

Abstract
The lateral amygdala (LA) is thought to be critical for the specific acquisition of conditioned fear, and the emotionally charged memories related to fear are thought to require a form of synaptic plasticity related to long-term potentiation (LTP). Is LTP in the lateral amygdala enduring, and, if so, does it require gene expression and the synthesis of new protein? Using brain slices, we have examined the molecular-signaling pathway of LTP in the cortico-amygdala and the thalamo-amygdala pathways. We find that a single high-frequency train of stimuli induces a transient LTP (E-LTP); by contrast, five repeated high-frequency trains induce an enduring late phase of LTP (L-LTP), which is dependent on gene expression and on new protein synthesis. In both pathways the late phase of LTP is mediated by protein kinase A (PKA) and mitogen-activated protein kinase (MAPK). Application of the adenylyl cyclase activator forskolin induced L-LTP in both pathways, and this potentiation is blocked by inhibitors of protein synthesis. The late phase of LTP also is modulated importantly by β-adrenergic agonists. An inhibitor of β-adrenergic receptors blocks L-LTP; conversely, application of a β-adrenergic agonist induces the L-LTP. Immunocytochemical studies show that both repeated tetanization and application of forskolin stimulate the phosphorylation of cAMP response element-binding proteins (CREB) in cells of the lateral nucleus of the amygdala. These results suggest that PKA and MAPK are critical for the expression of a persistent phase of LTP in the lateral amygdala and that this late component requires the synthesis of new protein and mRNA.
Keywords: amygdala, L-LTP, PKA, MAPK, protein synthesis, CREB
One of the major insights of modern cognitive neuroscience is that memory is not unitary but has at least two distinct forms: a procedural (implicit) memory, which stores information about emotional states and about perceptual and motor strategies, and a declarative (explicit) memory, which stores information about facts and events (Milner et al., 1998). Studies of implicit memory for the acquisition of fear, such as behavioral sensitization and classical conditioning in the invertebratesAplysia and Drosophila, reveal that both behavioral long-term memory and its neural representation require a cascade of gene expression that is triggered by cAMP response element-binding proteins (CREB) and that leads to the growth of new synaptic connections (Yin and Tully, 1996; Bailey et al., 1998; Milner et al., 1998). This cascade of gene activation is initiated by the joint activation of protein kinase A (PKA) and mitogen-activated protein kinase (MAPK) (Martin et al., 1997; Ghirardi et al., 1992;Impey et al., 1998a). These results from invertebrates raise the question of whether PKA and MAPK also are involved in the implicit memory of fear in mammals.
One of the best-studied forms of implicit memory of fear in mammals is cued conditioning, a form of fear conditioning that requires, for its expression, the lateral amygdala. This form of fear conditioning is produced by the pairing of a neutral tone as a conditioning stimulus (CS) with a shock as an unconditioning stimulus (US). The lateral amygdala is thought to be a cellular site for the convergence of the tone CS and the shock US (LeDoux, 1995; Maren and Fanselow, 1996). This convergence of the CS and US is thought to induce fear conditioning by increasing the synaptic strength of the CS pathway within the lateral nucleus by a long-term potentiation-like (LTP-like) mechanism (Rogan et al., 1995, 1997; McKenan and Schinnick-Gallagher, 1997).
One of the characteristic features of conditioned fear is persistence, yet so far the only aspects of LTP that have been examined in the lateral amygdala are transient. This raised the following questions: Does LTP in the lateral amygdala have an enduring phase (>3 hr) (Y.Huang et al., 1996)? If so, does it require the expression of genes and the synthesis of new protein as does the behavioral conditioned fear? Does LTP in the lateral nucleus lead to the activation of CREB? (Impey et al., 1998a; Schafe et al., 1999).
Previously, we have described some of the signaling mechanisms contributing to the induction and expression of the early phase of LTP (E-LTP) in the amygdala (Huang and Kandel, 1998). In this paper we have extended this analysis and focused on the signaling pathway for the enduring form of LTP (L-LTP). We have done so in the pathways to the lateral nucleus from the auditory cortex and from the auditory thalamus. Each of these two pathways is thought to be required for memory storage related to fear (LeDoux, 1995; Davis, 1997; Quirk et al., 1997).
MATERIALS AND METHODS
Sprague Dawley rats (4–5 weeks old) were decapitated. The whole brain was isolated and placed in ice-cold artificial CSF (aCSF); a block containing the amygdala was taken. Transverse slices (400 μm) were cut and transferred to an interface chamber. The slices were perfused constantly with ACSF at a rate of 2 ml/min and bubbled with 95% O2/5% CO2. The composition of ACSF was as follows (in mm): 124 NaCl, 1.3 MgSO4, 4 KCl, 1.0 Na2HPO4, 2.0 CaCl, 26 NaHCO3, and 10 d-glucose. In most of the experiments picrotoxin (10 μm) was present in the perfusion solution. The temperature of the slices was maintained at 29°C. Experiments were started at least 3 hr after dissection. Extracellular recordings were made by a tungsten electrode (3MEG, A-M Systems, Everett, WA). Stimuli were delivered through bipolar stainless steel electrodes (Microprobe). To record field potentials in the cortico-amygdala pathway, we placed the stimulating electrode in the external capsule, which contained fibers from the auditory cortex to the lateral amygdala. To record field potentials in the thalamo-amygdala pathway, we placed the stimulating electrode in the thalamic afferent fiber to the lateral amygdala, which is located in the ventral part of the striatum just above the central nucleus of the amygdala (see Fig. 1A). The test stimuli for basal synaptic response was 0.017 Hz (0.05 msec pulse duration). For the baseline field potential recording 50% of the maximum amplitude was used. LTP was elicited by one or five trains of tetanus (100 Hz, 1 sec at 3 min interval) with the same intensity and pulse duration as the test stimuli.
Fig. 1.

Monosynaptic field potential recording in the cortico-amygdala pathway. A, Schematic illustration of a coronal brain slice containing the amygdala. For the cortico-amygdala pathway a stimulating electrode was placed in the external capsule (S1); to stimulate the thalamo-amygdala pathway, we placed a stimulating electrode in the fibers from the internal capsule (S2). Field recordings were made from the lateral amygdala (R).B, The synaptic potential is blocked reversibly by kynurenic acid (KYN; 10 mm).Left, Before; middle, during KYN;right, 90 min after KYN was washed out.C, The synaptic potential recorded during a 50 Hz tetanus. The synaptic response followed the tetanus in a one-for-one manner without failure. D, LTD and LTP in the amygdala induced by different frequencies of stimulations. For 1–20 Hz, 900 pulses were applied; for 100 Hz, 100 pulses were applied. The changes in synaptic response were measured by the amplitude of field potential 15 min after the conditioning stimulus. E, E-LTP is induced by a single train of tetanus (100 Hz, 1 sec, indicated by thearrow). The LTP decayed to baseline in ∼40 min (n = 7; mean ± SEM). F, L-LTP is induced by five trains of tetanus (100 Hz, 1 sec at 3 min interval). The LTP is enduring and lasts at least 3 hr (n = 6; mean ± SEM).
For bath application the following drugs were made and stored as concentrated stock solutions and diluted 1000-fold when applied to the perfusate. The concentration of stock solution included 1 mm KT5720 (dissolved in DMSO; Biomol, Plymouth Meeting, PA), 50 mm forskolin, and 50 mm1.9-dideoxyforskolin (dissolved in DMSO; Sigma, St. Louis, MO); 20–40 mm anisomycin (Sigma); 40 mm actinomycin D (dissolved in DMSO; Sigma); 15 mm isoproterenol [Research Biochemicals (RBI), Natick, MA]; 1 mm propranolol (RBI); 10 mm timolol (RBI); and 20–50 mm PD98059 (dissolved in DMSO; Biomol). Emetine (100 mm dissolved in DMSO; Sigma), 100 mm DRB (dissolved in DMSO; IGN), and 10 mm kynurenic acid (Kyn; RBI) were dissolved directly in the perfusion solution. Because all drugs dissolved in DMSO were diluted 1000-fold when they were applied to the perfusion solution, the final concentration of DMSO was 0.1%. For immunocytochemistry studies the brain slices containing amygdala were maintained at 31°C, and the physiological and pharmacological treatment was applied 90 min after slice dissection. Then the slices were fixed in 4% paraformaldehyde in PBS for 1 hr at room temperature and permeabilized with 0.5% Triton X-100 for 1 hr at room temperature; free aldehydes were quenched with 50 μm NH4CL for 20 min. To block nonspecific staining, we treated the slices with 10% goat serum for 1 hr. Then the slices were incubated with rabbit anti-phospho-CREB antibody (diluted 1:100 in 10% goat serum/PBS; Upstate Biotechnology, Lake Placid, NY) for 24 hr at 4°C. After extensive washing in PBS, the slices were incubated in Cy3 goat anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) diluted 1:200 in 10% goat serum/PBS. Images were taken on a Bio-Rad confocal microscope (Richmond, CA) mounted on a Zeiss Axiovert microscope (Oberkochen, Germany) with 5 and 40× objectives. Optical sections were taken at depth of ∼100 μm into the slices, using identical settings of the laser light. z-Series with 3.78 μm of each step were collected and projected. The number of images is 10. For quantification of immunocytochemistry the averaged pixel intensity of a constant area was measured in the desired region of each slice. The amygdala region was located anatomically in its relation to the rhinal fissure, caudate putamen, external capsule, etc. at the section level of bregma −2.8 (Paxinos and Watson, 1986).
RESULTS
E-LTP and L-LTP in the pathway from the auditory cortex to the lateral amygdala
Orthodromic stimuli were applied to the external capsule, which carries axons from the auditory cortex to the amygdala. These stimuli elicited a simple negative field potential that has a constant latency of ∼3 msec and a duration of 5–15 msec. This field potential is blocked reversibly by glutamate antagonists (kynurenic acid, Kyn), consistent with glutamate being the transmitter at this synapse and consistent with the field potential reflecting glutaminergic synaptic currents (Fig. 1B). Indeed, we have found previously that the intracellularly recorded EPSPs have properties similar to those of the field potential (Huang and Kandel, 1998). As is the case with the EPSP, the field potential appears to reflect the activity of a monosynaptic connection from the auditory cortex and can follow presynaptic stimulation reliably without failure even at a frequency of 50 Hz (Fig. 1C). Consistent with its being monosynaptic, the field potential is sensitive to stimulation frequencies that increase or decrease transmitter release and thus shows LTP facilitation and depression [long-term synaptic depression (LTD)] as a function of frequency of presynaptic stimulation in the range of 1–100 Hz (Fig. 1D).
In each of the three major pathways of the hippocampus—the perforant, mossy fiber, and Schaffer collateral pathways—two temporally and mechanistically distinct forms of LTP have been delineated by simply varying the number of stimulus trains. One 100 Hz train elicits a short-lasting form of LTP lasting up to 3 hr, whereas three or more trains elicit a long-lasting form of LTP that endures up to 28 hr (Y.Huang et al., 1996; S. Patterson, D. Winder, and E. Kandel, personal communication). We have found similarly that it is possible to induce two forms of LTP in the lateral nucleus of the amygdala. A single train (100 Hz, 1 sec) induces a transient potentiation that decays to baseline within 40 min (Fig. 1E). By contrast, five such trains spaced 3 min apart induce an enduring LTP that lasts stably for at least 3 hr (Fig. 1F).
The expression of L-LTP in the lateral nucleus of the amygdala requires new protein synthesis and transcription
Previous studies of the perforant, the mossy fiber, and the Schaffer collateral pathways demonstrated that in each case the enduring late phase of LTP (L-LTP) requires the synthesis of new protein (Y. Huang et al., 1995, 1996). To determine whether such a requirement also exists for L-LTP in the amygdala, we perfused into the slices of the amygdala an inhibitor of protein synthesis (anisomycin, 20 μm) 15 min before and 60–90 min after tetanus. We found that the inhibitor had no effect on E-LTP produced by a single train (Fig.2A), but profoundly inhibited L-LTP produced by five trains (Fig. 2B). The inhibition was surprisingly rapid in its onset and was evident soon after the last tetanus. Thus, the inhibition of L-LTP in the lateral nucleus of the amygdala was even more dramatic than the inhibition of L-LTP by anisomycin in the hippocampus (Y. Huang et al., 1996). By 30 min after the tetanus, anisomycin had reduced L-LTP to 112 ± 8% of the control level (n = 7), and by 1 hr it had reduced it to the control level. Thus 3 hr after tetanus synaptic strength had returned to 101 ± 6% of baseline in anisomycin-treated slices (n = 7). This reduction is significantly different from control experimental slices not treated with anisomycin, which were 146 ± 8% at 30 min and 162 ± 14% (Fig. 2B) at 3 hr after the tetanus (n = 5; p < 0.005; Student'st test) in each case. To exclude the possibility that the rapid decay of LTP produced by anisomycin is attributable to a nonspecific effect of the drug that is unrelated to inhibiting protein synthesis, we also used emetine, another inhibitor of protein synthesis (Nguyen et al., 1994; Nguyen and Kandel, 1996). We again found that exposure to 100 μm emetine (for 30–50 min before and 60 min after tetanus) also inhibited E-LTP but with somewhat slower kinetics. In the presence of emetine LTP was 129 ± 9% (n = 7) at 1 hr after tetanus, which was smaller but not significantly different from control experiments (146 ± 8%;n = 5; p > 0.05). However, by 3 hr the LTP had decayed to 106 ± 8% at 3 hr (n = 6), which was significantly different from control L-LTP at 3 hr (p < 0.001; Fig. 2B). A control perfusion of 100 μm emetine for 2 hr had no effect on basal synaptic transmission (Fig.2D).
Fig. 2.
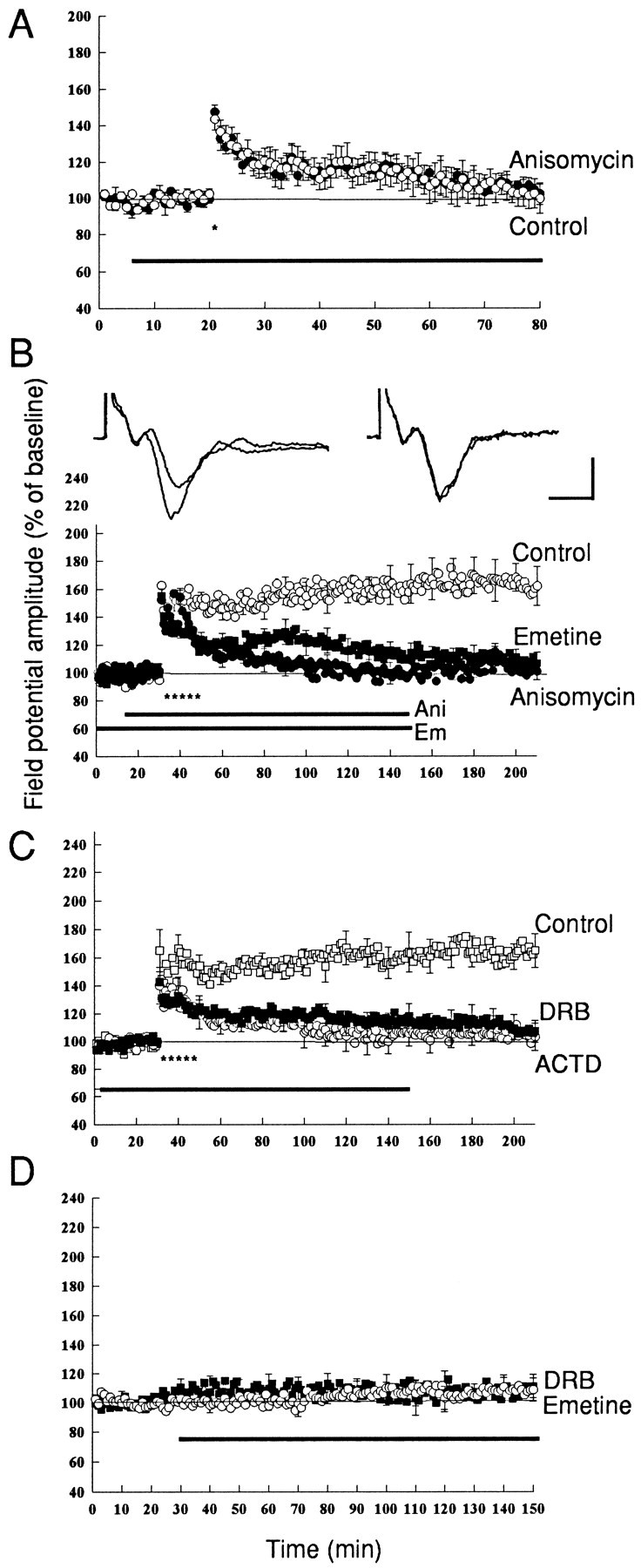
Inhibitors of protein and mRNA synthesis block L-LTP in the amygdala. A, Anisomycin has no effect on E-LTP induced by a single tetanus (100 Hz, 1 sec, indicated by theasterisk). Open circles, Control experiments (n = 5); filled circles, LTP in the presence of anisomycin (20 μm; n = 6). B, Anisomycin blocks L-LTP induced by five train of tetanus. Anisomycin (20 μm) or emetine (100 μm) was applied before the tetanus and perfused for 60–90 min. Open circles, Control experiments (n = 5); filled circles, LTP in the slices treated with anisomycin (n = 6);filled squares, LTP in slices treated with emetine (n = 7). Representative field potentials before and 3 hr after tetanus in control (left) and in anisomycin-treated slices (right) are shown at the top of this panel. Calibration: 5 msec, 0.5 mV. C, Transcriptional inhibitors block L-LTP. ACTD (40 μm) or DRB (100 μm) was applied 30–60 min before the tetanus and perfused for 2 hr. LTP decayed to the baseline in ∼90 min after tetanus in the presence of ACTD (open circles;n = 6) and DRB (filled squares; n = 7). D, Emetine (100 μm, open circles;n = 4) and DRB (100 μm, filled squares; n = 4) have no effect on the baseline synaptic response.
We next asked: Does L-LTP in the lateral amygdala also require gene transcription? To inhibit transcription, we exposed slices to actinomycin D (ACTD; 40 μm) for 30 min before the tetanus and found that it also caused a depression of L-LTP. At 3 hr after tetanus L-LTP was 106 ± 10% of control, which was significantly different from 165 ± 12% (p < 0.005) recorded in control experiments without ACTD (Fig. 2C). To exclude the nonspecific effect of ACTD on LTP, we also used another inhibitor of mRNA synthesis, 5,6-dichloro-1-β-d-ribofuranosyl benzimidazole (DRB; Nguyen et al., 1994; Nguyen and Kandel, 1996). Similarly to ACTD, LTP was reduced in the presence of DRB (100 μm) to 107 ± 8% of control at 3 hr. This again was significantly different from the control experiment without DRB (Fig. 2C) (n = 7;p < 0.005). A control perfusion of DRB (100 μm) for 2 hr has no effect on basal synaptic transmission (Fig. 2D).
The expression of L-LTP in amygdala requires both PKA and MAPK
Studies in Aplysia and Drosophila suggest that the molecular switch required to convert short- to long-term synaptic facilitation and short- to long-term memory involves cAMP-mediated activation of CREB-1 and relief from repression of CREB-2 (Bartsch et al., 1995; Yin and Tully, 1996; Abel et al., 1998; Milner et al., 1998). Studies of long-term facilitation in Aplysiafurther indicate that PKA recruits MAPK and that both translocate to the nucleus where their coordinated action is required for the activation of CREB-1 (Martin et al., 1997). In hippocampal neurons MAPK activation also is thought to be necessary to couple PKA to CREB phosphorylation (Roberson et al., 1999). Is the expression of L-LTP in amygdala also dependent on PKA? If so, is new protein synthesis in the amygdala during L-LTP also mediated by a cAMP/PKA signaling pathway and does it also recruit MAP kinase?
We first tested the requirement for PKA for the late phase of LTP by inhibiting PKA. We found that perfusion of KT5720 (1 μm), an inhibitor of PKA, caused a rapid blockade of LTP so that at 1 hr LTP was only 110 ± 9% (n = 6), which was significantly different from 150 ± 8% in the controls not exposed to the inhibitor (p < 0.01;n = 6) (Fig.3A). In the presence of the PKA inhibitor LTP relaxed to baseline in ∼90 min so that 3 hr after tetanus the LTP was 101 ± 8%, which was significantly different from control (160 ± 14%) at 3 hr (p < 0.01). Conversely, the adenylate cyclase activator forskolin induced a long-lasting potentiation that occluded both E- and L-LTP (Huang and Kandel, 1998; Wang et al., 1999). We therefore next tested the effect of anisomycin on the potentiation produced by forskolin and again found that anisomycin completely blocked the forskolin-induced potentiation. In the presence of anisomycin the potentiation induced by forskolin was reduced at 1 hr from a control of 170 ± 7% (n = 6) to 103 ± 2% (n = 6; p < 0.001) and was reduced to only 111 ± 13% at 3 hr after the application of forskolin as compared with 192 ± 10% in control experiments (Fig. 3B). These results suggest that the activation of cAMP signaling pathway in the lateral amygdala induces new protein synthesis.
Fig. 3.
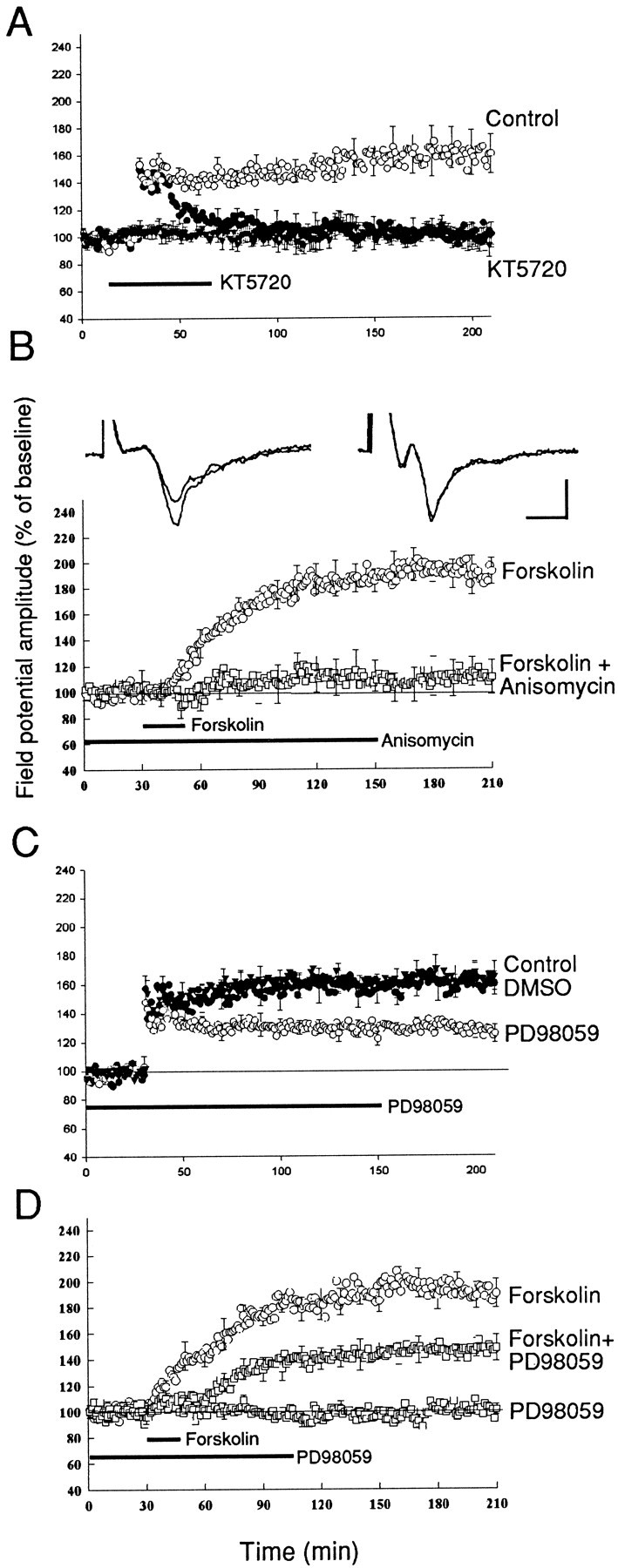
PKA and MAPK mediate the protein synthesis-dependent component of L-LTP. A, PKA inhibitor blocks L-LTP. In the presence of KT5720 (1 μm) LTP induced by five trains of tetanus (100 Hz, 1 sec at 3 min interval) was depressed shortly after tetanus and decayed to the baseline in 60–90 min (filled circles; n = 6). KT5720 (1 μm) has no effect on the baseline synaptic response (filled triangles; n= 5). B, Forskolin-induced synaptic potentiation is blocked by anisomycin. A brief application of forskolin (50 μm) induced a large potentiation (open circles; n = 6). Anisomycin (20 μm) was perfused 30–60 min before the application of forskolin and then perfused for 2 hr. Forskolin failed to induce any significant potentiation in the presence of anisomycin (open squares; n = 6). Representative field potentials before and 3 hr after forskolin application in forskolin alone (left) and forskolin with anisomycin (right) are shown at the topof this panel. Calibration: 5 msec, 0.5 mV. C, MAPK inhibitor blocks L-LTP. PD98059 (20 μm) was applied 30–60 min before tetanus and perfused for 2 hr. L-LTP was significantly depressed in the slices that were treated with PD98059 (open circles; n = 6) as compared with the control experiments (filled circles;n = 6). The application of 0.1% DMSO had no effect on amygdala L-LTP (filled triangles;n = 4). D, Forskolin-induced synaptic potentiation was depressed by a MAPK inhibitor. PD98059 (50 μm) was applied 30–60 min before the application of forskolin and perfused for 90 min. Forskolin induced only a weak synaptic potential in the presence of PD98059 (n = 6). The application of PD98059 (50 μm) had no effect on the baseline synaptic response (n = 5).
In Aplysia, in Drosophila, and in the mammalian hippocampus CREB-regulated transcription is important for long-term memory. During long-term facilitation in Aplysia and during L-LTP in the hippocampal CA1 region the late phase of long-term synaptic facilitation is associated with increased phosphorylation of CREB. Using an antibody that recognizes the phosphorylated form of CREB (phospho-CREB), we examined CREB phosphorylation during the late phase of amygdala LTP in two ways. First, we perfused the slices with 50 μm forskolin for 15 min and fixed them 60 min after the application of forskolin. Here we found an increase in phospho-CREB immunoreactivity in the lateral amygdala as compared with a vehicle (0.1% DMSO) control. A representative comparison and a quantification of the average changes of phospho-CREB immunoreactivity are shown on Figure 4.
Fig. 4.
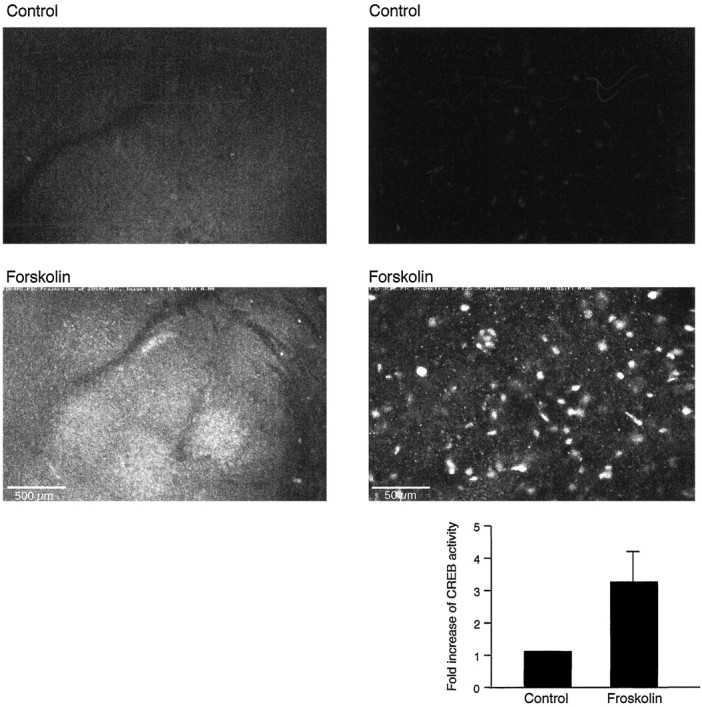
Forskolin stimulates CREB phosphorylation in the amygdala. Forskolin (50 μm) or vehicle (0.1% DMSO) was applied 90 min after slice dissection and perfused for 15 min; the slices were fixed 60 min after forskolin application. An increase in phospho-CREB immunoreactivity in the lateral amygdala was obtained in forskolin-treated slices (n = 8; bottom panels) as compared with the vehicle-treated slices (n = 8; top panels).Left, Lower magnification. Scale bar, 500 μm.Right, Higher magnification. Scale bar, 50 μm.
Second, we applied five tetanic trains (100 Hz at 3 min interval) to induce L-LTP and used low-frequency stimulation as a control (0.016 Hz). We then fixed the slices 10–15 min after either the last tetanus or at end of the low frequency of stimulation. We again found that there were clear differences in phospho-CREB immunoreactivity between the tetanic and nontetanic treatment (Fig.5). The increased phosphorylation of CREB by forskolin and by five tetani indicates that L-LTP in amygdala is associated with the activation of CREB.
Fig. 5.
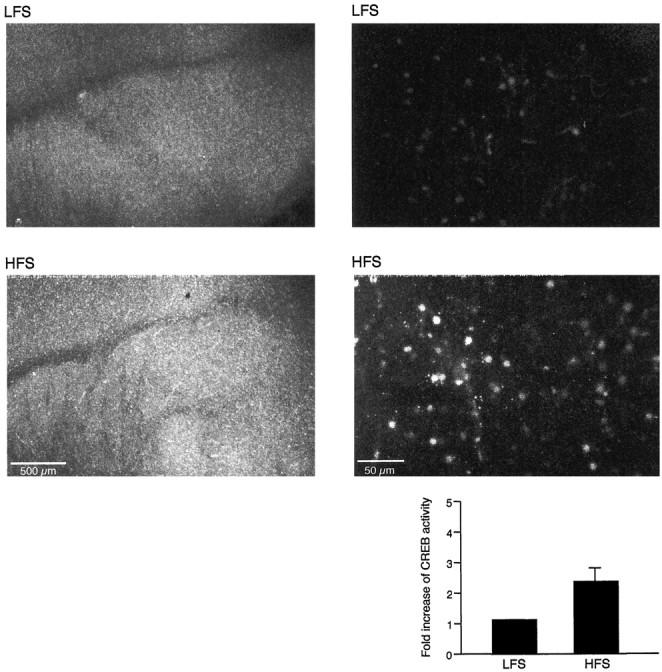
High-frequency stimulation stimulates CREB phosphorylation in the amygdala. High-frequency stimulation (HFS; five trains of tetanus, 100 Hz, at 3 min interval) or low-frequency (LFS; 0.016 Hz) stimulation was applied 90 min after slice dissection; the slice was fixed 10–15 min later. An increase in phospho-CREB immunoreactivity in the lateral amygdala was obtained in HFS-stimulated slices (n = 6;bottom panels) in both high-magnification (right) and low-magnification (left) images as compared with low frequency (n = 6;top panels). Scale bars: right, 50 μm;left, 500 μm.
In Aplysia and in rodent hippocampus the increase in intracellular cAMP also activates the MAPK, much as it does in PC12 cells (Yao et al., 1995; Martin et al., 1997). Thus, inAplysia sensory neurons PKA recruits MAPK, and injection of antibody to MAPK blocks long-term facilitation (Martin et al., 1997). MAPK also is involved in LTP in the CA1 region of hippocampus (English and Sweatt, 1997; Atkins et al., 1998; Impey et al., 1998a). Does MAPK play a similar role in amygdala LTP? To explore these ideas, we perfused the MAPK inhibitor PD98059 (20 μm) into the bath for 1 hr before the tetanus and found that it reduced L-LTP significantly, but not completely. L-LTP was reduced to 125 ± 6% (n = 6) 3 hr after tetanus, which is significantly different from 160 ± 12% in control experiments (see Fig. 3C) (n = 5; p < 0.025). By contrast, MAPK inhibitors have no effect on E-LTP induced by a one train tetanus (118 ± 3% at 15 min and 114 ± 6% at 30 min, n = 5, compared with 120 ± 4 and 118 ± 5% in control experiments, n = 5; p> 0.5) (data not shown). The synaptic potentiation induced by forskolin also was blocked by the MAPK inhibitor. In the presence of the inhibitor PD98059 (50 μm), forskolin induced only a weak potentiation 146 ± 7% (n = 5), which was approximately one-half of the forskolin-induced potentiation in control experiments (186 ± 9%,n = 6; p < 0.01) (see Fig.3D). The partial blockade induced by the MAPK inhibitor could be attributable to the lower affinity of the MAPK inhibitor or could reflect the fact that MAPK is only one of the downstream components activated by the cAMP/PKA signaling pathway in amygdala LTP.
L-LTP in amygdala recruits activation of the β-adrenergic receptor
Fearful stimuli recruit activity in the neurons of the locus ceruleus and consequently in the axons that form the noradrenergic pathways of the brain. The amygdala receives strong and direct projections from the locus ceruleus via the dorsal noradrenergic bundle (Lindrall and Bjorklund, 1974; Uprichard et al., 1980). Behavioral studies have demonstrated that β-adrenergic receptors play a crucial role in amygdala-dependent memory (Liang et al., 1990; McGaugh, 2000). Consistent with the functional importance of this innervation, β-adrenergic receptors are present throughout the amygdala complex (Rainbow et al., 1984; Ordway et al., 1988). Because activation of β-adrenergic receptor increases cAMP levels by means of the receptor-coupling protein GαS (Winder and Conn, 1993), we examined the role of β-adrenergic receptors in amygdala LTP. We first examined the effect of the β-adrenergic agonist isoproterenol (ISO), which enhances the EPSP in endopiriform pathway to the basal amygdala (C. Huang et al., 1996), a pathway that is unrelated to fear conditioning (McKenan and Schinnick-Gallagher, 1997). We asked whether isoproterenol also can potentiate the cortico-amygdala pathway concerned with fear. We found that brief applications of isoproterenol (15 μm, 15 min) produced a synaptic potentiation that lasted for at least 3 hr (143 ± 5% at 3 hr;n = 7) (Fig.6A). This potentiation could be prevented by coapplication of isoproterenol with the β-adrenergic antagonist timolol (10 μm; Fig. 6A). This potentiation also could be prevented by coapplication of either the PKA inhibitor KT5720 or the MAPK inhibitor PD98059. In the presence of KT5720 (2 μm) the application of isoproterenol (15 μm) induced no potentiation (108 ± 10% at 90 min; n = 5) (Fig. 6C). In the presence of the MAPK inhibitor PD98059 (20–50 μm) application of isoproterenol produced only a slight potentiation and decayed to the baseline in 40–50 min (101 ± 7% at 90 min; n = 5) (Fig.6B). The blockade of isoproterenol potentiation by inhibitors of PKA and MAPK indicates that β-adrenergic agonist-induced potentiation is mediated by PKA and MAPK. Adrenergically induced L-LTP also could be blocked by the mRNA synthesis inhibitor ACTD. In the presence of ACTD (40 μm) isoproterenol-induced potentiation at 3 hr was only 118 ± 7% (n = 7), which was significantly different from control (p < 0.05; Fig. 6D). These results indicate that isoproterenol-induced synaptic potentiation involves adrenergically stimulated gene expression, mediated by PKA.
Fig. 6.

β-Adrenergic receptor is involved in L-LTP of the amygdala. A, Brief application of β-adrenergic agonist isoproterenol (ISO; 15 μm, 15 min) induces a long-lasting synaptic potentiation in the amygdala. Representative field potentials before and 3 hr after ISO application are shown at the top of this panel. Calibration: 5 msec, 0.5 mV. B, Brief application of β-adrenergic agonist isoproterenol (15 μm) induces a synaptic potentiation (open circles; n = 6). In the presence of PD98059 (20–50 μm) no potentiation is induced by isoproterenol (open squares;n = 5). C, In the presence of KT5720 (2 μm) the application of isoproterenol induces no synaptic potentiation (n = 5). D, The ISO-induced synaptic potentiation was depressed by the mRNA synthesis inhibitor ACTD (40 μm). ACTD was perfused 60 min before the application of isoproterenol (15 μm) and was perfused for the next 2 hr. The late component of isoproterenol-induced potentiation was depressed (open squares; n = 6). E, L-LTP in amygdala that was induced by five trains of tetanus was blocked by the β-adrenergic receptor antagonist propranolol (1 μm).F, The D1 receptor antagonist SCH (1 μm) attenuates LTP in amygdala, but there is no significant difference between LTP in SCH-treated slices (open squares;n = 6) and control slices (open circles; n = 6; p > 0.5).
We next examined the effect of a β-adrenergic inhibitor on the expression of tetanus-induced LTP. We found that propranolol (1 μm), a specific inhibitor of β-adrenergic transmission, blocked the L-LTP induced by five trains. In the presence of propranolol the time course of LTP was similar to that in the presence of PKA inhibitor: LTP was reduced at 3 hr from 158 ± 15 to 102 ± 9% (n = 6; p < 0.01) (Fig. 6E). By contrast, an antagonist of the dopamine D1 receptor SCH23390 (1 μm), which depressed L-LTP in the Schaffer collateral pathway (Huang and Kandel, 1995), attenuated L-LTP only slightly, and this attenuation was not significantly different from control (p > 0.5; Fig. 6F). Taken together, the results with agonist and antagonist indicate that the β-adrenergic receptor is capable of activating the cAMP signaling pathway and thereby modulating the effectiveness of electrically induced L-LTP.
PKA-mediated L-LTP in thalamo-LA pathway of the amygdala
In addition to an indirect pathway from auditory cortex, the lateral amygdala also receives direct auditory input from the auditory thalamus (LeDoux, 1995). LTP in the thalamo-LA pathway correlates directly with fear conditioning (McKenan and Schinnick-Gallagher, 1997;Rogan and LeDoux, 1997). The mechanism for the induction of LTP in this pathway was studied recently (Weisskopf and LeDoux, 1999; Weisskopf et al., 1999). However, little is known about the molecular mechanism of L-LTP in this pathway. We therefore also examined L-LTP in this pathway by stimulating the fibers emerging from the internal capsule that carry efferents from the medial geniculate nucleus (Fig.7A) (see also Weisskopf and LeDoux, 1999; Weisskopf et al., 1999). Consistent with its also being a monosynaptic connection, we found that the field potential in this pathway also can follow a 50 Hz stimulation without failure (Fig.7B).
Fig. 7.

L-LTP in thalamo-LA pathway. A, Schematic illustration of the recording and stimulation site in the thalamo-LA pathway, as described in Figure 1. The traceon the bottomright of the panel is the sample recording of a field potential in this pathway before and after LTP. B, Synaptic response during a 50 Hz tetanus stimulation in thalamo-LA pathway. The synaptic response followed tetanus in a one-for-one manner without failure, consistent with a monosynaptic response. C, A single train of tetanus (100 Hz, 1 sec, indicated by the asterisk) induces E-LTP (n = 6). D, Multiple trains of tetanus induce L-LTP in thalamo-LA pathway (n = 6), and this L-LTP is depressed by the protein synthesis inhibitor anisomycin (20 μm;n = 6). E, L-LTP in the thalamo-LA pathway is depressed by the PKA inhibitor KT5720 (1 μm; n = 6). F, L-LTP in the thalamo-LA pathway is depressed by the MAPK inhibitor PD98059 (20 μm; n = 5). G, Brief application of forskolin (50 μm; 15 min) induces a long-lasting synaptic potentiation in the thalamo-LA pathway (n = 6). Representative field potentials before and 3 hr after forskolin application are shown at thetop of this panel. Calibration: 5 msec, 0.5 mV.H, Forskolin-induced potentiation occludes the tetanus-induced LTP. At 90 min after the application of forskolin the stimulus intensity was reduced to obtain a new baseline, and five trains of tetanus were applied. The tetanus induced only a weak LTP (n = 5).
In this pathway a single tetanus (100 Hz, 1 sec) induces E-LTP that relaxed back to the baseline in 40 min. By contrast, five tetanic trains induced L-LTP that lasted >3 hr (Fig. 7C,D). This L-LTP also is depressed significantly by protein synthesis inhibition (Fig. 7D). In the presence of anisomycin (20 μm) LTP at 3 hr is 112 ± 4% (n = 6) as compared with a control value at 3 hr of 149 ± 11% (n = 6; p < 0.01) (Fig. 7D). PKA also plays a critical role in this pathway. The PKA inhibitor KT5720 (1 μm) dramatically reduced L-LTP that was induced by five trains. At 1 hr after the tetani, L-LTP was only 110 ± 6% (n = 6) and by 3 hr LTP had decayed to baseline (103 ± 5%). By contrast, the controls were 148 ± 12% at 1 hr and 149 ± 11% at 3 hr (n = 5;p < 0.01) (Fig. 7E). KT5720 (1 μm) had no effect on baseline synaptic response in this pathway (94% ± 6% at 2 hr; n = 4). Consistent with the role for PKA, forskolin also induced L-LTP in the thalamo-LA pathway. Brief perfusion of forskolin (50 μm) induced a potentiation of 187 ± 23% (n = 8) that lasted at least 3 hr (Fig. 7G). The forskolin-induced synaptic potentiation occluded the LTP induced by tetanus. After forskolin-induced potentiation five trains of tetanus induced only a weak and short-lasting potentiation (116 ± 9% at 1 hr; n = 5) (Fig. 7H). By contrast, the inactive forskolin analog (1,9-dideoxy forskolin) did not induce synaptic potentiation (112 ± 14% at 2 hr; n = 4). These results indicate the L-LTP in the thalamo-amygdala pathway also is mediated by PKA. This form of L-LTP also requires MAPK. The MAPK inhibitor PD98059 (20 μm) reduced L-LTP to 114 ± 6% (n = 5) at 3 hr, which is significantly different from the L-LTP seen in controls (150 ± 14%; n = 5; p < 0.025) (Fig.7F). Although it has been reported that the induction of LTP in the thalamo-amygdala pathway is mediated by L-type voltage-gated calcium channels, the second messenger responsible for the maintenance of LTP is still unknown (Weisskopf et al., 1999). Our results demonstrated that for the late phase of LTP, at least, thalamo-amygdala and cortico-amygdala share a similar molecular mechanism.
DISCUSSION
Whereas there is now a beginning understanding of the molecular-signaling pathways for learned fear in invertebrates such asAplysia and Drosophila, we know little of the molecular-signaling pathways that are important for learned fear in mammals. By contrast, there is a quite good understanding of some of the key anatomical and functional components of the neural circuitry that are important for learned fear. Central to that circuitry is the amygdala (Maren and Faselow, 1996; Davis, 1997). One form of learning in the amygdala is the classical conditioning of fear produced by pairing a neutral tone (CS) with a fear-inducing shock (US). Auditory information for conditioning reaches the lateral nucleus of amygdala via two routes: directly from the medial geniculate nucleus of thalamus (the thalamo-LA pathway) and indirectly from the auditory cortex to lateral amygdala (cortico-LA pathway) (Davis, 1997; LeDoux, 1995). The formation and storage of conditioned fear is dependent on synaptic plasticity in both pathways (Rogan and LeDoux, 1995, 1997; Brambilla et al., 1997; McKenan and Schinnick-Gallagher, 1997). Earlier work in the amygdala focused only on the early phase of LTP (Chapman and Bellavangee, 1992; Watanabe et al., 1995; Huang and Kandel, 1998; Li et al., 1998; Weisskopf et al., 1999). There have been no previous studies of the late phase of LTP (L-LTP) in either of these two pathways.
Here we report that in both the thalamic input and the cortical input five repeated trains give rise to a protein synthesis-dependent late phase of LTP in the lateral nucleus of the amygdala. In both pathways L-LTP requires PKA-mediated gene expression, and both the repeated tetanus and the activation of adenyl cyclase by forskolin lead to late CREB phosphorylation in the amygdala. These cellular results are consistent with recent behavioral studies showing that (1) inhibitors of PKA, MAP kinase, or protein synthesis attenuate fear conditioning; (2) fear conditioning leads to an increase phosphorylation of CREB and increased injection of CRE-mediated gene expression in the lateral amygdala; and (3) CREB antisense in the amygdala impairs long-term memory (Lamprecht et al., 1997; Ding et al., 1998; Impey et al., 1998b;Schafe et al., 1999).
Studies on learned fear in Aplysia and Drosophilaindicate that cAMP, PKA, and CREB are parts of signal transduction pathways that are critical for the switch from short-term to long-term memory. The cAMP—PKA—CREB pathway is also important for explicit forms of long-term memory and synaptic plasticity in hippocampus (Bourtchouladze et al., 1994; Y. Huang et al., 1996; Abel et al., 1998; Impey et al., 1998a; Milner et al., 1998; Silva et al., 1998). Earlier studies in Aplysia indicated that the recruitment of MAP kinase is critical for the action of PKA (Martin et al., 1997). We find a similar recruitment in the amygdala. Moreover, we found that the activation of β-adrenergic receptor coupling with the PKA and MAPK signaling pathway is required for the maintenance of L-LTP. β-Adrenoreceptors are present throughout the amygdala and the noradrenergic fiber projected to the amygdala via the dorsal noradrenergic bundle. It is possible that the increased glutamate release following high-frequency stimulation facilitates the NE release via spillover on the presynaptic glutamate receptor of noradrenergic terminal, leading to the release of NE and the synergistic activation on common target cells of β-adrenergic and glutamate receptor (Whitton, 1997).
On the basis of this and our previous studies as well as the studies of others (Huang and Kandel, 1998; Huang et al., 1999; Weisskopf et al., 1999), we propose a working model for the molecular mechanisms underlying LTP in the amygdala. The induction and early expression of amygdala LTP represent a hybrid of both mossy fiber and Schaffer collateral LTP; it requires postsynaptic depolarization and Ca2+ influx into the postsynaptic cell (Huang and Kandel, 1998; Weisskopf and LeDoux, 1999; Weisskopf et al., 1999) that are similar in mechanism to the induction of LTP in the Schaffer collateral pathway (Nicoll and Malenka, 1995). However, in addition, E-LTP in the amygdala requires a PKA-mediated presynaptic mechanism (Huang and Kandel, 1998; Huang et al., 1999), which is similar to mossy fiber LTP (Weisskopf et al., 1994; Huang et al., 1995;Nicoll and Malenka, 1995) (but see also Yekel et al., 1999). For the expression of late phase, however, the amygdala pathways and both the two hippocampal pathways seem to use a common set of pathways that recruit cAMP MAPK and PKA-mediated gene activation (Y. Huang et al., 1996). Thus, on the mechanistic level the storage of long-term memory related to fear in both invertebrates and vertebrates may use common cellular signaling mechanisms and patterns of gene transcription. Moreover, these mechanisms for implicit memory storage that we have encountered here are similar to those delineated in the hippocampus, where they are used for explicit memory storage.
In the hippocampus substantial progress in analyzing memory storage has been made with genetically modified mice. By now applying a similar genetic approach to the amygdala, it should prove possible to analyze which aspects of LTP are important for the development of amygdala dependent memory of fear. The studies of the molecular-signaling pathway for amygdala LTP that we report here provide a necessary molecular background for beginning a genetic analysis.
Footnotes
This research was supported by grants from the National Alliance for Research on Schizophrenia and Depression, The Charles A. Dana Foundation, the G. Harold and Leila Y. Mathers Charitable Trust, Cure Autism Now Foundation, and the Howard Hughes Medical Institute. We thank S. Siegelbaum, D. Winder, and R. D. Hawkins for their comments on an earlier draft of this manuscript and H. Ayers and M. Pellan for typing this manuscript.
Correspondence should be addressed to Dr. Eric R. Kandel, Center for Neurobiology and Behavior, Columbia University College of Physicians and Surgeons, 722 West 168th Street, New York, NY 10032. E-mail:erk5@columbia.edu.
Dr. Martin's present address: Departments of Psychiatry and Biobehavioral Sciences, University of California, Los Angeles, Los Angeles, CA 90095-1761.
REFERENCES
- 1.Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- 2.Atkins CM, Selcher JC, Petzaitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 3.Bailey C, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1998;13:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- 5.Bourtchouladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 6.Brambilla R, Gnesutta N, Minichiella L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Arnaud CR, Grant SGN, Chapman PF, Lipp H-P, Sturani E, Klein R. A role for the Ras signaling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 7.Chapman PF, Bellavangee L. Induction of long-term potentiation in basolateral amygdala does not depend on NMDA receptor activation in amygdala. Synapse. 1992;6:271–278. doi: 10.1002/syn.890110406. [DOI] [PubMed] [Google Scholar]
- 8.Davis M. Neurobiology of fear response: the role of the amygdala. J Neuropsychiatry Clin Neurosci. 1997;9:382–402. doi: 10.1176/jnp.9.3.382. [DOI] [PubMed] [Google Scholar]
- 9.Ding C, Lee Y-L, Davis MD. Role of PKA and CaM kinases in fear conditioning assessed with fear-potentiated startle using local infusion of Rp-8-Br-cAMP, KN-62, or KN-93 into the amygdala. Soc Neurosci Abstr. 1998;22:365.9. [Google Scholar]
- 10.English JD, Sweatt D. A requirement for the mitogen-activated protein kinase C cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 11.Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N. Role of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron. 1992;9:479–489. doi: 10.1016/0896-6273(92)90185-g. [DOI] [PubMed] [Google Scholar]
- 12.Huang C-C, Hsu K-S, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or O-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y-Y, Kandel ER. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci USA. 1995;92:2446–2450. doi: 10.1073/pnas.92.7.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y-Y, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y-Y, Li X-C, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and the macromolecular synthesis-dependent late phase. Cell. 1995;79:69–74. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y-Y, Nguyen PV, Abel T, Kandel ER. Long-lasting form of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y-Y, Janz R, Kandel ER, Südhof TC. Genetic evidence for a presynaptic component to amygdala LTP. Soc Neurosci Abstr. 1999;25:690.8. [Google Scholar]
- 18.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Christophe J, Chan G, Storm DR. Crosstalk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998a;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 19.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998b;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 20.Lamprecht R, Hazri S, Dudai Y. cAMP response element-binding protein in the amygdala is required for long-, but not short-term, conditioned taste aversion memory. J Neurosci. 1997;17:8443–8450. doi: 10.1523/JNEUROSCI.17-21-08443.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeDoux JE. Emotion: clues from the brain. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Weiss SRB, Chuang D-M, Post RM, Rogawski M. Bidirectional synaptic plasticity in the rat basolateral amygdala: characterization of an activity-dependent switch sensitive to the presynaptic metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid. J Neurosci. 1998;18:1662–1670. doi: 10.1523/JNEUROSCI.18-05-01662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang KC, McGaugh JL, Yao H-Y. Involvement of amygdala pathways in the influence of post-training inter-amygdala norepinephrine and peripheral epinephrine on memory storage. Brain Res. 1990;508:225–233. doi: 10.1016/0006-8993(90)90400-6. [DOI] [PubMed] [Google Scholar]
- 24.Lindrall O, Bjorklund A. The organization of the ascending catecholamine neurons systems in the rat brain. Acta Physiol Scand Suppl. 1974;412:1–48. [PubMed] [Google Scholar]
- 25.Maren S, Fanselow M. The amygdala and fear conditioning: has the nut been cracked? Neuron. 1996;16:237–240. doi: 10.1016/s0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- 26.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus at presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 27.McGaugh JL. Memory—a century at consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 28.McKenan MG, Schinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 29.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:465–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen PV, Kandel ER. A macromolecular synthesis-dependent late phase of long-term potentiation requiring cAMP in medial perforant pathway of rat hippocampal slices. J Neurosci. 1996;16:3189–3198. doi: 10.1523/JNEUROSCI.16-10-03189.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 33.Ordway GA, Gambarna C, Frazer A. Quantitative autoradiography of central β-adrenoceptor subtypes: comparison of the effects of chronic treatment with desipramine or centrally administered l-isoproterenol. J Pharmacol Exp Ther. 1988;247:379–389. [PubMed] [Google Scholar]
- 34.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, 2nd Ed. Academic; New York: 1986. [Google Scholar]
- 35.Quirk G, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike firing in auditory cortex and lateral amygdala. Neuron. 1997;18:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 36.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of β1- and β2-adrenergic receptors in rat brain. Proc Natl Acad Sci USA. 1984;81:1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element-binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 39.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 40.Schafe GE, Nadel NV, Sullivan GM, Harris H, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- 41.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 42.Uprichard DC, Reisine TD, Mason ST, Fibiger HC, Yamamura HI. Modulation of rat brain α- and β-adrenergic receptor population by lesion of dorsal noradrenergic bundle. Brain Res. 1980;187:143–154. doi: 10.1016/0006-8993(80)90500-4. [DOI] [PubMed] [Google Scholar]
- 43.Wang SJ, Chen L-L, Gean PW. Cross modulation of synaptic plasticity by β-adrenergic and 5-HT1A receptor in the rat basolateral amygdala. J Neurosci. 1999;19:570–577. doi: 10.1523/JNEUROSCI.19-02-00570.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe Y, Ikegaya Y, Satio H, Abe K. Roles of GABA, NMDA, and muscarinic receptors in induction of long-term potentiation in the medial and lateral amygdala in vitro. Neurosci Res. 1995;21:317–322. doi: 10.1016/0168-0102(94)00867-f. [DOI] [PubMed] [Google Scholar]
- 45.Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- 46.Weisskopf MG, Castillo P, Zalutsky R, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 47.Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA in dependent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitton PS. Glutamatergic control over brain dopamine release in vivo and in vitro. Neurosci Biobehav Rev. 1997;21:481–488. doi: 10.1016/s0149-7634(96)00034-6. [DOI] [PubMed] [Google Scholar]
- 49.Winder DG, Conn P. Activation of metabotropic glutamate receptors increase cAMP accumulation in hippocampus by potentiating responses to endogenous adenosine. J Neurosci. 1993;13:38–44. doi: 10.1523/JNEUROSCI.13-01-00038.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao H, Labudda K, Rim C, Capodieci P, Ioda M, Stork PJS. Cyclic adenosine monophosphate can convert epidermal growth factor into a differentiation factor in neuronal cells. J Biol Chem. 1995;270:20748–20753. doi: 10.1074/jbc.270.35.20748. [DOI] [PubMed] [Google Scholar]
- 51.Yekel MF, Kapur A, Johnson D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;7:625–233. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin JC, Tully T. CREB and the formation of long-term memory. Curr Opin Neurobiol. 1996;2:264–268. doi: 10.1016/s0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]