

Abstract
Background:
The incidence of esophageal adenocarcinoma (EAC) has risen dramatically over the past half century, and the underlying reasons are incompletely understood. Broad shifts to the upper gastrointestinal microbiome may be partly responsible. The goal of this study was to describe alterations in the esophageal microbiome that occur with progression from Barrett’s esophagus (BE) to EAC.
Methods:
A case-control study of patients with and without BE who were scheduled to undergo upper endoscopy. Demographic, clinical, and dietary intake data were collected, and esophageal brushings were collected during the endoscopy. 16S rRNA gene sequencing was performed to characterize the microbiome.
Results:
A total of 45 patients were enrolled and included in the analyses (16 controls; 14 BE without dysplasia (NDBE); 6 low grade dysplasia (LGD); 5 high grade dysplasia (HGD); and 4 EAC). There was no difference in alpha diversity between non-BE and BE, but there was evidence of decreased diversity in patients with EAC as assessed by Simpson index. There was an apparent shift in composition at the transition from LGD to HGD, and patients with HGD and EAC had decreased Firmicutes and increased Proteobacteria. Additionally, patients with HGD or EAC had increased Enterobacteriaceae and Akkermansia muciniphila and reduced Veillonella. In the study population, patients taking proton pump inhibitors had increased Streptococcus and decreased Gram-negative bacteria overall.
Conclusions:
Shifts in the BE-associated microbiome were observed in patients with HGD and EAC, with increases in certain potentially pathogenic bacteria.
Impact:
The microbiome may play a role in esophageal carcinogenesis.
INTRODUCTION
The incidence of esophageal adenocarcinoma (EAC) has increased 10-fold since the late 1960s (1), and Barrett’s esophagus (BE) incidence likely began to rise as early as the 1950s. Known modifiable risk factors for EAC do not adequately explain these incidence trends. GERD prevalence began to rise in the 1970s (2,3), and modeling studies suggest that only a minority of EAC cases are attributable to GERD (4). The obesity epidemic did not begin until 1980, and obesity may only account for a small fraction of the rise in EAC (5).
Helicobacter pylori infection is associated with a 30-40% reduced risk of BE and EAC (6), and H. pylori prevalence has plummeted since the mid-20th century (7). When present, H. pylori dominates the gastric microbiome, and its absence results in major shifts to gastric microbiome composition (8,9). Thus, dramatic changes in the upper GI microbiome in western populations likely occurred at the same time that BE and subsequently EAC began to rise in incidence. Any role of the microbiome the development of EAC is likely complex and multi-factorial, and may represent a co-factor in the development of BE, the progression from BE to EAC, or both.
There is ample evidence that elements of the microbiome can directly contribute to the development of colon cancer (10). However, the role of the microbiome in the progression of Barrett’s esophagus (BE) to EAC has not been well described. In health, the esophageal microbiome is broadly similar in composition to the oral microbiome, with a high relative abundance of the phylum Firmicutes (11). Previously published data suggest that the esophageal microbiome in patients with reflux esophagitis or BE is heavily populated with Gram-negative bacteria, which may contribute to a chronic inflammatory, pro-neoplastic state (12,13). More recent analyses of EAC surgical resections have shown that the tumor-associated microbiome demonstrates decreased microbial richness and diversity compared to non-dysplastic BE and normal squamous tissue (14).
In order to understand the potential role of the microbiome in esophageal carcinogenesis, knowledge of microbiome alterations that occur along the neoplastic pathway from BE to EAC is needed. The current study aimed to elucidate shifts in the esophageal microbiome that occur in the setting of progression from Barrett’s esophagus to associated dysplasia and adenocarcinoma.
MATERIALS AND METHODS
Study Population
This was a case-control study of patients ≥18 years old, enrolling subjects without or with a diagnosis of Barrett’s esophagus who were scheduled to undergo upper endoscopy for clinical indications. Analysis of the salivary microbiome in these patients has been previously reported (15). Subjects were prospectively enrolled over 18 months at a single academic medical center (Columbia University Medical Center, New York, NY). Barrett’s esophagus subjects had histologically confirmed BE measuring ≥2 cm, had never received endoscopic therapy, and were taking at least once daily proton pump inhibitors (PPI) for the prior month. BE subjects were categorized based on worst prior or current confirmed pathology: no dysplasia (NDBE), low grade dysplasia (LGD), high grade dysplasia (HGD), or adenocarcinoma (EAC). Controls were patients with no prior history of BE and were included if taking at least once daily PPI or no acid suppression (PPIs or H2-receptor antagonists) for the prior month. Other details of the exclusion criteria have been described previously. (15)
Demographics, clinical data, and anthropometric measures were collected. History of reflux symptoms was assessed using by questionnaire (16), and dietary fat and fiber intake over the preceding 4 weeks was used using a food frequency questionnaire (17,18). All participants provided written informed consent. The Institutional Review Board of Columbia University approved the study on February 25, 2015.
Sample Collection
Details of the sample collections have been described previously. (15,19) The microbiome was sampled by brushing the squamous esophagus as well as BE tissue (BE patients) or gastric cardia, within 1 cm of the squamo-columnar junction (controls). Sampling of any nodules, masses, or other focal lesions was avoided, in case grossly altered topography affected bacterial colonization. Biopsies were also taken from the mid-BE segment or gastric cardia for subsequent gene expression analyses.
Microbiome Characterization
After DNA extraction from esophageal brushings, the V4 hypervariable ribosomal RNA region was amplified using primers 515F and 806R (20). Sequencing of the 16S rRNA gene V4 region was performed, and sequence data were uploaded to the NCBI Sequence Read Archive (BioProjectID PRJNA517734). Greengenes was used as reference database (21). Clustering of taxonomic units was made at 97% sequence similarity using USEARCH. The functions classify.seqs and classify.otu (both with default settings) from the mothur project (22) were used to make taxonomic assignments to OTUs. FastTree version 2.1.7 was used to generate a phylogenetic tree of the contigs (23). Using mothur and the phylogenetic tree, weighted and unweighted UniFrac distances as well as diversity indices were calculated (24).
Semi-quantitative PCR (SsoAdvanced Universal SYBR Green Supermix, Bio-Rad, Hercules, CA) was also performed from esophageal brushing DNA for Enterobacteriaceae to further assess key findings from 16S rRNA gene sequencing analyses using previously published primer pairs (25). ΔΔCt values were calculated, using as a reference the Ct value for Eubacteria for the corresponding sample. qPCR for Eubacteria represents the entire bacterial DNA in the sample; thus, the ΔΔCt values were analogous to relative abundance data from 16S rRNA gene sequencing.
Statistical Analyses
Continuous variables were analyzed using t-tests and rank sum tests, and categorical variables were analyzed using Fisher’s exact tests. ANOVA or Kruskal-Wallis tests were used to compare continuous variables across multiple categories. The main analyses for this study were of brushings from Barrett’s mucosa (BE patients) or gastric cardia (controls). Within-individual correlations were assessed between paired swabs from esophageal squamous lining and from paired swabs from BE or cardia by calculating Spearman rank correlation coefficients at the genus level for all genera with non-zero read counts in both of the paired swabs. There were high correlations between paired swabs from the same site within the same individual (esophageal squamous, mean rho 0.85, SD 0.15; BE or cardia, mean rho 0.86, SD 0.12). For the purpose of these analyses, the mean relative abundance for each taxon from paired swabs was calculated from each sampling site. Of note, there was also high within-individual correlation between esophageal squamous and BE or cardia brushings (mean rho 0.82, SD 0.13).
Alpha diversity was assessed by observed OTUs and Shannon and Simpson indices. Pair-wise weighted and unweighted UniFrac beta diversity was calculated using functions implemented in QIIME. Non-parametric permutational MANOVA, as implemented in the FATHOM Toolbox for MATLAB, was used to compare beta diversity measures between BE vs. controls and between NDBE/LGD vs. HGD/EAC groups. Principal coordinate analyses for these tests were also performed using functions implemented in the FATHOM Toolbox for MATLAB. Differentially abundant taxa between groups were identified using linear discriminant analysis effect size (LEfSe) (https://huttenhower.sph.harvard.edu/galaxy/). Functional composition of the esophageal microbiome was assessed using predicted metabolic pathways derived by phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) analysis (26). Analyses were performed focused on the relative abundance of Gram-negative bacteria; Gram-negative genera and species were identified using a reference list assembled by our group (Supplementary Table S1), and the relative abundances of these taxa were summed for each sample. Additional analyses were performed on relative abundance of Streptococcus, the most abundant genus in the esophagus; alterations in the relative abundance of this genus have been associated with a variety of esophageal conditions. (13,27,28)
Upon visual observation of relative abundance of phyla across levels of BE and associated neoplasia, it appeared that there were shifts in relative abundance of Firmicutes and Proteobacteria, the two most abundant phyla in the esophageal samples, with the transition from low- to high-grade dysplasia. (Supplementary Figure S1) Thus, additional analyses were performed with BE subjects categorized as NDBE/LGD or HGD/EAC. Multivariable linear regression analyses were performed to assess for covariates independently associated with relative abundance of differentially abundant phyla and other select taxa. Full models were created including all covariates with a univariate p-value <0.10. Variables with the highest p-value and >0.15 were then sequentially removed to generate a final reduced model. Statistical significance was defined as p<0.05. Analyses were performed using Stata 14.1 (StataCorp) and MATLAB (The MathWorks, Inc.).
RESULTS
A total of 45 subjects were enrolled and had brushings collected for analysis. The characteristics of the subjects are shown in Table 1. There were 16 non-BE subjects and 29 subjects with BE (14 without dysplasia, 6 LGD, 5 HGD, and 4 intramucosal EAC).
Table 1.
Characteristics of patients who underwent upper endoscopy and had microbiome analyses, comparing those without to those with Barrett’s esophagus (BE).
| Non-BE (n=16) | BE (n=29) | p | |
|---|---|---|---|
| Age, mean (SD) | 60.1 (14.9) | 63.6 (11.7) | 0.39 |
| Sex, male | 9 (56%) | 25 (86%) | 0.04 |
| WHR, mean (SD) | 0.95 (0.08) | 0.97 (0.05) | 0.37 |
| GERD | 10 (63%) | 27 (93%) | 0.02 |
| Ever smoker | 7 (44%) | 19 (66%) | 0.21 |
| PPI use | 6 (38%) | 29 (100%) | <0.001 |
| Aspirin use | 3 (19%) | 11 (38%) | 0.31 |
| Dietary fiber*, grams per day; mean (SD) | 15.2 (5.3) | 16.5 (4.5) | 0.42 |
| Dietary fat*, % daily calories; mean (SD) | 33.6 (2.3) | 34.3 (3.2) | 0.46 |
SD: standard deviation; WHR: waist-to-hip ratio; GERD: gastro-esophageal reflux disease; PPI: proton pump inhibitor
Dietary data missing in 1 subject.
Microbiome Analyses
There were no significant differences in alpha diversity comparing BE to non-BE patients, both in terms of richness and evenness. (Supplementary Figure S2) There was decreased diversity assessed by Simpson index, but not by Shannon index or observed OTUs, across levels of BE-associated neoplasia (NDBE, LGD, HGD, and EAC). (Supplementary Figure S3) In post hoc pairwise comparisons, the Simpson index in EAC was significantly reduced compared to NDBE (p=0.006), LGD (p=0.01), and HGD (p=0.01). None of the other pairwise comparisons were significant. On beta-diversity analyses there was no evidence of significant clustering comparing BE vs. controls. (Supplementary Figure S4)
The most abundant phyla in the samples from BE and gastric cardia were Firmicutes (46.2%), Proteobacteria (22.9%), Bacteroidetes (19.6%), Actinobacteria (5.6%), and Fusobacteria (5.1%). BE subjects had significantly reduced relative abundance of Bacteroidetes compared to controls (16.3% vs. 25.5%, p=0.04), although there was no association after adjusting for patient characteristics. (Supplementary Table S2)
There were no overall differences in relative abundance of phyla across levels of BE-associated neoplasia. However, upon visual inspection of the results, it appeared that there was a shift in composition with regard to Firmicutes and Proteobacteria, the two predominant phyla, with the transition from LGD to HGD. (Supplementary Figure S1) Thus, subsequent analyses were performed with BE subjects categorized as (NDBE or LGD) and (HGD or EAC). Compared to NDBE/LGD, subjects with HGD/EAC had decreased relative abundance of Firmicutes (38.3% vs. 55.0%, p=0.04) and increased relative abundance of Proteobacteria (32.1% vs. 17.7%, p=0.04). (Figure 1) In multivariable analyses, HGD/EAC remained independently associated both with increased Firmicutes (p=0.03) and decreased Proteobacteria (p=0.01). (Supplementary Table S2) On beta-diversity analyses there was no evidence of significant clustering comparing HGD/EAC vs. NDBE/LGD. (Supplementary Figure S4)
Figure 1.
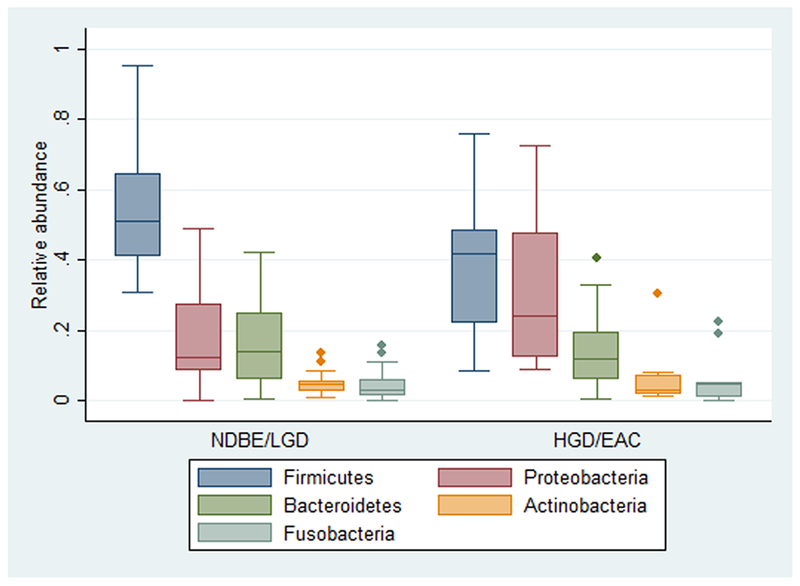
Relative abundance of the major phyla comparing subjects with no dysplasia or LGD to those with HGD or EAC. Compared to NDBE/LGD subjects, those with HGD or EAC had decreased Firmicutes (p=0.04) and increased Proteobacteria (p=0.04).
Taxonomic Differences
As compared to controls, subjects with BE had increased relative abundance of Sphingomonas and an unclassified species of Campylobacter. Non-BE subjects had increased relative abundance of various taxa including Prevotella pallens, Porphyormonas endodontalis, and Aggregatibacter segnis. (Supplementary Table S3) Based on the observations that there was a shift with transition from LGD to HGD at the phylum level, additional differences in relative abundance of taxa were assessed by LEfSe with subjects again categorized as NDBE/LGD and HGD/EAC. (Figure 2A) Patients with NDBE/LGD had significantly increased Veillonella. Several taxa were increased in the HGD/EAC subjects, notably in Enterobacteriaceae and Verrucomicrobiaceae, specifically Akkermansia muciniphila. (Figure 2B)
Figure 2.
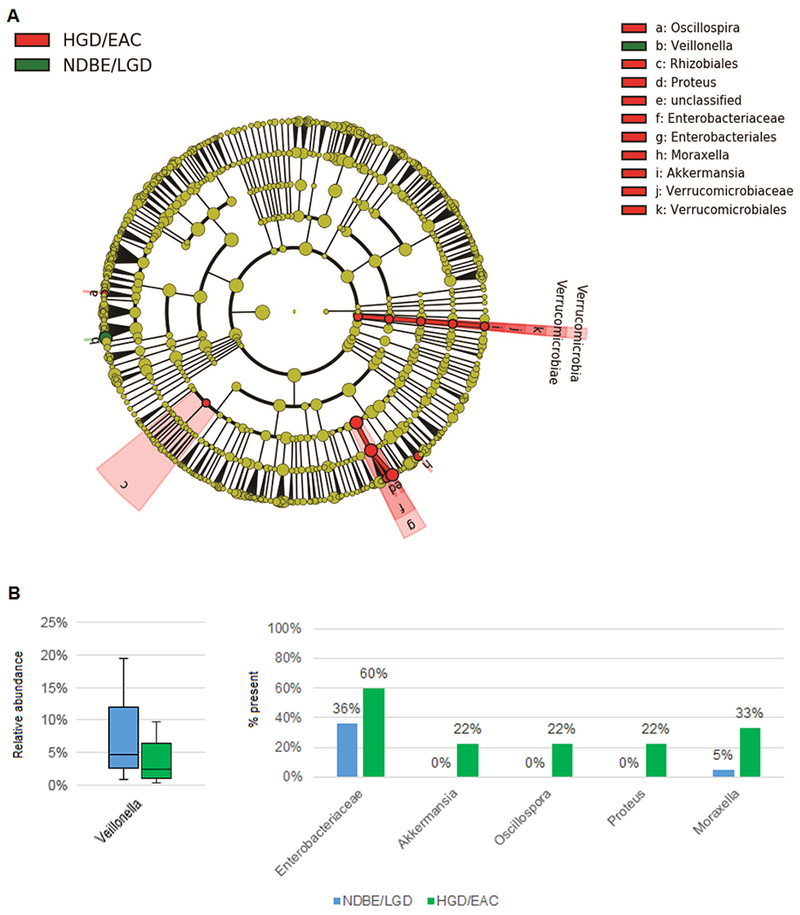
(A) Cladogram from LEfSe analyses of differentially abundant taxa comparing BE patients without dysplasia (NDBE) or LGD vs. HGD or EAC. (B) Subjects with HGD or EAC had reduced relative abundance of Veillonalla (left), and had increased proportion of samples with presence of the other differentially abundant taxa (right), which were relatively rare. (Presence defined as having any reads, except for Enterobacteriaceae, which was defined as relative abundance >0.1%.)
As members of Enterobacteriaceae can promote gut inflammation and neoplasia, the data on this family was examined in greater detail. Compared to NDBE/LGD, patients with HGD/EAC were more likely to be smokers (p=0.03) and had higher dietary fat intake (p=0.05). After adjusting for these two factors, HGD/EAC remained significantly associated with the relative abundance of Enterobacteriaceae (p=0.02). (Supplementary Table S2) Two subjects had very high relative abundance of Enterobacteriaceae; one of these had HGD and a relative abundance of 38.3%, and one had intramucosal EAC and a relative abundance of 30.4%. These findings were replicated in the esophageal squamous brushings, where these two subjects again had the highest relative abundance of Enterobacteriacea in the study population. For each of these subjects, a single distinct OTU drove the high relative abundance. On further evaluation of these OTUs using NCBI BLAST, one matched predominantly to species in the genera Klebsiella and Enterobacter, and the other matched to species in genera including Escherichia and Shigella.
Esophageal and cardia biopsies were then analyzed by qPCR to assess whether they harbored differences compared to brushings in relative abundance of Enterobacteriaceae. There was no significant difference by qPCR comparing patients with NDBE/LGD and HGD/EAC (median ΔΔCt 12.5 vs. 12.8, respectively; p=0.57).
Gram-Negative Bacteria
In brushings the mean relative abundance of Gram-negative bacteria in all of the subjects was 54.7% (SD 23.0). There was no significant difference in the relative abundance of Gram-negative bacteria comparing non-BE controls to BE subjects (61.6% vs. 50.9%, p=0.14). There were also no significant alterations in the relative abundance of Gram-negative bacteria across levels of BE-associated neoplasia (ANOVA p=0.66). In the entire study population (BE and non-BE), PPI users had decreased relative abundance of Gram-negative bacteria compared to PPI non-users (51.1% vs. 67.3%; p=0.05). (Figure 3A)
Figure 3.
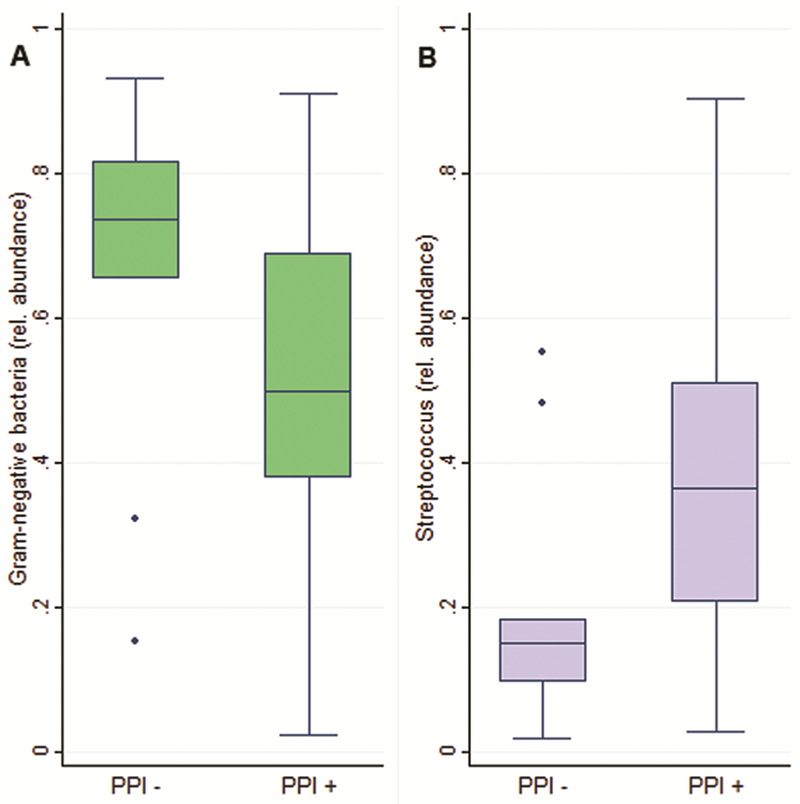
Compared to controls not taking PPIs, patients taking PPIs had: (A) reduced relative abundance of Gram-negative bacteria (p=0.05), and (B) increased relative abundance of Streptococcus (p=0.03).
Streptococcus
The mean relative abundance of Streptococcus in the study population was 32.6% (SD 20.9%). There was no significant difference in the relative abundance of Streptococcus comparing BE patients to non-BE controls (35.7% vs. 26.9%, p=0.18) and no significant overall alteration in the relative abundance of Streptococcus across levels of BE-related neoplasia (ANOVA p=0.51). With regard to PPI use, all subjects (BE and non-BE) on PPIs had greater relative abundance of Streptococcus compared to controls not on PPIs (36.2% vs. 19.9%, p=0.03). (Figure 3B)
Functional Profiling
PICRUSt analyses were performed to assess for functional alterations to the esophageal microbiome. Several gene pathways were significantly altered comparing patients with BE to non-BE controls. (Supplementary Figure S5A) Controls had increased RNA degradation and vitamin B6 metabolism, whereas BE patients had increased glycerolipid metabolism. Compared to patients with NDBE or LGD, those with HGD or EAC exhibited increased glycerophospholipid metabolism and decreased other glycan degradation. (Supplementary Figure S5B)
DISCUSSION
In the current study, we assessed the Barrett’s esophagus microbiome with progression to dysplasia and adenocarcinoma. We observed a shift in composition with progression, notably at the transition from LGD to HGD. This was manifested by significant clustering in beta diversity analyses, as well as alterations to the two predominant phyla, with reductions in Firmicutes and increases in Proteobacteria.
There is little previous data describing esophageal microbiome changes that occur in the development of EAC. Elliott et al. reported microbiome alterations comparing esophageal squamous samples from non-BE controls, Barrett’s samples from patients without dysplasia, and tumor tissue from patients with EAC (14). The authors noted that EAC tumors had decreased alpha diversity compared to BE, and in the present study there was some evidence of a decline in diversity with progression. However, many of the specific taxonomic alterations were distinct. This may be explained in part by the fact that the EAC tumor-microbiome was analyzed in this prior study (14), whereas in the current study sampling were performed only of normal appearing Barrett’s mucosa, avoiding any nodules or lesions, in patients with EAC. Also in the current study, there were high within-individual correlations between squamous and BE or cardia brushings, but the across-group alterations were less marked in squamous as compared to BE or cardia (data not shown). Finally, the EAC subjects in the current study all had very early lesions (T1a), and thus microbiome alterations in these patients would not have been caused by stasis due to tumor obstruction.
The increased relative abundance of Enterobacteriaceae in esophageal brushings from patients with HGD and EAC has potential biological significance. Certain species within Enterobacteraiceae harbor the pks genomic island and can produce colibactin, a genotoxin that induces DNA damage (29). Colibactin-producing E. coli promote tumor growth in xenograft mouse models (30), modify the tumor microenvironment (31), and have been found in high abundance in colonic biofilms in patients with familial adenomatous polyposis (32). Members of the family Enterobacteriaceae have also been implicated in gut inflammation in inflammatory bowel disease (33–35). Thus, it is plausible that increased levels of Enterobacteriaceae in Barrett’s esophagus may promote progression to EAC, either directly via colibactin or other bacterial products or indirectly by triggering an immune response and local inflammation.
Interestingly, the Enterobacteriaceae findings from 16S analyses of esophageal brushings were not replicated by qPCR of esophageal biopsies. However, the two subjects with high relative abundance of Enterobacteriaceae had similar findings in the squamous esophagus, in line with prior work demonstrating that there is little within-individual variability in the microbiome in the squamous and Barrett’s lining in patients with BE (36). Further, our group previously showed that patients with HGD or EAC have increased Enterobacteriaceae in saliva, and that there is strong within-individual correlation between the salivary and esophageal microbiome (15). Thus, possible explanations for the discrepant findings are that esophageal brushings are superior to biopsies for microbiome assessment, as previously reported by Gall et al (36), and that Enterobacteriaceae may reside predominantly within the esophageal biofilm rather than within the mucosa (37).
The increased relative abundance of Akkermansia muciniphila in subjects with HGD or EAC was also notable. In the colon, A. muciniphila has been associated with many beneficial effects related to obesity and metabolic syndrome (38). However, depending on the context, this species also can degrade mucins and thin the mucus layer (39), potentially leading to increased interaction between pathobionts and the underlying epithelium. In this fashion, the presence of A. muciniphila could conceivably lead to increased Barrett’s tissue inflammation and promote progression to EAC.
Yang et al previously described a microbiome associated with reflux esophagitis and BE that was characterized by decreased relative abundance of Streptococcus and increased relative abundance of Gram-negative bacteria (13). In the current study, there were no differences in relative abundance of Streptococcus or in overall Gram-negative bacteria comparing non-dysplastic BE to controls (data not shown) or with progression from BE to EAC. However, controls not taking PPIs had increased Gram-negative bacteria and decreased Streptococcus compared to subjects on PPIs, and our group has previously demonstrated that PPIs cause significant increases in Streptococcus in the distal gut (40). If Gram-negative bacteria in the esophagus promote chronic inflammation and increase the risk of BE and EAC (12), then PPIs may provide a chemoprotective effect by reducing overall levels of Gram-negative bacteria. However, the PPI results from the current study should be interpreted with caution, as the PPI users were a mix of BE and non-BE patients.
The current study has several strengths. There were patients from all stages of BE-associated neoplasia, which permitted the ascertainment of microbiome shifts prior to the development of EAC. During the endoscopy only flat BE tissue was sampled, avoiding lesions so as to minimize confounding by the presence of bacteria that may have been mere colonizers due to an altered tumor macro- and micro-environment. Care was taken with regard to exclusion criteria to minimize the effects of certain factors on the microbiome such as antibiotics and immunosuppressants. Detailed clinical information and dietary intake data were recorded and assessed in the analyses.
There were also certain limitations. The sample size was relatively small, and the study may have been underpowered to detect additional important microbiome alterations associated with neoplastic progression in BE. The current study describes associations with various stages of BE neoplasia but no information on causative effects on progression. However, the findings provide key hypothesis-generating data for follow-up functional studies.
In conclusion, there were pronounced shifts in the microbiome in Barrett’s esophagus associated with progression to EAC, particularly at the transition from low- to high- grade dysplasia. Notably, patients with HGD and EAC had increased relative abundance of Enterobacteriaceae, and members of this family have been implicated in gut inflammation and carcinogenesis. Further studies are indicated to identify specific bacteria that may promote the development of EAC, and also whether therapies targeting the microbiome can be developed to modify the risk of EAC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
The authors were supported in part by a Columbia Physician’s and Surgeon’s Dean’s Research Fellowship (EJS), a Career Development Award from NIDDK (K23 DK111847; DEF), a U54 award from NCI (U54 CA163004; JAA), a R01 from NIAID (AI116939; ACU), and the Price Family Foundation (JAA).
The authors would like to acknowledge the New York Genome Center (New York, NY) for performing the 16S rRNA gene sequencing on the samples collected as part of this study. The authors would also like to acknowledge Nora C. Toussaint (ETH Zurich, NEXUS Personalized Health Technologies, Zurich, Switzerland; New York Genome Center, New York, NY, USA) for assistance with processing and analyses of the sequencing data.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
REFERENCES
- 1.Abrams JA, Sharaiha RZ, Gonsalves L, Lightdale CJ, Neugut AI. Dating the rise of esophageal adenocarcinoma: analysis of Connecticut Tumor Registry data, 1940-2007. Cancer Epidemiol Biomarkers Prev 2011;20(1):183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.el-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer and reflux disease. Gut 1998;43(3):327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedenberg FK, Hanlon A, Vanar V, Nehemia D, Mekapati J, Nelson DB, et al. Trends in gastroesophageal reflux disease as measured by the National Ambulatory Medical Care Survey. Dig Dis Sci 2010;55(7):1911–7. [DOI] [PubMed] [Google Scholar]
- 4.Hazelton WD, Curtius K, Inadomi JM, Vaughan TL, Meza R, Rubenstein JH, et al. The Role of Gastroesophageal Reflux and Other Factors during Progression to Esophageal Adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2015;24(7):1012–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong CY, Nattinger KJ, Hayeck TJ, Omer ZB, Wang YC, Spechler SJ, et al. The impact of obesity on the rise in esophageal adenocarcinoma incidence: estimates from a disease simulation model. Cancer Epidemiol Biomarkers Prev 2011;20(11):2450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer prevention research 2008;1(5):329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis 1993;168(1):219–21. [DOI] [PubMed] [Google Scholar]
- 8.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A 2006;103(3):732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abreu MT, Peek RM Jr. Gastrointestinal malignancy and the microbiome. Gastroenterology 2014;146(6):1534–46.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan CA, Garrett WS. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol 2016;70:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A 2004;101(12):4250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L, Francois F, Pei Z. Molecular pathways: pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(8):2138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology 2009;137(2):588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott DRF, Walker AW, O’Donovan M, Parkhill J, Fitzgerald RC. A non-endoscopic device to sample the oesophageal microbiota: a case-control study. Lancet Gastroenterol Hepatol 2017;2(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snider EJ, Compres G, Freedberg DE, Giddins MJ, Khiabanian H, Lightdale CJ, et al. Barrett’s esophagus is associated with a distinct oral microbiome. Clin Transl Gastroenterol 2018;9(3):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Locke GR, Talley NJ, Weaver AL, Zinsmeister AR. A new questionnaire for gastroesophageal reflux disease. Mayo Clinic proceedings 1994;69(6):539–47. [DOI] [PubMed] [Google Scholar]
- 17.Thompson FE, Midthune D, Subar AF, Kahle LL, Schatzkin A, Kipnis V. Performance of a short tool to assess dietary intakes of fruits and vegetables, percentage energy from fat and fibre. Public health nutrition 2004;7(8):1097–105. [DOI] [PubMed] [Google Scholar]
- 18.Thompson FE, Midthune D, Subar AF, McNeel T, Berrigan D, Kipnis V. Dietary intake estimates in the National Health Interview Survey, 2000: methodology, results, and interpretation. J Am Diet Assoc 2005;105(3):352–63; quiz 487. [DOI] [PubMed] [Google Scholar]
- 19.Nobel YR, Snider EJ, Compres G, Freedberg DE, Khiabanian H, Lightdale CJ, et al. Increasing Dietary Fiber Intake Is Associated with a Distinct Esophageal Microbiome. Clin Transl Gastroenterol 2018;9(10):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nossa CW, Oberdorf WE, Yang L, Aas JA, Paster BJ, Desantis TZ, et al. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World journal of gastroenterology : WJG 2010;16(33):4135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26(19):2460–1. [DOI] [PubMed] [Google Scholar]
- 22.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 2009;75(23):7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 2010;5(3):e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology 2005;71(12):8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nature medicine 2015;21(7):808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology 2013;31(9):814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris JK, Fang R, Wagner BD, Choe HN, Kelly CJ, Schroeder S, et al. Esophageal microbiome in eosinophilic esophagitis. PLoS One 2015;10(5):e0128346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benitez AJ, Hoffmann C, Muir AB, Dods KK, Spergel JM, Bushman FD, et al. Inflammation-associated microbiota in pediatric eosinophilic esophagitis. Microbiome 2015;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, et al. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014;63(12):1932–42. [DOI] [PubMed] [Google Scholar]
- 31.Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014;5(5):675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018;359(6375):592–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright EK, Kamm MA, Teo SM, Inouye M, Wagner J, Kirkwood CD. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: a systematic review. Inflamm Bowel Dis 2015;21(6):1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell host & microbe 2015;18(4):489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell host & microbe 2010;8(3):292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gall A, Fero J, McCoy C, Claywell BC, Sanchez CA, Blount PL, et al. Bacterial Composition of the Human Upper Gastrointestinal Tract Microbiome Is Dynamic and Associated with Genomic Instability in a Barrett’s Esophagus Cohort. PLoS One 2015;10(6):e0129055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackett KL, Siddhi SS, Cleary S, Steed H, Miller MH, Macfarlane S, et al. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett’s and oesophageal carcinoma: association or causality? Aliment Pharmacol Ther 2013;37(11):1084–92. [DOI] [PubMed] [Google Scholar]
- 38.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog 2017;106:171–81. [DOI] [PubMed] [Google Scholar]
- 39.Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med 2016;8(339):339ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freedberg DE, Toussaint NC, Chen SP, Ratner AJ, Whittier S, Wang TC, et al. Proton Pump Inhibitors Alter Specific Taxa in the Human Gastrointestinal Microbiome: A Crossover Trial. Gastroenterology 2015;149(4):883–5 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.