

ABSTRACT
Chromosomal instability (CIN) causes structural and numerical chromosome aberrations and represents a hallmark of cancer. Replication stress (RS) has emerged as a driver for structural chromosome aberrations while mitotic defects can cause whole chromosome missegregation and aneuploidy. Recently, first evidence indicated that RS can also influence chromosome segregation in cancer cells exhibiting CIN, but the underlying mechanisms remain unknown. Here, we show that chromosomally unstable cancer cells suffer from very mild RS, which allows efficient proliferation and which can be mimicked by treatment with very low concentrations of aphidicolin. Both, endogenous RS and aphidicolin-induced very mild RS cause chromosome missegregation during mitosis leading to the induction of aneuploidy. Moreover, RS triggers an increase in microtubule plus end growth rates in mitosis, an abnormality previously identified to cause chromosome missegregation in cancer cells. In fact, RS-induced chromosome missegregation is mediated by increased mitotic microtubule growth rates and is suppressed after restoration of proper microtubule growth rates and upon rescue of replication stress. Hence, very mild and cancer-relevant RS triggers aneuploidy by deregulating microtubule dynamics in mitosis.
KEYWORDS: Chromosomal instability, chromosome segregation, mitosis, replication stress
Introduction
Chromosomal instability (CIN) is a major hallmark of human cancer and contributes to the generation of genetic heterogeneity and the clonal evolution of tumors [1,2]. Two forms of CIN are prevalent in human cancer. First, whole chromosome instability (W-CIN) is defined by gains and losses of whole chromosomes during mitosis leading to the generation of whole chromosome aneuploidy. Second, structural chromosome instability (S-CIN), which leads to structural aberrations on chromosomes including translocations, deletions and amplifications [3].
Various defects in mitosis affecting the mitotic spindle or centrosomes or chromatid cohesion have been associated with whole chromosome missegregation and thus, with W-CIN in cancer cells [4,5]. In addition, abnormally increased microtubule plus end assembly rates during mitosis can account for whole chromosome missegregation in cancer cells by facilitating the generation of erroneous merotelic microtubule-kinetochore attachments resulting in so-called lagging chromosomes during anaphase, a pre-stage of whole chromosome missegregation [6,7]. Importantly, our previous work has shown that an abnormal increase in microtubule dynamics in mitosis provides a mechanistic basis for W-CIN in colorectal cancer (CRC) cells [6,8,9].
On the other hand, S-CIN and structural chromosome aberrations can be the result of different cancer-related defects including impaired DNA repair and abnormalities during DNA replication. In fact, DNA replication stress (RS), a condition, which is defined as slowed or stalled replication forks during S-phase of the cell cycle, appears to be a major source for S-CIN [10]. RS is frequently detected in cancer cells and can be caused by different mechanisms including oncogene activation, shortage of nucleotides, unresolved obstacles at the replication fork, which hinders timely progression of the forks or also conflicts between DNA replication and transcription [11,12]. Experimentally, replication stress can be induced by inhibiting DNA polymerase by the natural compound aphidicolin and this mean has been extensively used to investigate the mechanisms and consequences of replication stress [13–16]. High aphidicolin concentrations or severe endogenous replication stress results in temporarily or even terminally arrested replication forks. If not repaired, those forks can collapse, which can be associated with the induction of DNA damage. To prevent this, cells use intra-S phase checkpoint mechanisms that involve the function the ATR and Chk1 kinases and others, which contribute to a halt of the cell cycle and to stabilize arrested forks in order to allow subsequent repair [17]. In contrast, mild replication stress slows down replication fork progression, which can remain unrecognized by the checkpoints. This situation can result in an unscheduled entry into mitosis in the presence of under-replicated DNA. The consequences of RS on mitosis under those conditions remain incompletely understood, but is of high relevance for cancer since cancer cells often suffer from RS, but still progress through the cell cycle [18,19]. One of the first consequences of mild RS in mitosis that was observed is the instability of defined genomic loci known as common fragile sites (CFSs). These loci may represent difficult to replicate DNA sequences that are hypersensitive to RS. CFSs are highly prone to breakage and thus, are hotspots for chromosomal rearrangements in cancer [20]. These sites and other under-replicated DNA might also be subject to mitotic DNA synthesis (MiDAs) in order to complete DNA replication even in mitosis to rescue a deleterious impact of RS on mitosis [21]. If this is not sufficient, cells may attempt to segregate their sister chromatids with partially unreplicated DNA and this can results in the formation of stretched single-stranded DNA, which is too fine to be stained by DNA intercalating dyes. Instead, these so-called ultra-fine bridges (UFB) recruit the single stranded DNA binding protein RPA and DNA helicases including the Bloom (BLM) and PICH (Plk1-interaction checkpoint helicase) helicases [22]. How chromosome segregation in mitosis is accomplished in the presence of under-replicated DNA and how UFBs are finally resolved in order to proceed through mitosis is currently little understood. It seems likely that UFBs may result in chromosome breakage rather than leading to whole chromosome missegregation. Intriguingly, however, recent work suggested that there might be a link between RS and whole chromosome missegregation during mitosis [23]. In fact, it was shown that chromosomally unstable cancer cells showing whole chromosome missegregation and W-CIN often suffer from RS, but the mechanisms linking RS to mitotic chromosome missegregation remains unknown.
In our work we provide evidence that only very mild levels of RS, which escape checkpoint control, are detectable in cancer cells exhibiting W-CIN. This very mild RS does not result in the formation of UFBs, but instead triggers whole chromosome missegregation and evolving aneuploidy constituting a W-CIN phenotype. Importantly, we show that these very mild RS levels induce abnormally increased microtubule plus end growth rates within mitotic spindles, a mitotic defect previously described to be associated with W-CIN in cancer cells. In fact, we demonstrate that RS-induced increased microtubule growth rates are responsible for whole chromosome missegregation and the induction of aneuploidy in response to RS.
Material and methods
Cell lines and treatments
HCT116, SW480, SW620 and HT29 colorectal cancer cell lines were obtained from ATCC (USA) All cell lines were grown in RPMI1640 medium supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (Sigma, Germany) and cultured at 37°C and 5% CO2. To induce DNA replication stress, cells were treated with increasing concentrations (0–1000 nM) of aphidicolin (Santa Cruz, USA). To induce DNA damage cells were treated with 600 nM adriamycin (Santa Cruz, USA) for 24 hours. In order to rescue abnormal microtubule polymerization rates, cells were treated with 0.2 nM Taxol for 24 hours (Sigma, Germany) [6].
Cell proliferation assay
To determine cell proliferation, 5,000 cells were transferred into a well of a 12-well plate on day 0 and incubated in medium with various aphidicolin concentrations. Confluency of the cells was measured at different time points using a Celigo Cytometer (Cyntellect, USA) and cell proliferation calculated using the Celigo software.
FACS and determination of mitotic index
FACS analyses to determine cell cycle distribution was performed as described [24]. The proportion of cells entering mitosis upon aphidicolin treatment was determined by FACS analysis using a FACS Canto II flow cytometer (Beckton Dickinson, Germany). Cells were treated with increasing concentrations of aphidicolin for 20 hours and with 2 µM Dimethylenaston (DME; Calbiochem, Germany) for 16 hours. Cells were harvested, fixed and stained using MPM-2 antibodies (Merck, Germany) and the mitotic index was calculated as described [24].
Measurement of microtubule plus-end assembly rates
Microtubule plus end growth rates were determined by tracking Eb3-GFP in living cells [25]. Cells were transfected 48 hour prior to the measurement with pEGFP-EB3 (kindly provided by L. Wordeman, University of Washington, USA), seeded onto glass bottom dishes (Ibidi, Germany) and treated with Dimethylenastron (2 µM, Calbiochem, Germany) for 2 hours before measurements. Live-imaging was performed using a Deltavision ELITE microscope (GE Healthcare, USA) equipped with an Olympus x60 1.42 NA objective and a PCO Edge sCMOS camera (PCO, Germany) and images were recorded every 2 sec while cells were incubated at 37°C and 5% of CO2. Images were deconvolved using SoftWorx 5.0/6.0 software (Applied Precision, Canada) and average assembly rates were calculated for 20 individual microtubules per cell. 30 cells were analyzed in 3 independent experiments.
DNA combing assays
To detect DNA replication fork progression we used DNA combing [26]. Unsynchronized cells were either not treated or pre-treated with increasing concentrations of aphidicolin for 1 hour before incubating with medium containing aphidicolin and 5-chloro-2ʹ-deoxyuridine (CldU, 100 µM; Sigma, Germany) for 30 min followed by incubation of medium containing aphidicolin and 5-iodo-2ʹ-deoxyuridine (IdU, 100 µM; Sigma, Germany) for additional 30 min. Labeled DNA track lengths were measured by DNA combing analyses carried out as part of an EasyComp Service by Genomic Vision (France). At least 300 DNA tracks were analyzed per sample.
Nucleoside supplementation
Nucleoside supplementation for replication stress rescue experiments was performed by 48 hour treatments of the cells with medium containing 20 µM 2ʹ-Deoxyadenosine monohydrate (Santa Cruz, USA), 20 µM 2ʹ-Deoxycitidine hydrochloride (Santa Cruz, USA), 20 µM Thymidine (Santa Cruz, USA) and 20 µM 2ʹ-Deoxyguanosine monohydrate (Santa Cruz, USA) as described previously [27].
Detection of lagging chromosomes and acentric chromosome fragments
To detect lagging chromosomes, cells were accumulated in anaphase by a thymidine block protocol with 2 mM thymidine treatment for 20 hours followed by release in fresh medium for 8–11.5 hours. Anaphase cells were analyzed by immunofluorescence microscopy detecting anaphase spindles, chromosomes and centromeres using anti-alpha-tubulin antibodies (1:700; Santa Cruz, USA), Hoechst 33342 staining (1:20,000; Biomol, Germany) and anti-centromere protein C antibodies (anti-CENP-C; 1:1,000; MBL, USA), respectively. For immunofluorescence microscopy experiments, cells were fixed and permeabilized by adding 2% PFA for 5 min at room temperature and subsequently incubated with methanol for 5 min at −20°C. Images were taken by an AF6000 microscope (Leica, Germany) equipped with a DFC360FX camera (Leica, Germany). Imaging was performed using the LAS AF 2.7.3.9 software (Leica, Germany). Only CENP-C positive chromosomes clearly separated from the two DNA masses were defined as lagging chromosomes. CENP-C negative DNA, separated from the two DNA masses was defined as acentric chromosome fragments. In both cases at least 100 anaphase cells were determined for each sample.
Determination of ultra-fine anaphase bridges
To detect UFBs, anaphase cells were analyzed by immunofluorescence microscopy detecting BLM positive bridges using anti-BLM antibodies (C-18; 1:500, Santa Cruz, USA). Cells were fixed and permeabilized by adding 2% PFA for 5 min at room temperature and methanol for 5 min at −20°C, respectively. Images were taken by an AF6000 microscope (Leica, Germany) equipped with a DFC360FX camera (Leica, Germany). Imaging was performed using the LAS AF 2.7.3.9 software (Leica, Germany). At least 100 anaphase cells were determined for each sample.
Karyotype analysis
Single-cell clones were generated and cultured for 30 generations in the presence of increasing aphidicolin concentrations and additional 0.2 nM Taxol when indicated. Chromosome spread analysis and chromosome counting of individual cells was performed as described previously [28]. Chromosome fragments were also visualized by chromosome spread analyses.
Determination of centrosome amplification
Single cell clones derived from HCT116 cells and treated with aphidicolin for 30 generations were used to determine centrosome numbers by immunofluorescence microscopy using anti-alpha-tubulin antibodies (1:700, Abcam, UK) and anti-gamma-tubulin antibodies (GTU88, 1:500, Sigma, USA). As a control, HCT116 cells were transfected with a plasmid expressing Flag-tagged Plk4 (pCMV-Flag-Plk4; a kind gift from Ingrid Hoffmann, German Cancer Research Center, Heidelberg) and induced centrosome amplification was quantified. At least 100 cells were analyzed for each sample and cells showing more than 2 centrosomes were considered as cells with supernumerary centrosomes.
Western blotting
Cells were lysed in lysis buffer (1% Triton-X 100, 1% Sodium Deoxychelat, 0.1% SDS, 150 mM NaCl, 10 mM EDTA, 20 mM Tris-HCl, protease inhibitor inhibitor cocktail (Roche, Switzerland) phosphatase inhibitor cocktail (Roche, Switzerland) and 2 M Urea). Protein lysates were sonified using a Bioruptor Sonicator (Deganode, Belgium). Proteins were resolved on 11% or 13% SDS polyacrylamide gels and blotted onto nitrocellulose membranes by semi-dry blotting procedure. The following antibodies and solutions were used: anti-actin (1:40,000; AC-15, Sigma, Germany), anti-CHK1 (6F5; 1:2,000; Thermo Fisher, USA) anti-phospho-CHK1 (1:2,000; phospho-Ser345, Thermo Fisher, USA) anti-RPA (32 kDa subunit, 1:2,000; Abcam, UK) anti-phospho-RPA (phospho-Ser33; 1:2,000; Bethyl, USA), anti-phospho-H2AX (phospho-Ser139, JBW301; 1:2,000; Merck, Germany), secondary antibodies conjugated to horseradish peroxidase (1:10,000; Jackson ImmunoResearch, USA). Proteins were detected by enhanced chemoluminescence. Representative examples of the western blot bands shown in the figures were repeated at least three times and used for quantification using ImageJ software (NIH, USA).
Statistical analysis
For all data mean values and standard error of the mean (s.e.m) were calculated using Graph Pad Prism 5.0 software (Graph Pad Software, USA). Statistical analysis was performed using two-sided unpaired t-tests and significances are indicated as: * = 0.01 < p < 0.05; ** = 0.001 < p < 0.01; *** = p < 0.001, **** = p < 0.0001.
Results
Only very mild replication stress allows long-term proliferation of cancer cells
It is well established that severe levels of RS, which are often associated with DNA damage, can be induced by high micro molar concentrations of the DNA polymerase inhibitor aphidocolin (APH) [13]. In contrast, treatment of cells with 200–400 nM of aphidicolin (APH) is a widely used condition to induce so-called mild replication stress. In fact, these low concentrations of APH, when used short-term, are commonly used to investigate the consequences of RS in mitosis [14–16,29]. We asked whether such low concentrations of APH represents a cancer-relevant condition and still allows long-term cell proliferation and survival of cancer cells. We treated chromosomally stable and near diploid HCT116 human colorectal cancer cells with increasing concentrations of APH and determined cell proliferation for up to 11 days. Interestingly, only concentrations of APH up to 50 nM allowed almost undisturbed cell proliferation in long-term. Slow proliferation was already observed in the presence of 100 nM APH while 200 nM APH completely prevented proliferation beyond 4 days (Figure 1a and Figure S1). As expected, proliferation impairment was associated with APH-dependent increasing cell cycle arrest in S- and G2-phase of the cell cycle as demonstrated by FACS analyses (Figure 1b). Consequently, this led to an impaired ability of cells to timely enter mitosis (Figure 1c). In support of this, we detected increasing phosphorylation of Chk1 and RPA proteins indicating cell cycle checkpoint activation already in response to 100–200 nM of APH while DNA damage assessed by phosphorylation of H2AX was not significantly induced by these rather low concentrations of APH (Figure 1d). Hence, RS induced by the commonly used 200–400 nM concentration range of APH or higher clearly causes cell cycle delay and arrest and prohibits long-term cell proliferation and therefore, might not reflect a cancer-relevant condition. Instead, only very mild RS mimicked by treatment with very low concentrations of APH in the range of 20–100 nM remains undetected by the checkpoint control, allows efficient cell cycle progression and long-term proliferation and thus, might represent a level of RS that is compatible with survival and proliferation of cancer cells.
Figure 1.
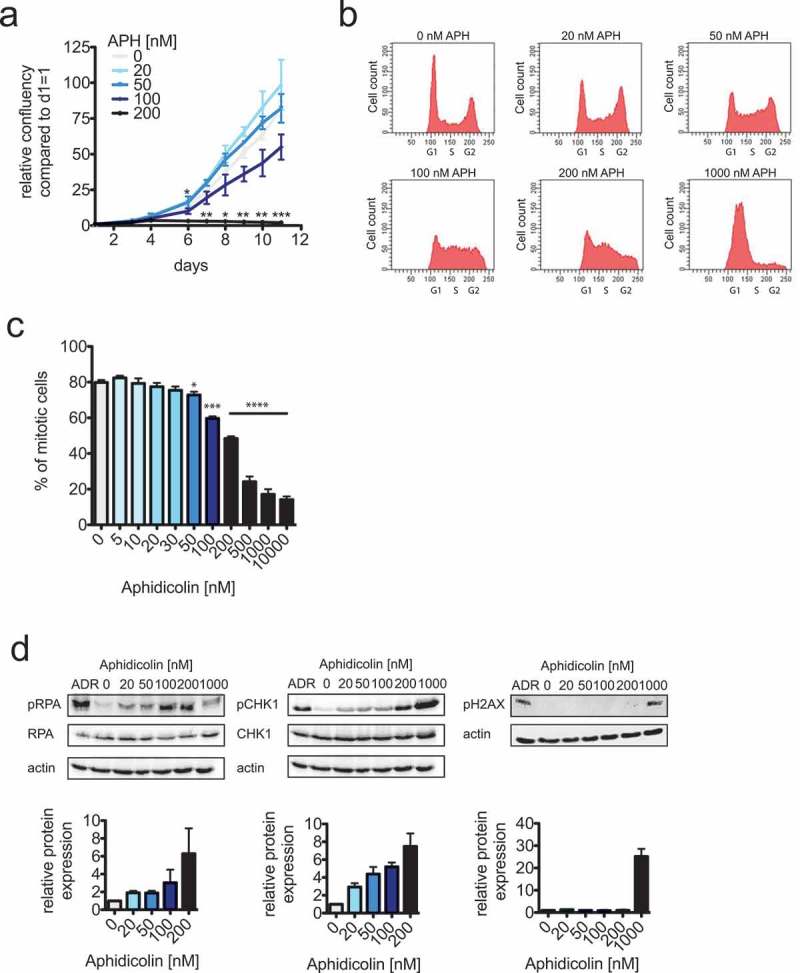
Only very mild replication stress allows efficient cell proliferation.
(a) Cell proliferation measurements of HCT116 cells treated with increasing concentrations of aphidicolin (APH) for up to 11 days. Cells were seeded at identical cell numbers, treated with increasing concentrations of aphidicolin and cell proliferation was quantified using a Celigo cytometer based on area confluency (n = 3 experiments, mean ± SEM, t-test). (b) Representative FACS profiles of HCT116 cells treated with increasing concentrations of aphidicolin (APH) for 24 hours. Cells were stained with propidiumiodide and the DNA content was determined.(c) Determination of mitotic entry of HCT116 cells treated with increasing concentrations of aphidicolin. Asynchronously growing cells were pre-treated with aphidicolin for 4 hours and subsequently together with dimethylenastrone for additional 20 hour to arrest cell cycle progression beyond prometaphase. Mitotic cells were quantified by FACS analyses detecting phosphorylated MPM2 epitopes (n = 3 experiments, mean ± SEM, t-test). (d) Western blot analyses to detect phospho-S33-RPA, phospho-S345-Chk1 and phospho-Ser139-H2AX as markers for S/G2 checkpoint activation and DNA damage. HCT116 cells were treated with the indicated aphidicolin concentrations or with 600 nM adriamycin (ADR) to induce DNA damage for 24 hours. Whole cell lysates were subjected to western blot detecting the indicated antigens. Representative western blots are shown and band intensities were quantified based on three independent experiments.
Chromosomally unstable cancer cells suffer from very mild replication stress
Previous work suggested that cancer cells exhibiting CIN suffer from RS [23]. To directly compare the level of RS in chromosomally unstable cancer cells and induced by very low concentrations of APH we performed DNA combing experiments after pulse labeling of newly replicated DNA to visualize and to quantify replication fork progression during S phase (Figure 2a) [26]. As expected, APH treatment reduced fork progression speed in a concentration dependent manner, already after treatment with very low concentrations of 20–50 nM APH. In addition, when compared to chromosomally stable HCT116 cells (average fork progression of 1.29 kb/min), three chromosomally unstable colorectal cancer cell lines (SW480, SW620 and HT29) showed approximately 32% reduced fork progression (average fork progression of 0.87 kb/min) indicating RS in CIN cells (Figure 2b). Interestingly, the level of RS in CIN cells was comparable to the level seen upon treatment with only 60–70 nM of APH indicating that CIN cells suffer from very mild RS, which does not significantly impact on cell proliferation (Figure 1). These quantitative results also indicate that RS levels induced by 200–400 nM of APH, which are commonly used e.g. to investigate the consequences of RS on mitosis [13–16], are significantly higher than the levels seen in chromosomally unstable cancer cells. Thus, for our studies investigating the consequences of RS on mitotic chromosome segregation we focused on conditions of very mild RS induced by 20–100 nM of APH.
Figure 2.
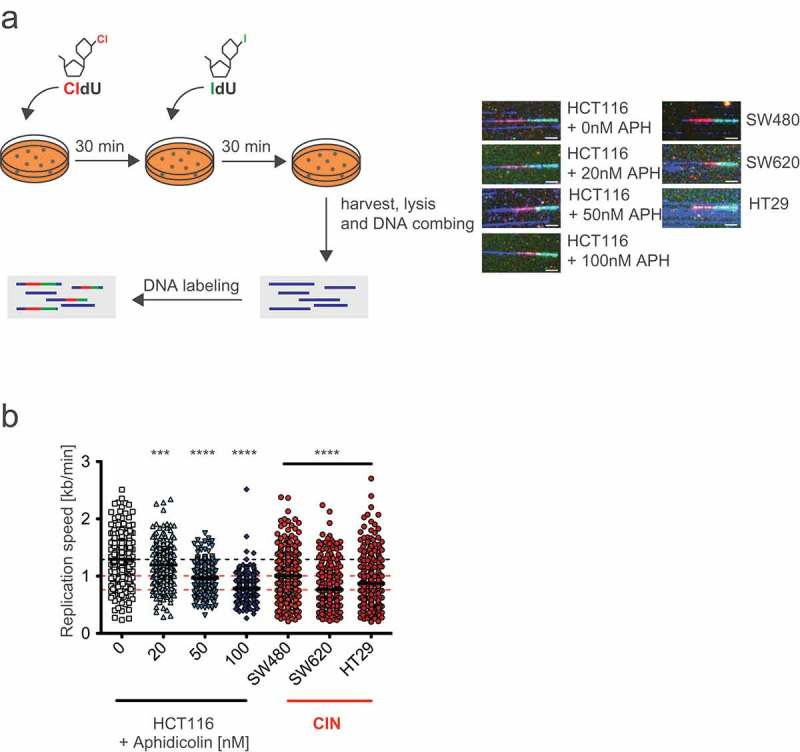
Chromosomally unstable cancer cells suffer from very mild replication stress that is mimicked by very low concentrations of aphidicolin.
(a) Principle and examples for DNA combing measurements of HCT116 cells treated with increasing concentrations of aphidocolin and of untreated CIN cells. Cells were pulse labeled consecutively with the nucleotide analogues CldU and IdU for 30 min each in the absence or presence of aphidicolin. Representative newly replicated and labeled DNA fibers are shown for each measurement (scale bar, 7.5 µm). (b) DNA combing measurements of single replicated DNA fibers of HCT116 cells treated with increasing concentrations of aphidocolin and of untreated CIN cells. Scatter dot plots show means and range of calculated fork progression rates (n < 300 fibers, t-test).
Very mild replication stress triggers abnormal microtubule growth rates in mitosis
Previous work has demonstrated a link between RS and whole chromosome missegregation in chromosomally unstable cancer cells [23], but it remained unknown how chromosome missegregation might be mediated in response to RS. It is well established that chromosome missegregation in CIN cells is associated with the generation of so-called lagging chromosomes during anaphase, which are the result of erroneous merotelic microtubule-kinetochore attachments [6,28,30,31]. Moreover, in chromosomally unstable colorectal cancer cells the generation of lagging chromosomes is dependent on an abnormal increase in microtubule plus end growth rates and thus, increased microtubule growth rates represent a mitotic defect closely associated with W-CIN in cancer cells [6,8,9]. Therefore, we considered a link between RS and increased microtubule growth rates in mitosis as a possible mechanism underlying chromosome missegregation in response to RS. By tracking the microtubule end-binding protein EB3 (EB3-GFP) in live cells [6,25] we systematically determined microtubule growth rates in HCT116 cells after treatment with increasing concentrations of APH as well as in three CIN cell lines known to be characterized by the induction of lagging chromosomes and aneuploidy [6] (Figure 3a). We found that all three CIN cell lines exhibit significantly increased microtubule growth rates when compared to the non-CIN cells (16.6 µm/min vs. 20.7 µm/min; Figure 3b). Importantly, abnormally increased microtubule growth rates were clearly induced in the chromosomally stable cells by mild replication stress in an APH concentration dependent manner. In agreement with previous results [6,8,9], normal microtubule growth rates were restored by treatment with sub-nanomolar concentrations of the microtubule binding drug Taxol that does not affect cell proliferation and survival (Figure 3b). Thus, we found an unexpected link between mild replication stress and an induction of abnormal microtubule growth rates during mitosis, which, in turn, might be responsible for whole chromosome missegregation in response to RS in cancer cells. It is of note that we previously demonstrated that the presence of increased microtubule growth rates is not associated with gross alterations of additional microtubule dynamics parameters including overall dynamicity, time of pausing and catastrophe rates [6].
Figure 3.
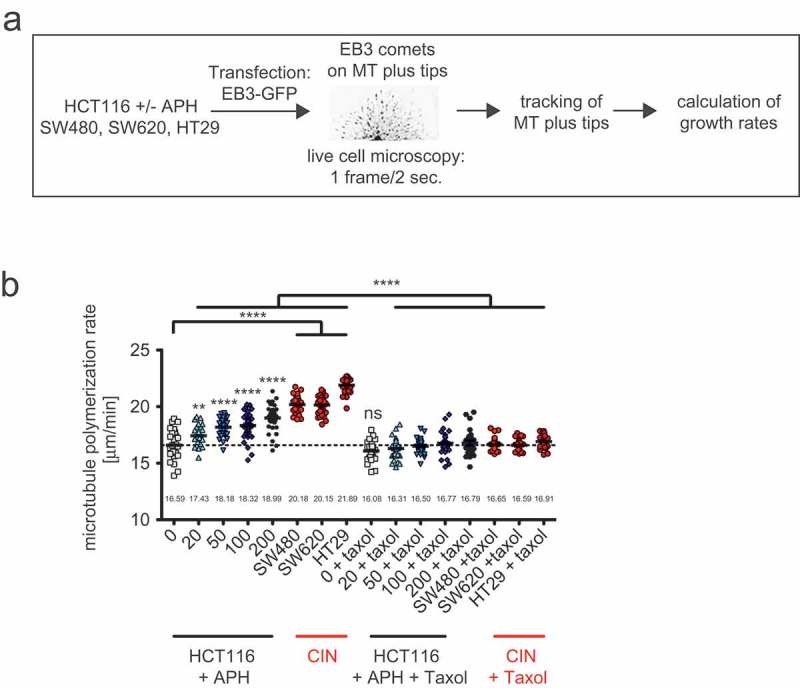
Mild replication stress induces abnormally increased microtubule growth rates within mitotic spindles.
(a) Schematic depiction of microtubule growth rates measurements. Cells were transfected in order to express GFP-tagged EB3, which localizes to growing microtubule plus tips. By live cell microscopy EB3 comets are tracked and growth rates are calculated based on 600 individual microtubules from 30 cells. (b) Mitotic microtubule plus end assembly rates in HCT116 cells after treatment with increasing concentrations of aphidicolin and in untreated CIN cells. Cells were treated with aphidicolin 24 hours before measurement. To restore normal microtubule growth rates cells were additionally pre-treated with 0.2 nM of Taxol. Scatter dot plots show average growth rates (20 microtubules/cell, mean ± SEM, t-test, n = 30 cells).
RS-triggered abnormal microtubule growth rates neither influence the formation of ultra-fine anaphase bridges nor the generation of acentric chromosome fragments
Next, we investigated the role of RS-induced abnormal microtubule growth rates on mitotic chromosome segregation. At least three different segregation abnormalities have been described to be induced by RS during mitosis: ultra-fine anaphase bridges (UFBs), acentric chromosome fragments and lagging chromosomes. UFBs cannot be visualized by DNA intercalating dyes, but are bound e.g. by DNA helicases such as the Bloom helicase (BLM) [22]. We detected UFBs in non-CIN cells after treatment with low concentrations of APH and in untreated CIN cells by immunostaining for BLM. As expected, increasing concentrations of APH elevated the proportion of cells exhibiting BLM-positive UFBs (Figure 4a). However, we could not observe an increase in UFBs in CIN cells when compared to non-CIN cells, although those CIN cells suffer from very mild RS comparable with a level induced by at least 50 nM of APH. Importantly, the formation of UFBs in response to APH treatment was not affected upon restoration of proper microtubule growth rates during mitosis by co-treatment with Taxol indicating that abnormal microtubule dynamics is not involved in the formation of UFBs after RS.
Figure 4.
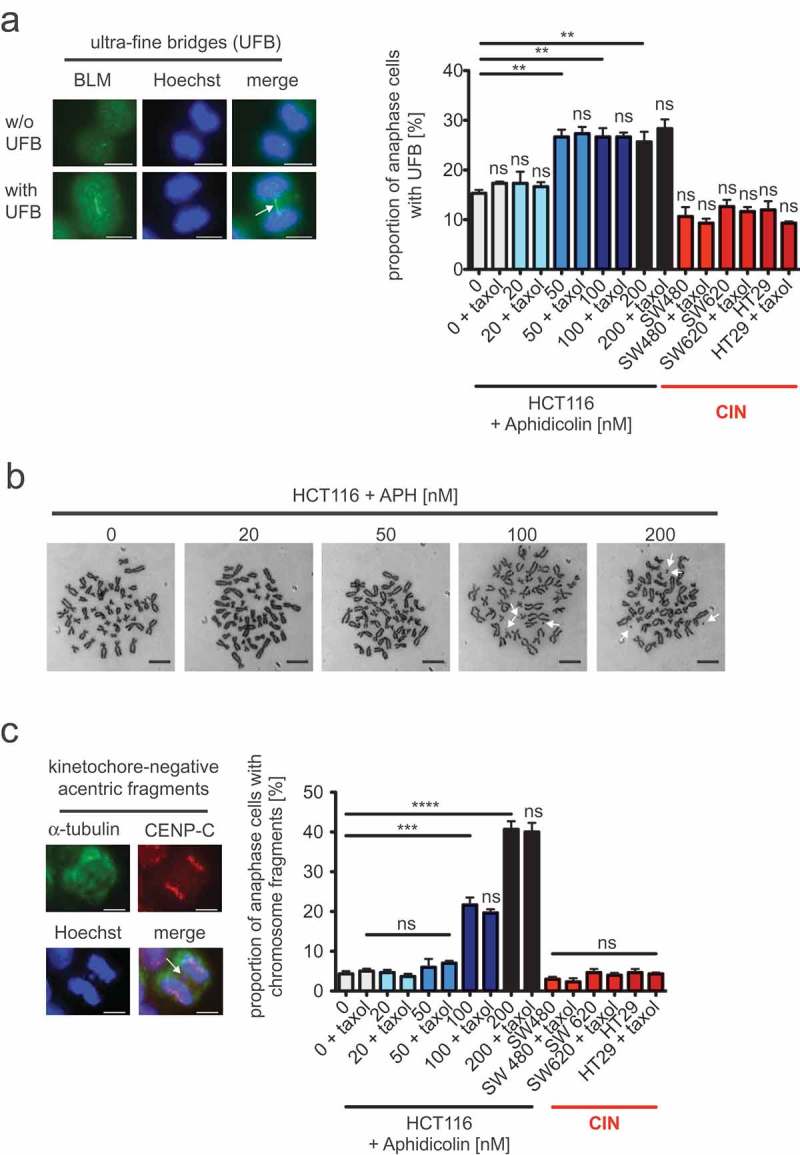
Mild replication stress-triggered abnormal microtubule growth rates neither influence the formation of ultra-fine anaphase bridges nor the generation of acentric chromosome fragments.
(a) Detection and quantification of cells exhibiting BLM-positive ultra-fine bridges. HCT116 cells were treated with increasing concentrations of aphidicolin for 16 hours before fixing and detecting ultra-fine bridges (arrow) using anti-BLM immunofluorescence (green) microscopy. Chromosomes were stained with Hoechst (blue). Representative examples of a cell with and without ultra-fine bridges are shown (scale bar, 7.5 µm). The proportion of cells exhibiting ultra-fine bridges were quantified and the graph shows mean values ± SEM (n = 300 anaphase cells from 3 independent experiments, t-test). (b) Examples of metaphase chromosome spreads from HCT116 cells treated with increasing concentrations of aphidicolin for 24 hours. Treated cells were arrested in mitosis and trypan-blue stained chromosome spreads were used to detect chromosome fragments as indicated by arrows. Representative examples are shown (scale bar, 10 µm). (c) Detection and quantification of anaphase cells exhibiting acentric chromosome fragments. HCT116 cells were treated with increasing concentrations of aphidicolin for 24 hours and acentric chromosome fragments were detected by immunofluorescence microscopy (chromosomes, Hoechst, blue; spindle, anti-alpha-tubulin, green; kinetochores, anti-CenpC, red; scale bar, 7.5 µm). A representative example of a cell with a kinetochore-negative chromosome fragment (arrow) is shown. Only kinetochore-negative chromosomes were quantified as acentric fragments and the graph shows the proportion of cells with acentric fragments (mean values ± SEM, n = 300 anaphase cells from 3 independent experiments, t-test).
Since RS might also contribute to chromosome breakage we investigated the occurrence of chromosome fragments in response to mild RS. In metaphase spreads from HCT116 cells treated with increasing concentrations of APH we found that chromosome fragments became indeed apparent, but only at higher APH concentrations (100–200 nM APH), but were hardly detectable at very mild RS conditions (20–50 nM APH; Figure 4b). To support this result, we detected acentric chromosome fragments as kinetochore-negative DNA in anaphase by immunofluorescence microscopy. Similar to the generation of UFBs, acentric chromosome fragments were clearly induced by mild RS triggered upon higher APH concentrations, but were neither dependent on microtubule growth rates nor commonly detected in the three CIN cell lines that are characterized by increased microtubule growth rates (Figure 4c). Thus, anaphase abnormalities typically associated with RS including UFBs and the generation of chromosome fragments are induced only at higher levels of RS and are not triggered by abnormal microtubule dynamics.
Abnormal microtubule growth rates in mitosis contribute to whole chromosome missegregation and W-CIN in response to RS
To investigate a possible link between RS, microtubule dynamics and whole chromosome missegregation we evaluated the generation of kinetochore-positive lagging chromosomes in anaphase cells. Lagging chromosomes represent pre-stages of whole chromosome missegregation and reflect a typical outcome of erroneous merotelic microtubule-kinetochore attachments in cancer cells [7]. Importantly, our previous work demonstrated that lagging chromosomes arise in response to abnormally increased microtubule growth rates [6,8,9]. We detected lagging chromosomes as kinetochore (CenpC marker)-positive chromatids that lag between the groups of segregated chromatids in anaphase cells. Similar to CIN cells we found a clear induction of lagging chromosomes in non-CIN cells upon treatment with increasing concentrations of APH, i.e. upon induction of RS (Figure 5a). It is of note that in contrast to UFBs (Figure 4a) lagging chromosomes are already significantly induced upon induction of very mild RS with 20 nM of APH (Figure 5a). Since lagging chromosomes can also result from supernumerary centrosomes [32] we also quantified centrosome numbers in response to RS, but we could not find an induction of supernumerary centrosomes (Fig. S2). In contrast, we found that the induction of lagging chromosomes in non-CIN cells by RS was suppressed when abnormally increased microtubule growth rates were restored to normal levels by applying sub-nanomolar concentrations of Taxol (Figures 5(a), 3(b)). These results suggest that increased microtubule dynamics in response to very mild RS can trigger perpetual whole chromosome missegregation, which is expected to cause an induction of numerical karyotype variability reflecting the W-CIN phenotype. To test this directly, we generated single cell clones derived from chromosomally stable HCT116 cells that were grown for 30 generations in the absence or presence of low APH concentrations to induce mild RS and additionally with or without Taxol to restore proper microtubule growth rates. Subsequently, independent single cell clones were subject to karyotype analyses and chromosome copy numbers per cell were determined (Figure 5b). In fact, in line with the induction of lagging chromosomes very mild RS mediated by 20–100 nM of APH induced a high numerical karyotype variability within 30 generations (Figure 5c) and Fig. S3 and supplemental Table 1), which is comparable to the level of karyotype instability typically seen in CIN cells [6]. It is remarkable that already a treatment with 20 nM of APH, which induce very mild RS, resulted in CIN-like aneuploidy. Thus, even very mild RS is sufficient to induce W-CIN in otherwise chromosomally stable cancer cells. Most importantly, similar to lagging chromosomes during anaphase also the resulting karyotype variability was reduced when the increased microtubule growth rates were restored to normal levels during the karyotype evolvement period (Figure 5c and Figure S3 and supplemental Table 1). These results indicate that very low level of RS increases microtubule growth rates in mitosis thereby triggering the generation of lagging chromosomes and the induction of W-CIN.
Figure 5.
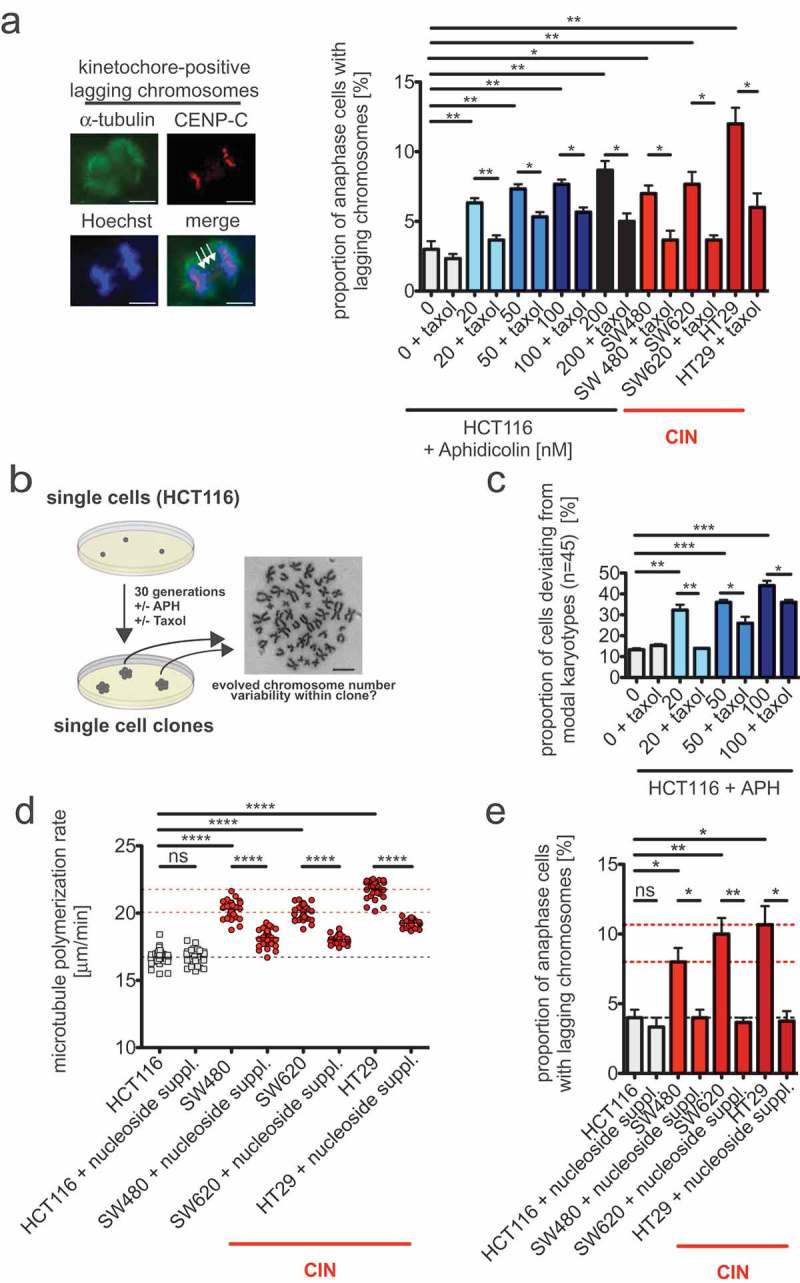
Abnormal microtubule growth rates in mitosis contribute to whole chromosome missegregation and W-CIN in response to mild replication stress.
(a) Detection and quantification of anaphase cells exhibiting lagging chromosomes fragments. HCT116 cells were treated with increasing concentrations of aphidicolin for 24 hours and lagging chromosomes were detected by immunofluorescence microscopy (chromosomes, Hoechst, blue; spindle, anti-alpha-tubulin, green; kinetochores, anti-CenpC, red; scale bar, 7.5 µm). A representative example of a cell with a kinetochore (CenpC)-positive lagging chromosome (arrows) is shown. Only kinetochore-positive chromosomes were quantified as lagging chromosomes and the graph shows the proportion of anaphase cells with laggings (mean values ± SEM, n = 300 anaphase cells from 3 independent experiments, t-test). (b) Schematic depiction of the generation of single cell clones used for determination of induced numerical karyotype variability as a measure for W-CIN. Single cell clones were grown in the continuous presence or absence of aphidicolin or Taxol for 30 generations and karyotype variability within the individual cell clones were determined by chromosome counting from metaphase spreads. A representative example of a chromosome spread is shown (scale bar, 10 µm). (c) Quantification of the proportion of cells within single cell clones harboring a karyotype with chromosome numbers deviating from the modal (modal number: 45 chromosomes in HCT116 cells). Single cell clones were treated with or without aphidicolin or Taxol and analyzed after 30 generations in culture. The graph shows mean values ± SEM for three independent single cell clones (n = 50 cells). (d) Mitotic microtubule plus end assembly rates using HCT116 and three CIN cell lines after nucleoside supplementation. Cells were treated with nucleosides for 48 hours before measurement. Scatter dot plots show average growth rates (20 microtubules/cell, mean ± SEM, t-test, n = 30 cells). (e) Quantification of anaphase cells exhibiting lagging chromosomes fragments after nucleoside supplementation. Asynchronously growing cells were treated with nucleosides for 48 hours before lagging chromosomes were detected by immunofluorescence microscopy. The graph shows the proportion of anaphase cells with laggings (mean values ± SEM, n = 300 anaphase cells from 3 independent experiments, t-test).
Finally, we wished to test whether endogenous mild replication stress in cancer cells exhibiting CIN is indeed responsible for abnormal microtubule growth rates and for chromosome missegregation. As an established mean to rescue RS we used nucleoside supplementation of the growth medium as described [23,27] and determined microtubule growth rates in mitotic CIN cells by live cell microscopy. Upon rescue of RS all three CIN cancer cell lines showed a significant suppression of increased microtubule growth rates (Figure 5d). Moreover, rescue from RS also led to substantial suppression of lagging chromosomes, and thus, of whole chromosome missegregation in these CIN cancer cells (Figure 5e). Together, these results indicate that not only experimentally aphidicolin-mediated RS, but also endogenous mild RS in CIN cells can trigger abnormal microtubule growth rates in mitosis, which, in turn, can act as a trigger for whole chromosome missegregation and for W-CIN in response to RS.
Discussion
Evolving chromosome aberrations contribute to tumor evolution and might even act as a driving force for tumorigenesis and tumor progression [33,34]. In cancer cells, structural and numerical chromosome aberrations causing deletions, amplification, re-arrangements and whole chromosome aneuploidy, respectively, are frequently detected concomitantly suggesting that both forms of chromosome aberrations might be somehow linked. There is indeed first evidence indicating that this might be the case. It was shown that chromosome missegregation during mitosis can occasionally result in chromatids trapped in the cleavage furrow during cytokinesis, which can cause DNA damage leading to structural chromosome alterations [35]. It has also been demonstrated that stable whole chromosome aneuploidy might be linked to the generation of structural chromosome aberrations by triggering replication stress (RS) during S-phase of the cell cycle [36]. RS is considered as being a major source for structural chromosome aberrations in cancer, but is remains unclear how exactly RS causes structural chromosome aberrations and how e.g. aneuploidy can cause RS [11]. On the other hand, it was shown that RS might contribute to whole chromosome missegregation in cancer cells exhibiting W-CIN. But again, a mechanistic explanation of how RS may contribute to W-CIN remained unknown [23]. Nevertheless, these previous studies provide first evidence for a cross-talk between RS and mitotic chromosome missegregation and vice versa.
In our work presented here we set out to address the question of how RS may affect whole chromosome missegregation in mitosis. Interestingly, we revealed that very mild levels of RS are sufficient to trigger an abnormal increase in microtubule growth rates within mitotic spindles and this can act as a trigger for whole chromosome missegregation and hence, for the induction of evolving aneuploidy. In our previous work we have established that increased microtubule growth rates are specifically and frequently detectable in aneuploid cancer cells exhibiting W-CIN. Moreover, we demonstrated that rescue of this abnormal microtubule behavior is sufficient to suppress ongoing chromosome missegregation in CIN cancer cells, thereby establishing a causal link between abnormal microtubule dynamics and CIN in cancer cells [6,8,9]. Intriguingly, increased microtubule growth rates in CIN cells are not associated with a change of microtubule dynamics parameters per se, but cause transient mispositioning of the mitotic spindle, which facilitates the generation of erroneous merotelic microtubule-kinetochore attachments leading to the generation of lagging chromosomes in anaphase [6]. Upon induction of very mild RS we also observe the generation of lagging chromosomes and those were suppressed when normal microtubule growth rates were restored. Thus, RS-induced abnormally increased microtubule growth rates can mediate whole chromosome missegregation in mitosis. It is currently not known how RS, which occurs during S-phase, increases microtubule growth rates in the subsequent mitosis. It is known, however, that several microtubule plus end binding proteins can contribute to tip growth behavior of microtubules. Perhaps most significant, the microtubule polymerase ch-TOG/CKAP5 (XMAP215 in Drosophila) acts as a processive microtubule polymerase at plus tips and mediates growth [37]. Accordingly, partial repression of ch-TOG leads to reduced microtubule plus end growth rates and was used to restore normal growth rates in CIN cancer cells [6]. It is interesting to note that ch-TOG was originally identified as a protein frequently overexpressed in cancer (colonic and hepatic tumor over expressed gene, ch-TOG) [38] and based on this, higher levels of ch-TOG might confer W-CIN in cancer cells though increasing microtubule growth rates. Indeed, overexpressing ch-TOG in non-CIN cells results in increased microtubule growth rates and the generation of lagging chromosomes [6]. It is tempting to speculate that RS might cause a hyper-activation of ch-TOG, but the mechanism for such a scenario remains currently unclear. Future work in our laboratory will address this important point.
It is remarkable that already very low levels of RS induced by very low concentrations of aphidicolin are sufficient to induce increased microtubule growth rates, lagging chromosomes and aneuploidy. These very mild RS conditions were also detectable in colorectal cancer cells exhibiting W-CIN. Rescue from RS resulted in suppression of abnormal microtubule growth rates and restored proper chromosome segregation demonstrating that very mild RS represents indeed an important mediator of aneuploidy in chromosomally unstable cancer cells. These very low levels of RS remain undetected by the cellular checkpoint pathways and allow even long-term proliferation of cells, which is a prerequisite for a RS condition present in cancer cells. Intriguingly, these very mild RS conditions, although sufficient to trigger aneuploidy, are not sufficient to induce gross DNA damage, the generation of acentric chromosome fragments or the induction of ultra-fine bridges in anaphase. Especially the latter have been suggested to be a typical outcome of RS when transmitted into mitosis. Ultra-fine bridges represent very thin, possibly single-stranded, DNA stretches that appear as a result of unfinished replication business [22]. How ultra-fines bridges are resolved in mitosis is not clear, but might involve mitotic DNA synthesis to finish up DNA replication [21] and DNA helicases such as BLM and PICH to untangle intertwined DNA strands [22]. Since we detected BLM-positive ultra-fines bridges only at higher concentrations of aphidicolin (>100 nM) and not at high frequency in cancer cells with W-CIN, it is possible that their generation requires higher levels of RS raising the question whether ultra-fine bridges are always relevant in CIN cancer cells. In fact, previous studies detecting ultra-fine bridges often used induction conditions by the use of 200–400 nM of aphidicolin [13–16], which, based on our directly measurements present here, severely reduces replication fork speeds typically not seen in chromosomally unstable cancer cells. Based on these observations it appears that very mild RS when transmitted into mitosis causes primarily lagging chromosomes and whole chromosome missegregation mediated by increased microtubule dynamics. Ultra-fine bridges, however, become apparent only at higher (still mild) levels of RS. Whether unresolved ultra-fine bridges also contribute to whole chromosome missegregation remains to be revealed. Since our work showed that chromosome missegregation induced by higher levels of RS cannot be fully suppressed upon restoration of proper microtubule growth rates, it is possible that mechanisms in addition to abnormal microtubule dynamics can contribute to whole chromosome missegregation at higher levels of RS. These could also involve unresolved ultra-fine bridges.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [FOR2800].
Acknowledgments
We thank Petra Beli and Zuzana Storchova for comments on the manuscript. We are grateful to Matthias Dobbelstein for the access to the Celigo cytometer and to Ingrid Hoffmann for providing plasmids. This work was supported by the Deutsche Forschungsgemeinschaft (FOR2800).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Burrell RA, McGranahan N, Bartek J, et al. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. [DOI] [PubMed] [Google Scholar]
- [2].Bakhoum SF, Landau DA.. Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb Perspect Med. 2017;7:a029611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McGranahan N, Burrell RA, Endesfelder D, et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bastians H. Causes of chromosomal instability. In: Recent results in Cancer Research. 2015;200:95–113. doi:10.1007/978-3-319-20291-4_5. [DOI] [PubMed] [Google Scholar]
- [5].Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ertych N, Stolz A, Stenzinger A, et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer. Nat Cell Biol. 2014;16:779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gregan J, Polakova S, Zhang L, et al. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011;21:374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ertych N, Stolz A, Valerius O, et al. CHK2-BRCA1 tumor-suppressor axis restrains oncogenic Aurora-A kinase to ensure proper mitotic microtubule assembly. Proc Natl Acad Sci U S A. 2016;113:1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Luddecke S, Ertych N, Stenzinger A, et al. The putative oncogene CEP72 inhibits the mitotic function of BRCA1 and induces chromosomal instability. Oncogene. 2016;35:2398–2406. [DOI] [PubMed] [Google Scholar]
- [10].Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. [DOI] [PubMed] [Google Scholar]
- [11].Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Magdalou I, Lopez BS, Pasero P, et al. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol. 2014;30:154–164. [DOI] [PubMed] [Google Scholar]
- [13].Glover TW, Berger C, Coyle J, et al. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 1984;67:136–142. [DOI] [PubMed] [Google Scholar]
- [14].Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 2009;11:761–768. [DOI] [PubMed] [Google Scholar]
- [15].Chan KL, Palmai-Pallag T, Ying S, et al. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. [DOI] [PubMed] [Google Scholar]
- [16].Lukas C, Savic V, Bekker-Jensen S, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. [DOI] [PubMed] [Google Scholar]
- [17].Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst). 2009;8:1038–1046. [DOI] [PubMed] [Google Scholar]
- [18].Fragkos M, Naim V. Rescue from replication stress during mitosis. Cell Cycle. 2017;16:613–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gelot C, Magdalou I, Lopez BS. Replication stress in mammalian cells and its consequences for mitosis. Genes (Basel). 2015;6:267–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Debatisse M, Rosselli F. A journey with common fragile sites: from S phase to telophase. Genes Chromosomes Cancer. 2019;58:305–316. [DOI] [PubMed] [Google Scholar]
- [21].Minocherhomji S, Ying S, Bjerregaard VA, et al. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y, Nielsen CF, Yao Q, et al. The origins and processing of ultra fine anaphase DNA bridges. Curr Opin Genet Dev. 2014;26:1–5. [DOI] [PubMed] [Google Scholar]
- [23].Burrell RA, McClelland SE, Endesfelder D, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vogel C, Kienitz A, Hofmann I, et al. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene. 2004;23:6845–6853. [DOI] [PubMed] [Google Scholar]
- [25].Stepanova T, Slemmer J, Hoogenraad CC, et al. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci. 2003;23:2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herrick J, Bensimon A. Introduction to molecular combing: genomics, DNA replication, and cancer. Methods Mol Biol (Clifton, NJ) 2009;521: 71–101. [DOI] [PubMed] [Google Scholar]
- [27].Wilhelm T, Magdalou I, Barascu A, et al. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc Natl Acad Sci U S A. 2014;111:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Stolz A, Ertych N, Bastians H. Loss of the tumour-suppressor genes CHK2 and BRCA1 results in chromosomal instability. Biochem Soc Trans. 2010;38:1704–1708. [DOI] [PubMed] [Google Scholar]
- [29].Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. Embo J. 2007;26:3397–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cimini D, Howell B, Maddox P, et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bakhoum SF, Thompson SL, Manning AL, et al. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009;11:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. [DOI] [PubMed] [Google Scholar]
- [34].McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–3266. [DOI] [PubMed] [Google Scholar]
- [35].Janssen A, van der Burg M, Szuhai K, et al. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. [DOI] [PubMed] [Google Scholar]
- [36].Passerini V, Ozeri-Galai E, de Pagter MS, et al. The presence of extra chromosomes leads to genomic instability. Nat Commun. 2016;7:10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brouhard GJ, Stear JH, Noetzel TL, et al. XMAP215 is a processive microtubule polymerase. Cell. 2008;132:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Charrasse S, Coubes P, Arrancibia S, et al. Expression of the tumor over-expressed ch-TOG gene in human and baboon brain. Neurosci Lett. 1996;212:119–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.