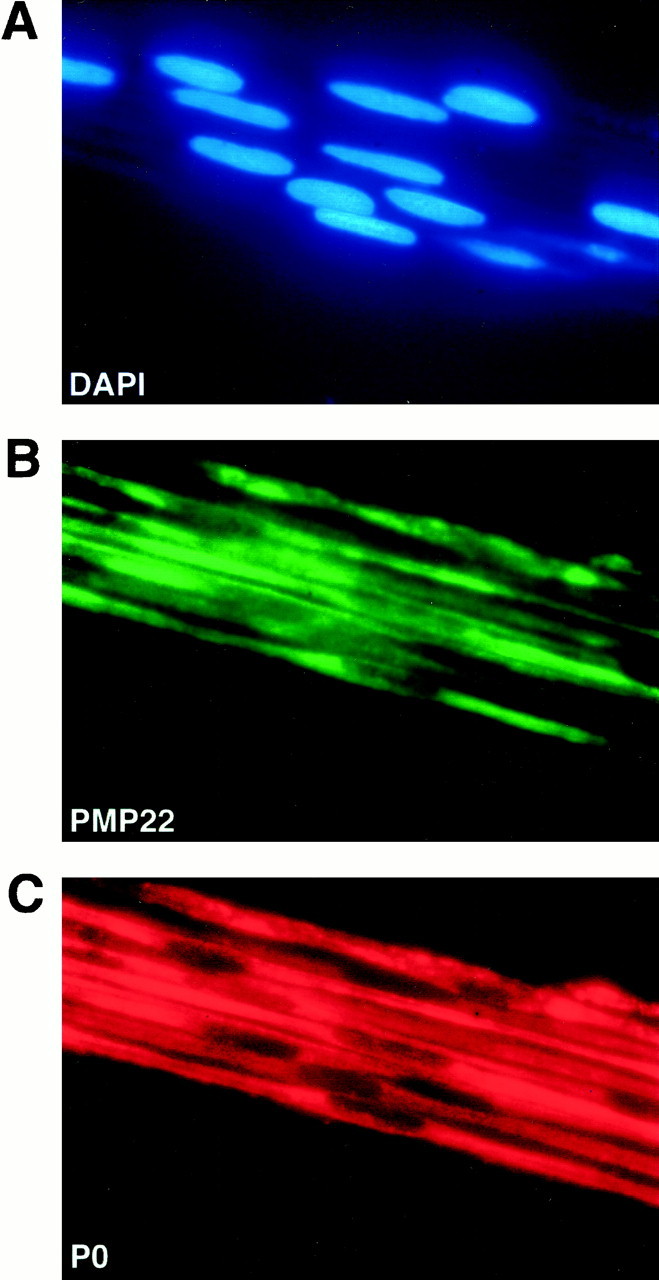
Immunolocalization of PMP22 and P0 in a myelinated sciatic nerves. Teased fibers were prepared from sciatic nerves of homozygous transgenic rats at the age of 5 weeks. By double-immunofluorescence staining, strong expression of PMP22 (B) and P0 (C) was confirmed at the protein level and colocalized to individual SCs.A, SC nuclei stained with DAPI.
The early SC markers were less uniformly expressed. In wild-type nerves, LNGFR/p75 immunoreactivity was restricted to a few presumably nonmyelinating SCs (Fig. 5A,arrows), whereas in the unmyelinated nerve of homozygotes, most SCs stained positive (Fig. 5B). Similarly, SCIP immunoreactivity was barely detectable in SC nuclei of wild-type rats (Fig. 5C), but SCIP expression was obvious in many SCs of homozygotes (Fig. 5D).
Fig. 5.

Expression of p75/LNGFR and SCIP/Tst-1/Oct-6 by SCs arrested at the promyelin stage. Longitudinal cryostat sections of sciatic nerves from 5-week-old wild-type (A,B) and homozygous PMP22-transgenic (C,D) rats were stained with antibodies against p75/LNGFR (A, C) and for SCIP/Tst-1/Oct-6 (B, D). In wild-type nerve, p75/LNGFR is expressed by only a few, presumably nonmyelinating SCs (arrows in A), and SCIP is not detectable (B). In contrast, the majority of SCs in the homozygous mutant express p75/LNGFR (C), and a fraction can be stained for SCIP (D). Scale bar (in D), 50 μm.
To determine whether early and late markers are possibly coexpressed by the same SC, we performed double immunolabeling. On longitudinal sections of nerves from wild-type rats, the staining of SCs for LNGFR/p75 and P0 was mutually exclusive (Fig.6A,B,arrows). In homozygotes, however, LNGFR/p75 and intracellular P0 immunoreactivity were found frequently in the same transgenic SC (Fig. 6C,D, arrowheads). Is such a colocalization of early and late markers also observed in normal development, i.e., at an earlier time point? To approach this question, we analyzed wild-type sciatic nerves immediately after birth, when the majority of SCs are at the promyelinating stage (Friede and Samorajski, 1968). By immunostaining of cryosections, we observed prominent immunoreactivity for LNGFR/p75 (Fig.7C), a small number of SCIP-positive cells (Fig. 7D), and a weak P0 immunoreactivity scattered throughout the nerve section (Fig.7B), but no overlap. These data suggest that the coexpression of early and late marker proteins, as shown in Figure 6, is a unique feature of PMP22 transgenic SCs.
Fig. 6.

Abnormal coexpression of early and late Schwann cell markers at 5 weeks of age. Longitudinal cryostat sections of sciatic nerves from wild-type (A, B) and homozygous PMP22 transgenic (C, D) rats were stained for p75/LNGFR (A, C) and P0 (B, D). Note that, in wild-type rats, p75/LNGFR is expressed by cells that do not stain for P0 (arrows in A and B). In contrast, p75/LNGFR and P0 are clearly coexpressed by some transgenic SCs (arrowheads in C andD). Scale bar (in B), 20 μm.
Fig. 7.

Promyelinating stage SCs in normal rat development. Transverse cryostat sections were taken from sciatic nerves of neonatal wild-type rat (when promyelinating SCs are prominent). Phase contrast is shown in A, and immunolabeling was with antibodies against P0 (B), p75/LNGFR (C), and SCIP (D). Virtually all SCs show staining for p75/LNGFR (C), whereas SCIP immunoreactivity is present in only a fraction of cells (D). The level of P0 immunoreactivity (B) is still very low, and the staining of epineurium is unspecific. Scale bar (inD), 20 μm.
Does PMP22 expression occur in proliferating Schwann cells, which are by this criterion still immature? When DNA synthesis was monitored byin vivo BrdU incorporation in the sciatic nerve of homozygous rats at 5 weeks of age and in age-matched controls, we found no obvious difference in the number of labeled cells. Thus, nearly all homozygous SCs had withdrawn from the cell cycle. When BrdU-labeled sections were double-stained for PMP22, we never observed double-positive cells, confirming that abundant PMP22 expression marks only postmitotic Schwann cells (data not shown).
PMP22 trafficking
Because impaired intracellular trafficking is a common disease mechanism in PMP22 point mutation-based myelination defects in mouse and human (Naef et al., 1997; D'Urso et al., 1998; Naef and Suter, 1999; Tobler et al., 1999), we checked for signs of abnormal trafficking of wild-type PMP22 when overexpressed. A reliable colocalization of PMP22 with subcellular antigen was difficult in the spindle-shaped Schwann cells of a teased fiber (data not shown). As an indicator, we therefore compared PMP22 in sciatic nerve extracts from homozygous and hemizygous animals and wild-type controls after deglycosylation with PNGase and EndoH. PNGase cleaves the N-linked oligosaccharides, irrespective of their composition, from mature glycoproteins. EndoH removes specifically the high mannose-containing, noncomplex oligosaccharides that must be modified by mannosidase II, a resident enzyme of the medial Golgi compartment, before complex glycosylation (Kornfeld and Kornfeld, 1985). Thus, EndoH-resistant glycoproteins have passed the medial Golgi and are more “mature” than EndoH-sensitive glycoproteins.
As shown in Figure 8, PMP22 is PNGase-sensitive but EndoH-resistant in both wild-type and transgenic animals. PNGase removes the sugar moieties of the 22 kDa molecule (A), leaving the 18 kDa PMP22 core protein behind. Treatment of the same extracts with EndoH revealed that PMP22, as well as P0, are resistant and thus almost exclusively contain complex oligosaccharides (a low amount of EndoH-sensitive protein is detectable in wild-type nerve extracts). Thus, when overexpressed in homozygous rats (Fig. 8, lanes hz1, hz2), PMP22 and P0 are not aberrantly retained in the endoplasmic reticulum. They have reached at least a medial Golgi compartment and can be biochemically detected as a mature protein. This is in marked difference to Trembler mutant PMP22, which has been analyzed in transfected cells (Naef et al., 1997). We also note that PMP22 was readily found in homozygous nerve extracts by Western blot analysis, whereas deglycosylated P0 protein was only poorly detectable, even when five times more protein extract was used for the analysis (Fig. 8B). Although it could be argued that the P0-specific antibody has a low affinity to SDS-deactivated protein, the differences between wild-type and mutant levels also suggest that, in the absence of myelination, PMP22 accumulates in the affected Schwann cells to a higher level than P0 glycoprotein.
Fig. 8.

Intracellular processing of PMP22 in transgenic SCs proceeds normally. Deglycosylation profile and Western blot analysis demonstrate that virtually all PMP22 reaches the Golgi compartment of SCs, also when transgenically overexpressed. Protein extracts from sciatic nerves of 5-week-old wild-type (wt), hemizygous (tg), and homozygous (hz1, hz2) rats were treated with PNGase F (A) or EndoH (B). Protein detection was with antibodies against PMP22 and P0. Control samples (−) were incubated without the addition of enzyme.Lanes for the PNGase treatment contained 2.5 μg, andlanes for the EndoH treatment contained 10 μg of protein. Molecular weights are indicated to the right(in kilodaltons). Note that PMP22 and P0 are completely EndoH-resistant in overexpressing SCs, indicating that normal complex glycosylation has occurred and that PMP22 and P0 did not accumulate before the medial Golgi compartment.
DISCUSSION
We have shown previously that transgenic rats, defined by an experimentally increased PMP22 gene dosage, share the major clinical and pathological features of human CMT1A (Sereda et al., 1996). Here, we have analyzed the underlying disease mechanism more specifically by further increasing the PMP22 gene dosage and examining the gain-of-function phenotype in rats bred to homozygosity. We have found that such homozygously mutant rats are more severely and more uniformly affected, both at the cellular level and clinically compared with hemizygous animals. In homozygous rats, virtually all SC–axon units are arrested at the promyelinating stage, lacking the multilayered myelin sheath. This finding suggests that the underlying PMP22 disease mechanisms operate after axon recognition but before the onset of myelin assembly. It does not rule out the theoretical possibility that also PMP22 overexpression in motoneurons (Parmantier et al., 1995) contributes to the pathology.
Surprisingly, the molecular differentiation of SCs arrested at the promyelinating stage is not blocked. In fact, the PMP22 transgenic SCs contain transcripts of the major myelin protein genes at approximately the same or higher levels than wild-type SCs. Because the encoded myelin proteins have no myelin domain as a target area in which to accumulate, they must be degraded and their steady-state level is lower than in the myelinated wild-type nerve. Nevertheless, the corresponding proteins were clearly immunodetectable, most prominently in intracellular compartments.
At the single cell level, myelin proteins were coexpressed in some SC with early developmental markers that are normally absent from differentiated myelin-forming cells (i.e., LNGFR/p75 and SCIP/Tst-1/Oct-6). However, the number of SCIP-expressing SCs in these homozygous rats was obviously lower than the number of SCs arrested at the promyelinating stage, suggesting that the failure to myelinate was not caused by the aberrant SCIP expression. Abnormal p75 LNGFR expression has been detected previously in nerve biopsies of CMT1A patients and in PMP22 transgenic mice (Hanemann et al., 1996; Magyar et al., 1996). It is possible that this unusual combination of antigenic markers is part of the altered phenotype of “differentiated” SCs in response to the lack of myelin and the continued “promyelin” type of axonal contact (Lemke and Chao, 1988). The uncoupling of molecular and morphological aspects of differentiation is intriguing and should be interpreted such that the promyelin stage in the life of an SC is not defined as a particular step in the genetic program but can be associated with different patterns of gene expression.
When taken together, the analysis of the CMT rat and its homozygous variant have provided new insight into the role of PMP22 in peripheral neuropathies. Although PMP22 overexpression can cause the complete failure to assemble myelin, it is not associated with a corresponding block of SC differentiation, at least not at the molecular level. The geometry of spindle-shaped SCs makes it difficult to obtain convincing colocalization data of PMP22 with typical subcellular marker antigens. However, given that PMP22 and P0 appear not to be aberrantly retained in the ER (as judged by deglycosylation analysis), a rather late membrane sorting and fusion event preceding myelin formation might be negatively affected by the excess of PMP22. According to this model, reduced myelin membrane deposition could further increase the intracellular pool of PMP22 (and other myelin proteins), possibly in a late Golgi–cell membrane compartment, and contribute to a complete halt of myelination. Myelin protein synthesis in the absence of real myelin assembly suggests further that most of the newly synthesized membrane proteins must be targeted to nonmyelin domains and are ultimately degraded (Notterpek et al., 1997). At highest magnification, we have observed SCs with abnormal vesicular structures of unknown origin (Fig. 2). These vesicles may represent autophagic vacuoles, swellings of the Golgi–trans-Golgi network, or abnormal membrane fusion products. Given their low frequency, however, they are unlikely to be required for the uniform SC pathology.
We cannot formally exclude the possibility that homozygous SCs remain “undifferentiated” in that they are lacking one or more unknown protein(s) required for myelin assembly. This is unlikely, however, given that (1) nearly all hemizygous SCs are competent to myelinate, and (2) myelin protein genes are expressed at equivalent mRNA levels in homozygous and hemizygous transgenic rats. One might also argue that unknown “inhibitory” proteins of myelin assembly (other than PMP22), which normally stabilize the promyelining stage, are not properly downregulated in the presence of early SC transcription factors (such as SCIP). However, there are many more SCs arrested at the promyelin stage (>99%) than SCs with immunodetectable SCIP (<20%; data not shown), suggesting that abnormal SCIP expression is a consequence rather than a cause of the continued arrest at the promyelining stage.
An important question is the relationship between the myelin pathology after PMP22 overexpression and the myelin pathology in autosomal-dominant mutants of this gene. In two reports, identical point mutations of the PMP22 gene were identified in mouse and man (Valentijn et al., 1992; Ionasescu et al., 1997). Since gene targeting studies have provided definite evidence that both Trembler and Trembler-J mice behave differently from mutants with a null allele (Adlkofer et al., 1995, 1997a,b), it has become clear that altered PMP22 protein products exert aberrant “gain-of-function” effects. However, follow-up studies using in vitro and ex vivo preparations have revealed an unexpected difference between the two natural mutants. In Trembler, the substitution G150D in the last transmembrane domain causes a transportation arrest of PMP22 in the ER (Naef et al., 1997; D'Urso et al., 1998), a situation reminiscent of the dysmyelinating mutations in the proteolipid protein/DM20 gene (Gow et al., 1994; Jung et al., 1996). In contrast, the substitution of Trembler-J (L16P) appears compatible with normal PMP22 transport to the intermediate compartment between ER and Golgi (Notterpek et al., 1997; Tobler et al., 1999). The present study assigns a similar gain-of-function effect to wild-type PMP22 when overexpressed.
Different cellular defects can lead into the same pathway of disease expression by SCs (Martini and Schachner, 1997). Onion bulb formation in the CMT rat is a prominent but unspecific sign of an SC derailment, because they are also seen in other myelin gene mutants, such as the P0- and Cx32-deficient mice (Giese et al., 1992; Martini et al., 1995;Anzini et al., 1997). Thus, in CMT1A, the question arises what aspects, if any, of the overexpression phenotype reflect the underlying PMP22 function. In this sense, the early defect of homozygous PMP22 transgenic rats is more informative than the CMT rat, because it lacks many unspecific features common to CMT1 diseases. Future studies are warranted to determine the kinetics of myelin protein synthesis and to determine the myelin assembly rate as a function of PMP22 gene dosage in this animal model.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant SFB317, the European Community Biomed-2 Program (to K.A.N.), Action Research (to I.G.), and a stipend of the DFG Graduiertenprogramm “Molekulare und Zelluläre Neurobiologie” (to S.N. and M.S.). We thank S. Scherer for helpful comments on this manuscript and H. Krischke, C. Zgraggen, and M. McCulloch for excellent technical assistance. Antibodies were kindly provided by J. Archelos (P0) and G. Lemke (SCIP). We also thank D. Cobay, M. Jung, M. Klugmann, M. Rossner, and M. H. Schwab for helpful discussions.
Correspondence should be addressed to Dr. Klaus-Armin Nave at his present address: Department of Neurogenetics, Max-Planck-Institute for Experimental Medicine, Hermann-Rein-Strasse 3, D-37075 Goettingen, Germany. E-mail: nave@sun0.urz.uni-heidelberg.de.
Dr. Sereda's present address: Department of Neurogenetics, Max-Planck-Institute for Experimental Medicine, D-37075 Göttingen, Germany.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in PMP-22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U. Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating tomaculous neuropathy. J Neurosci. 1997a;17:4662–4671. doi: 10.1523/JNEUROSCI.17-12-04662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adlkofer K, Naef R, Suter U. Analysis of compound heterozygous mice reveals that the Trembler mutation can behave as a gain-of-function allele. J Neurosci Res. 1997b;49:671–680. doi: 10.1002/(SICI)1097-4547(19970915)49:6<671::AID-JNR2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archelos JJ, Roggenbuck K, Schneider-Schaulies J, Linington C, Toyka KV, Hartung HP. Production and characterization of monoclonal antibodies to the extracellular domain of P0. J Neurosci Res. 1993;35:46–53. doi: 10.1002/jnr.490350107. [DOI] [PubMed] [Google Scholar]
- 6.Baechner D, Liehr T, Hameister H, Altenberger H, Grehl H, Suter U, Rautenstrauss B. Widespread expression of the peripheral myelin protein-22 gene (PMP22) in the neural and non-neural tissues during murine development. J Neurosci Res. 1995;42:735–741. doi: 10.1002/jnr.490420602. [DOI] [PubMed] [Google Scholar]
- 7.Bermingham JR, Scherer SS, O'Connell S, Arroyo E, Kalla KA, Powell FL, Rosenfeld MG. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 1996;10:1751–1762. doi: 10.1101/gad.10.14.1751. [DOI] [PubMed] [Google Scholar]
- 8.Brancolini C, Marzinotto S, Edomi P, Agostini E, Fiorentini C, Müller HW, Scheider C. Rho-dependent regulation of cell spreading by the tetraspan membrane protein Gas3/PMP22. Mol Biol Cell. 1999;10:2441–2459. doi: 10.1091/mbc.10.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavrier P, Zerial M, Lemaire P, Almendral J, Bravo R, Charnay P. A gene encoding a protein with zinc fingers is activated during G(0)/G(1) transition in cultured cells. EMBO J. 1988;7:29–35. doi: 10.1002/j.1460-2075.1988.tb02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Coetzee T, Suzuki K, Popko B. New perspectives on the function of myelin galactolipids. Trends Neurosci. 1998;21:126–130. doi: 10.1016/s0166-2236(97)01178-8. [DOI] [PubMed] [Google Scholar]
- 12.D'Urso D, Prior R, Greiner-Petter R, Gabreels-Festen AA, Müller HW. Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci. 1998;18:731–740. doi: 10.1523/JNEUROSCI.18-02-00731.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wulf P, Bernhardt RR, Suter U. Characterization of peripheral myelin protein 22 in zebrafish (zPMP22) suggests an early role in the development of the peripheral nervous system. J Neurosci Res. 1999;57:467–478. [PubMed] [Google Scholar]
- 14.Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF. Peripheral Neuropathy, Vol 2. Saunders; Philadelphia: 1993. [Google Scholar]
- 15.Fabbretti E, Edomi P, Brancolini C, Schneider C. Apoptotic phenotype induced by overexpression of wild-type gas3/PMP22: its relation to the demyelinating peripheral neuropathy CMT1A. Genes Dev. 1995;9:1846–1856. doi: 10.1101/gad.9.15.1846. [DOI] [PubMed] [Google Scholar]
- 16.Friede RL, Samorajski T. Myelin formation in the sciatic nerve of the rat: a quantitative electron microscopic, histochemical and autoradiographic study. J Neuropath Exp Neurol. 1968;27:546–570. [PubMed] [Google Scholar]
- 17.Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Disruption of the P0 gene in mice leads to abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]
- 18.Gow A, Friedrich VL, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- 19.Hagedorn L, Suter U, Sommer L. P0 and PMP22 mark a multipotent neural crest-derived cell type that displays community effects in response to TGF-beta family factors. Development. 1999;126:3781–3794. doi: 10.1242/dev.126.17.3781. [DOI] [PubMed] [Google Scholar]
- 20.Hanemann CO, Müller HW. Pathogenesis of Charcot-Marie-Tooth 1A (CMT1A) neuropathy. Trends Neurosci. 1998;21:282–286. doi: 10.1016/s0166-2236(97)01222-8. [DOI] [PubMed] [Google Scholar]
- 21.Hanemann CO, Gabreels-Festen AA, Müller HW, Stoll G. Low affinity NGF receptor expression in CMT1A nerve biopsies of different disease stages. Brain. 1996;119:1461–1469. doi: 10.1093/brain/119.5.1461. [DOI] [PubMed] [Google Scholar]
- 22.Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, Fontes M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5:563–569. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- 23.Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, Saberan-Djoniedi D, Figarella-Branger D, Pellissier JF, Thomas PK, Fontes M. Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. 1998;7:449–458. doi: 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- 24.Ionasescu VV, Searby CC, Ionasescu R, Chatkupt S, Patel N, Koenigsberger R. Dejerine-Sottas neuropathy in mother and son with same point mutation of PMP22 gene. Muscle Nerve. 1997;20:97–99. doi: 10.1002/(sici)1097-4598(199701)20:1<97::aid-mus13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 25.Jaegle M, Mandermakers W, Broos L, Zwart R, Karis A, Visser P, Grosveld F, Meijer D. The POU factor Oct-6 and Schwann cell differentiation. Science. 1996;273:507–510. doi: 10.1126/science.273.5274.507. [DOI] [PubMed] [Google Scholar]
- 26.Jung M, Sommer I, Schachner M, Nave K-A. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J Neurosci. 1996;16:7920–7929. doi: 10.1523/JNEUROSCI.16-24-07920.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 28.Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 1985;40:501–508. doi: 10.1016/0092-8674(85)90198-9. [DOI] [PubMed] [Google Scholar]
- 29.Lemke G, Chao M. Axons regulate SC expression of the major myelin and NGF receptor genes. Development. 1988;102:499–504. doi: 10.1242/dev.102.3.499. [DOI] [PubMed] [Google Scholar]
- 30.Lupski JR, de Oca Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 31.Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U. Impaired differentiation of SCs in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16:5351–5360. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manfioletti G, Ruaro ME, Del Sal G, Philipson L, Schneider C. A growth arrest-specific (gas) gene codes for a membrane protein. Mol Cell Biol. 1990;10:2924–2930. doi: 10.1128/mcb.10.6.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martini R, Schachner M. Molecular bases of myelin formation as revealed by investigations on mice deficient in glial cell surface molecules. Glia. 1997;19:298–310. [PubMed] [Google Scholar]
- 34.Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathie. Nat Genet. 1995;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- 35.Mirsky R, Jessen KR. Schwann cell development, differentiation and myelination. Curr Opin Neurobiol. 1996;6:89–96. doi: 10.1016/s0959-4388(96)80013-4. [DOI] [PubMed] [Google Scholar]
- 36.Monuki ES, Weinmaster G, Kuhn R, Lemke G. SCIP: a glial cell POU domain gene regulated by cyclic AMP. Neuron. 1989;3:783–793. doi: 10.1016/0896-6273(89)90247-x. [DOI] [PubMed] [Google Scholar]
- 37.Monuki ES, Kuhn R, Weinmaster G, Trapp BD, Lemke G. Expression and activity of the POU transcription factor SCIP. Science. 1990;249:1300–1303. doi: 10.1126/science.1975954. [DOI] [PubMed] [Google Scholar]
- 38.Naef R, Suter U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc Res Tech. 1998;41:359–371. doi: 10.1002/(SICI)1097-0029(19980601)41:5<359::AID-JEMT3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Naef R, Suter U. Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol Disease. 1999;6:1–14. doi: 10.1006/nbdi.1998.0227. [DOI] [PubMed] [Google Scholar]
- 40.Naef R, Adlkofer K, Lescher B, Suter U. Aberrant protein trafficking in trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol Cell Neurosci. 1997;9:13–25. doi: 10.1006/mcne.1997.0604. [DOI] [PubMed] [Google Scholar]
- 41.Nave K-A. Neurological mouse mutants: a molecular genetic analysis of myelin proteins. In: Kettenmann H, Ransom B, editors. Neuroglial cells. Oxford UP; New York: 1995. pp. 571–586. [Google Scholar]
- 42.Notterpek L, Shooter EM, Snipes GJ. Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parmantier E, Cabon F, Braun C, D'Urso D, Mueller HW, Zalc B. Peripheral myelin protein-22 is expressed in rat and mouse brain and spinal cord motoneurons. Eur J Neurosci. 1995;7:1080–1088. doi: 10.1111/j.1460-9568.1995.tb01095.x. [DOI] [PubMed] [Google Scholar]
- 44.Patel PI, Roa B, Welcher AA, Schoener-Scott R, Trask B, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 45.Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 46.Roa BB, Dyck PJ, Marks HG, Chance PF, Lupski JR. Dejerine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene. Nat Genet. 1993;5:269–273. doi: 10.1038/ng1193-269. [DOI] [PubMed] [Google Scholar]
- 47.Roach A, Boylan KB, Horvath S, Prusiner SB, Hood LE. Characterization of cloned cDNA representing rat myelin basic protein: absence of expression in brain of shiverer mutant mice. Cell. 1983;34:799–806. doi: 10.1016/0092-8674(83)90536-6. [DOI] [PubMed] [Google Scholar]
- 48.Sancho S, Magyar JP, Aguzzi A, Suter U. Distal axonopathy in peripheral nerves of PMP22 mutant mice. Brain. 1999;122:1563–1577. doi: 10.1093/brain/122.8.1563. [DOI] [PubMed] [Google Scholar]
- 49.Sereda M, Griffiths I, Pühlhofer A, Stewart H, Rossner MJ, Zimmermann F, Magyar JP, Schneider A, Hund E, Meinck H-M, Suter U, Nave K-A. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 50.Snipes GJ, Suter U. Molecular basis of common hereditary motor and sensory neuropathies in humans and in mouse models. Brain Pathol. 1995;5:233–247. doi: 10.1111/j.1750-3639.1995.tb00600.x. [DOI] [PubMed] [Google Scholar]
- 51.Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol. 1992;117:225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sommer L, Suter U. The glycoprotein P0 in early peripheral gliogenesis. Cell Tissue Res. 1998;292:11–16. doi: 10.1007/s004410051029. [DOI] [PubMed] [Google Scholar]
- 53.Spreyer P, Kuhn G, Hanemann CO, Gillen C, Schaal H, Kuhn R, Lemke G, Müller HW. Axon-regulated expression of a Schwann cell transcript that is homologous to a “growth arrest-specific” gene. EMBO J. 1991;10:3661–3668. doi: 10.1002/j.1460-2075.1991.tb04933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoffel W, Bosio A. Myelin glycolipids and their functions. Curr Opin Neurobiol. 1997;7:654–661. doi: 10.1016/s0959-4388(97)80085-2. [DOI] [PubMed] [Google Scholar]
- 55.Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA. 1992a;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992b;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 57.Tobler A, Notterpek L, Naef R, Taylor R, Suter U, Shooter EM. Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J Neurosci. 1999;19:2027–2036. doi: 10.1523/JNEUROSCI.19-06-02027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi ABY, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 59.Tso JY, Sun XH, Kao TH, Reece KS, Wu R. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNA: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 1985;13:2485–2502. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, van den Bosch NH, Zorn I, Gabreels-Festen AW, de Visser M, Bolhuis PA. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 61.Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, Lupski JR. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;4:382–384. doi: 10.1038/ng0498-382. [DOI] [PubMed] [Google Scholar]
- 62.Webster H. Development of peripheral nerve fibres In: Peripheral neuropathy, Ed 3 (Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, eds), pp 243–266. Saunders; Philadelphia: 1993. [Google Scholar]
- 63.Welcher AA, Suter U, De Leon M, Snipes GJ, Shooter EM. A myelin protein is encoded by the homologue of a growth arrest-specific gene. Proc Natl Acad Sci USA. 1991;88:7195–7199. doi: 10.1073/pnas.88.16.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zoidl G, Blass-Kampmann S, D'Urso D, Schmalenbach C, Müller HW. Retroviral-mediated gene transfer of the peripheral myelin protein PMP22 in SCs: modulation of cell growth. EMBO J. 1995;14:1122–1128. doi: 10.1002/j.1460-2075.1995.tb07095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zorick TS, Lemke G. SC differentiation. Curr Opin Cell Biol. 1996;8:870–876. doi: 10.1016/s0955-0674(96)80090-1. [DOI] [PubMed] [Google Scholar]
- 66.Zorick TS, Syroid DE, Arroyo E, Scherer SS, Lemke G. The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in SC differentiation. Mol Cell Neurosci. 1996;8:129–145. doi: 10.1006/mcne.1996.0052. [DOI] [PubMed] [Google Scholar]