
Fig. 3.
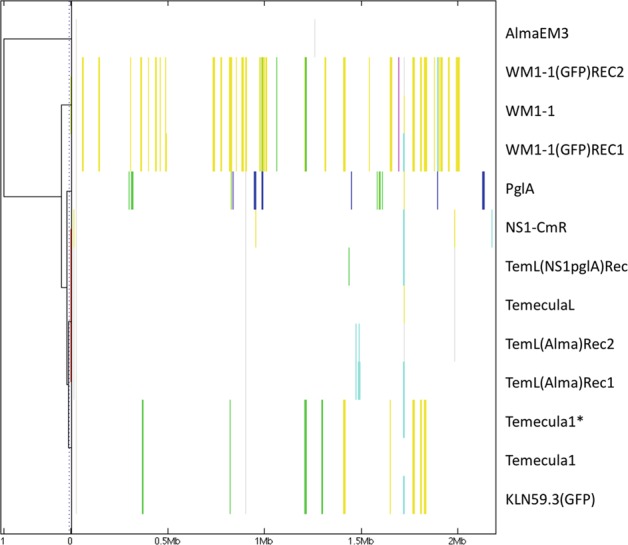
Detection of recombinogenic regions in the core genomes of X. fastidiosa in vitro recombinants and their parental strains using BratNextGen. Core-genome alignment obtained with progressiveMauve was used for genealogy reconstruction, and recombination was identified using BratNextGen algorithm. The estimation of recombination was performed with 20 iterations of the HMM estimation algorithm and 100 permutations with 5% significance threshold. The left panel corresponds to the proportion of shared ancestry (PSA) tree computed by BratNextGen. In PSA tree, strains are clustered according to the proportion of sequence shared, including recombinant regions. In the right panel, recombinant segments identified by BratNextGen for each strain are shown. The colors are arbitrarily assigned. The fragments of the same color present in the same column share the same origin. BratNextGen identified intrasubspecies recombination events (within fastidiosa subspecies), also indicating differences among different “Temecula” strains (see text for more details)