

Abstract
Background
The aim of this study was to identify biomarkers closely related to the pathogenesis and prognosis of oral squamous cell carcinoma (OSCC) by using weighted gene co-expression network analysis (WGCNA) based on integrative transcriptome datasets.
Material/Methods
Gene expression profiles of OSCC were downloaded from the Gene Expression Omnibus (GEO) database. Differentially expressed genes (DEGs) were obtained and we then performed with Gene ontology (GO) and pathway enrichment analysis as well as protein–protein interactions (PPI) network analysis. WGCNA was used to construct the co-expression network. Multipart results were intersected to acquire the candidate genes, and survival analysis was used to identify the hub genes.
Results
A total of 568 DEGs, including 272 upregulated genes and 296 downregulated genes, were identified. GO and pathway analyses revealed that these DEGs were mainly enriched in extracellular matrix (ECM), ECM organization, structural constituent of muscle, and ECM-receptor interaction. The PPI network of DEGs was established, comprising 428 nodes and 1944 edges. In the co-expression network, pink module was the key module, in which 34 genes with high connectivity were identified. After the intersection of multipart results, 24 common genes were chosen as the candidate genes, among which 7 hub genes (PLAU, SERPINE1, LAMC2, ITGA5, TGFBI, FSCN1, and HLF) were identified using survival analysis.
Conclusions
Seven potential biomarkers were identified as being closely related with the initiation and prognosis of OSCC and might serve as potential targets for early diagnosis and personalized therapy of OSCC.
MeSH Keywords: Carcinoma, Squamous Cell; Gene Expression Profiling; Biological Markers
Background
Oral cavity cancer is a global public health issue and is the sixth most common malignancy of humans, accounting for more than 300 000 new cases annually [1,2]. It has been estimated that approximately 300 400 new cases of oral cancer and 145 400 related fatalities occurred worldwide in 2012 [3]. Despite the relatively low incidence of oral cancer in China, the number of OSCC patients is still large due to the huge population base. It was reported that in 2010, there were over 34 000 new cases and 14 000 people died from oral cancer in China [4].
Oral cancer is growth of cancerous tissue in the oral cavity, and most oral malignancies (approximately 90%) are histologically subtyped as oral squamous cell carcinoma (OSCC) [5]. The main etiological factors are tobacco use, excessive alcohol consumption, chewing betel quid (especially in some Asian areas), and human papilloma virus 16 (HPV16) infection [6–9]. In the last few decades, great efforts have been made to fight OSCC. Despite substantive progress in surgical and medical treatments for OSCC, the overall 5-year survival rate has not significantly improved and remains approximately 50% [5]. Delay in diagnosis of OSCC patients in an early stage leads to progression to an advanced stage [10]. Most OSCC patients had a poor prognosis because of the advanced clinical stage at which they were diagnosed. One of the important reasons for failure in early diagnosis is insufficient research on the mechanisms at molecular levels underlying the carcinogenesis of OSCC. In-depth research on the molecular mechanisms in cancer initiation and prognosis of OSCC are needed, and this would also benefit OSCC patients at treatable stages. Therefore, it is of great importance to identify novel biomarkers for OSCC and reveal the molecular events contributing to OSCC pathogenesis.
The comparative analysis of differential gene expression between cancer tissues and normal controls will strengthen our exploration of the molecular pathogenesis and thus promote the identification of potential target genes and pathways for OSCC therapy. Previous bioinformatics studies about OSCC either utilized a single dataset with small sample size, or just directly merged multiple datasets, ignoring their batch effects and inherent heterogeneity [11,12]. As an alternative, the ComBat method can address these limitations, and can combine gene expression profiles from different datasets by removing the batch effects [13]. Furthermore, based on the theory that genes with high expression profile similarity may have closely related functional linkages or be involved in interacting pathways [14], the weighted gene co-expression network analysis (WGCNA) algorithm provides a systems biology approach to describe detailed characteristics at the level of genetic networks. WGCNA can establish free-scale gene co-expression networks to screen clusters (modules) of highly correlated genes and construct modules related to sample traits [15]. Zhang et al. used WGCNA to identify 2 modules and 10 hub genes associated with OSCC [16], but they only used a single dataset and the study lacked sufficient representativeness due to its limited sample size. Thus, integrating the data from different independent studies by ComBat method and the construction of a co-expression network based on this data will provide deeper insight into the molecular mechanisms of tumor genes associated with OSCC.
In the present study, we first integrated 5 gene expression profile datasets from GEO and removed the batch effects of these datasets by ComBat method. Then, we identified differentially expressed genes (DEGs) between OSCCs and normal samples, which were further assessed with Gene ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, as well as protein–protein interactions (PPI) network analysis. Weighted gene co-expression network analysis (WGCNA) was used to construct a co-expression network of relationships between genes to find network-centric genes. Subsequently, multipart results were intersected to obtain the candidate genes, and then the log-rank test of Kaplan-Meier analysis was performed to identify the hub genes for OSCC. Finally, we used other datasets to demonstrate the value of the hub genes.
Material and Methods
Date search strategy and selection criteria
The gene expression profile datasets were searched from the GEO database with the search terms “Oral squamous cell carcinoma” Or “OSCC” And “Homo sapiens” And “Expression profiling by array”. The datasets were eligible if they met the following criteria: (1) mRNA expression profiling by array; (2) datasets compared with normal control; (3) the number of samples more than 20; and (4) accessible gene expression profiles and platform information. The exclusion criteria were: (1) duplicated or non-relevant datasets; (2) non-coding RNA expression profiles; (3) methylation profiles; (4) datasets compared between cell lines; (5) datasets without normal control; (6) the number of samples less than 20; and (7) incomplete gene expression profiles or platform information. Moreover, a manual search of relevant OSCC datasets listed in the Materials and Methods section of the published articles was conducted. The dataset selection procedure is summarized in Figure 1. Six microarray gene expression datasets met the inclusion criteria (GSE30784, GSE13601, GSE37991, GSE31056, GSE9844, and GSE23558) were obtained from the GEO repository and the characteristics of these datasets are listed in Table 1. Five datasets with a larger sample sizes (GSE30784, GSE13601, GSE37991, GSE31056, and GSE9844) were used to peform an integrative transcriptome analysis, and the other dataset (GSE23558) was used to validate the results.
Figure 1.
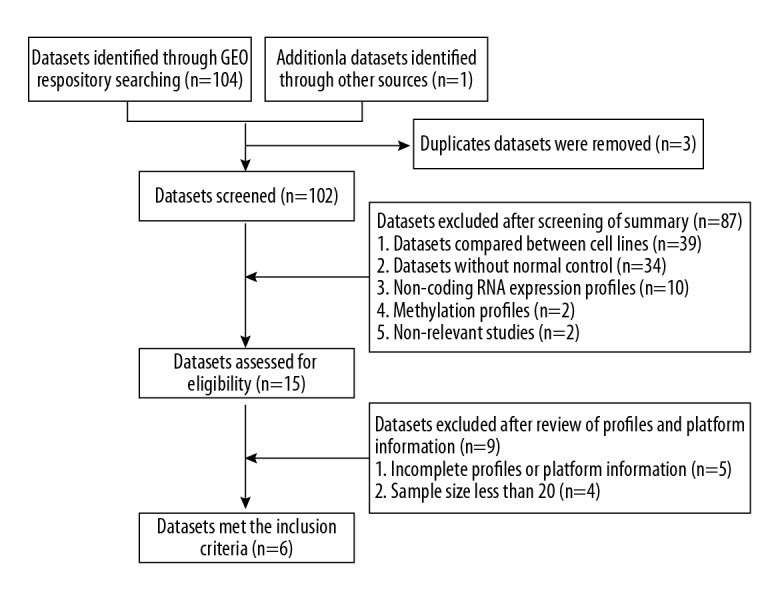
Flow chart of the datasets search and selection process.
Table 1.
Summary of datasets met the inclusion criteria.
| First author | GEO number | Sample size | Platform | |
|---|---|---|---|---|
| Normal | Tumor | |||
| Chen C | GSE30784 | 45 | 167 | GPL570; Affymetrix Human Genome U133 Plus 2.0 Array |
| Reis PP | GSE31056 | 73 | 23 | GPL10526; Affymetrix GeneChip Human Genome HG-U133 Plus 2 Array |
| Lee CH | GSE37991 | 40 | 40 | GPL6883; Illumina HumanRef-8 v3.0 expression beadchip |
| Estilo CL | GSE13601 | 26 | 31 | GPL8300; Affymetrix Human Genome U95 Version 2 Array |
| Ye H | GSE9844 | 12 | 26 | GPL570; Affymetrix Human Genome U133 Plus 2.0 Array |
| Ambatipudi S | GSE23558 | 5 | 27 | GPL6480; Agilent-014850 Whole Human Genome Microarray |
Data preprocessing and differential expression analysis
The raw data were preprocessed for background correction, log2 transformation, quantile normalization, and then converted into expression matrix by using Robust Multi-array Average (RMA) function of the affy R package [17]. Batch effects are inevitable when pooling microarray data across different laboratories, array types, or platforms. ComBat, combining location (mean) and scale (variance) adjustment with empirical Bayes, is a highly effective method of removing batch effects [13]. The batch effects when integrating the 5 datasets were removed by the ComBat function in the SVA R package. Afterwards, DEGs of the integrated datasets were screened through the limma R package with the cut-off criteria adjusted P<0.05 and |log 2-fold change (FC)| >1 based on the Benjamin and Hochberg (BH) procedure [18].
Gene ontology and pathway enrichment analysis
GO and KEGG pathway enrichment analyses were performed using the clusterProfiler package, which offers enrichGO and enrichKEGG methods for enrichment analysis and has the visualization function of profiles for genes and gene clusters [19]. P<0.05 was considered as the threshold value.
Protein–protein interactions (PPI) network construction
The online database STRING (http://string-db.org/) was employed to establish the PPI network of DEGs, with a confidence score ≥0.7. Cytoscape software (National Institute of General Medical Sciences of the National Institutes of Health, Bethesda, MD, USA) was then used to visualize and analyze the network. The Molecular Complex Detection (MCODE) plug-in of Cytoscape was used to find clustered sub-networks (highly interconnected regions) in the PPI network [20]. Degree ≥2, node score ≥0.2, K-core ≥2, and max depth=100 were used as cut-off criteria.
Construction of co-expression network
The integrated gene expression data after removing the batch effects were chosen for constructing the co-expression network using the “WGCNA” R package [15]. First, we constructed the Pearson correlation coefficient matrix between gene pairs based on the gene expression profile. Second, an appropriate soft threshold power (β) was selected in line with scale-free topology criteria. We turned the correlation coefficient matrix into the weighted adjacency matrix through a power function αij=|cor(xi, xj)| β (aij=weighted adjacency matrix, cor(xi, xj)=Pearson correlation coefficient matrix between gene pairs). Third, the adjacency matrix was transformed into a topological overlap matrix (TOM) by the fuction of TOM similarity, and the corresponding TOM-based dissimilarity (1-TOM) was calculated as well. Next, an average linkage hierarchical clustering dendrogram was built on the basis of TOM-based dissimilarity, and clustering module identification was achieved using dynamic tree cut with the minimum module size 50. Moreover, the dissimilarity of module eigengenes (MEs, the first principal component of one module) were calculated, and the cut line of 0.25 for module dendrogram was chosen as the module-merged standard. Finnaly, we calculated the relationships between module and trait, gene significance (GS), and module membership (MM) to find the key modules. GS, defined as the log10 transformation of the P value (lgP) in the linear regression between gene expression and trait information, was used to quantify associations of individual genes with a trait. MM was defined as the correlation between gene expression profile and the ME of a given module. The key modules were identified based on the correlation between MEs and trait.
Mining of candidate genes
Hub genes in key modules were regarded as genes with high module connectivity measured by the absolute value of the Pearson correlation and the clinical trait relationship (|MM| >0.8 and |GS| >0.2). The preprocessed level 3 RNA-seq expression data and clinical information for HNSC were downloaded from TCGA. A total of 499 patients with detailed survival data were included for subsequent survival analysis. The edgeR package was used to identify the DEGs for TCGA RNA-seq data under the cut-off criteria of false discovery rate (FDR) < 0.05 and |log2 FC| >1.5 [21]. We intersected multipart results (DEGs of 5 gene microarray data, DEGs of TCGA data, and hub genes in the co-expression network) to get the candidate genes.
Hub genes identification and validation
Log-rank test in Kaplan-Meier analysis was performed to demonstrate the effect of candidate genes expression on prognosis using survival R package. Candidate genes (log-rank P<0.05) were considered as the hub genes. Similarly, GSE23558 was used to verify expression of hub genes. The effect of hub gene expression on patient prognosis was verified by GSE41613.
Results
DEGs screening in OSCC
The integrated data (GSE30784, GSE13601, GSE37991, GSE31056, and GSE9844) were analyzed using the limma R package after preprocessing and removing batch effects. The heatmap was used to evaluate batch effects of the integrated data before and after using ComBat methods (Supplementary Figures 1, 2). Before removing batch effects, the cluster tree of the samples was divided into 5 categories. All samples of the 5 datasets in the cluster tree were evenly mixed together and the batch effects were removed after performing ComBat methods. A total of 568 DEGs, including 272 upregulated genes and 296 downregulated genes, were identified for subsequent analysis.
Gene ontology and pathway enrichment analysis
GO and KEGG pathway enrichment analyses were performed using the clusterProfiler R package. For the biological processes (BP), the top 5 enriched categories among DEGs were extracellular matrix (ECM) organization, extracellular structure organization, collagen metabolic process, muscle filament sliding, and actin-myosin filament sliding. In cellular component (CC) ontology, DEGs were significantly enriched in ECM, proteinaceous ECM, ECM component, contractile fiber componant, and contractile fiber componant. Molecular function (MF) analysis indicated that structural constituent of muscle, integrin binding, actin binding, heparin binding, and growth factor binding were the top 5 commonly enriched categories (Table 2). KEGG analysis showed that DEGs were mainly enriched in ECM-receptor interaction, focal adhesion, protein digestion and absorption, amoebiasis, and IL-17 signaling pathway (Table 2).
Table 2.
The top five significant enriched GO terms and KEGG pathways of DEGs in OSCC.
| Category | Term/pathway ID | Description | Count | P-value |
|---|---|---|---|---|
| BP | GO: 0030198 | Extracellular matrix organization | 64 | 1.46E-27 |
| BP | GO: 0043062 | Extracellular structure organization | 59 | 5.25E-27 |
| BP | GO: 0032963 | Collagen metabolic process | 29 | 1.73E-19 |
| BP | GO: 0030049 | Muscle filament sliding | 19 | 9.50E-19 |
| BP | GO: 0033275 | Actin-myosin filament sliding | 19 | 9.50E-19 |
| CC | GO: 0031012 | Extracellular matrix | 84 | 2.11E-40 |
| CC | GO: 0005578 | Proteinaceous extracellular matrix | 69 | 1.50E-34 |
| CC | GO: 0044420 | Extracellular matrix component | 31 | 5.15E-23 |
| CC | GO: 0043292 | Contractile fiber | 44 | 1.82E-22 |
| CC | GO: 0044449 | Contractile fiber part | 42 | 4.39E-21 |
| MF | GO: 0008307 | Structural constituent of muscle | 18 | 7.59E-16 |
| MF | GO: 0005178 | Integrin binding | 20 | 1.73E-10 |
| MF | GO: 0003779 | Actin binding | 39 | 6.54E-10 |
| MF | GO: 0008201 | Heparin binding | 23 | 7.87E-10 |
| MF | GO: 0019838 | Growth factor binding | 21 | 9.45E-10 |
| KEGG | hsa04512 | ECM-receptor interaction | 21 | 4.76E-11 |
| KEGG | hsa04510 | Focal adhesion | 28 | 1.34E-08 |
| KEGG | hsa04974 | Protein digestion and absorption | 19 | 6.94E-08 |
| KEGG | hsa05146 | Amoebiasis | 18 | 2.22E-07 |
| KEGG | hsa04657 | IL-17 signaling pathway | 16 | 3.48E-06 |
GO – Gene ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes; BP – biological processes; CC – cellular component; MF – molecular function.
PPI network construction and module selection
The PPI network of DEGs, including 428 nodes and 1944 edges, was constructed using the STRING online database and Cytoscape software (Figure 2). Then, the MCODE plug-in was applied for module selection of the PPI network and module 1, comprising 93 nodes and 818 edges, got the highest score (Figure 3). In module 1, the top 10 genes according to mocde scores were PLOD2, COL10A1, COL11A1, COL17A1, COL5A2, COL7A1, COL16A1, COL4A5, OAS1, and RSAD2.
Figure 2.

PPI network construction. PPI network of DEGs containing 428 nodes and 1944 edges was constructed. Upregulated and downregulated genes are shaded in red color and green, respectively. PPI – protein–protein interaction; DEGs – differentially expressed genes.
Figure 3.

Module analysis of PPI network. Module 1 with the highest score was identified by MCODE plug-in of Cytoscape software, including 93 nodes and 818 edges. Upregulated and downregulated genes are shaded red and green, respectively. PPI – protein–protein interaction; MCODE – Molecular Complex Detection.
Weighted co-expression network construction and identification of key modules
The integrated gene expression profiles in which we had been eliminated the batch effects were analyzed by WGCNA R package. The power of β=5 (scale-free R2=0.86) was set as the appropriate soft-thresholding value to satisfy the scale-free network criteria. A total of 16 modules, ranging in size from 77 to 1857 genes, were identified and labeled with different colors (Figure 4A). According to the correlation between MEs and trait, the pink module was the most correlated with trait of cancer and was considered as the key module (Figure 4B).
Figure 4.

WGCNA of the integrated data. (A) Dendrogram of the integrated gene expression data clustered based on a dissimilarity measure. (B) Heatmap of the correlation between module MEs and OSCC. The pink module was the most positively correlated with OSCC. (C) Scatter plot of MEs in the pink module. (D) Twenty-four genes all in 3 parts of results were identified as the candidate genes. The detailed data are shown in Supplementary Table 1. OSCC – oral squamous cell carcinoma; WGCNA – weighted gene co-expression network analysis; MEs – module eigengenes (the first principal component of one module).
L2 Candidate genes mining for OSCC
Based on the hub genes screening criteria in the key module (|GS|>0.2 and |MM|> 0.8), 34 genes in the pink module were regarded as hub genes in the co-expression network (Figure 4C, Supplementary Table 1). TCGA RNA-seq expression data were analyzed using the edgeR package. There were 6712 DEGs in total under the threshold value of FDR <0.05 and |log2FC| >1.5, in which 4060 upregulated and 2652 downregulated genes were identified. Using the intersect function in Venny 2.1.0. we identified 24 genes in all 3 parts of the results (DEGs of 5 gene microarray data, DEGs of TCGA data, and hub genes in the co-expression network) as the candidate genes (Figure 4D, Supplementary Table 1).
Hub genes identification and validation
Log-rank analysis was used to evaluate the difference in overall survival between high expression and low expression of these candidate genes. This procedure showed that 7 genes (PLAU, SERPINE1, LAMC2, ITGA5, TGFBI, FSCN1, and HLF) were significantly associated with the prognosis of OSCC patients, and these were defined as hub genes (Figure 5, Supplementary Figures 3, 4). Survival analysis demonstrated that OSCC patients with high expression of 6 hub genes (PLAU, SERPINE1, LAMC2, ITGA5, TGFBI, and FSCN1) had lower overall survival than those with low expression. In contrast, patients with high expression of gene HLF displayed remarkably longer overall survival compared to those with low expression.
Figure 5.

Hub genes identification by Kaplan-Meier survival analysis. Survival analysis indicated that PLAU, SERPINE1, LAMC2, ITGA5, TGFBI, FSCN1, and HLF were the hub genes. The survival analysis of the other 17 genes is shown in Supplementary Figures 3, 4.
GSE23558 was used to confirm the differences in expression of 7 hub genes between normal and OSCC tissues. The results suggested that the expression of hub genes was significantly higher in OSCC tissues than in normal tissues, except for gene HL, which had lower expression in OSCC tissues compared with normal tissues (Table 3). The GSE41613 dataset was successfully used to validate the effect of hub genes on OSCC patient prognosis (Figure 6).
Table 3.
Validation of seven real hub genes in GSE23558.
| Microarray datasets | Gene names | LogFC | Adjusted P value |
|---|---|---|---|
| PLAU | 2.98 | 1.90E-07 | |
| SERPINE1 | 4.42 | 7.13E-05 | |
| LAMC2 | 2.72 | 7.91E-05 | |
| GSE23558 | ITGA5 | 1.82 | 0.0038 |
| TGFBI | 1.75 | 0.0161 | |
| FSCN1 | 2.74 | 0.0004 | |
| HLF | −5.58 | 3.01E-06 |
FC – fold change.
Figure 6.

Survival analysis validation of the 7 hub genes by using GSE41613.
Discussion
The transformative process of normal stratified squamous oral mucosa into squamous cell carcinoma contains several steps and factors in which accumulated genetic alterations intervene with the normal functions of oncogenes and tumor suppressor genes [22]. However, the underlying molecular mechanisms involved in the process are unclear. In the present study, by integrating 5 individual cohorts of gene expression profile datasets and using multiple bioinformatics methods, we identified key pathways and 7 hub genes in OSCC tissues not found in the normal controls.
Based on the GO analysis, the most significant enrichment in biological process was extracellular matrix (ECM) organization, a process occurring at the cellular level, resulting in the assembly, arrangement of constituents, or disassembly of ECM. Through a literature research, we found that the process of ECM organization appears to be associated with cancer-associated fibroblasts (CAFs). Comparison of genes or proteins between CAFs and normal fibroblasts revealed that most of the enriched genes or proteins are related to ECM organization [23,24]. As a major element of tumor stroma, CAFs can promote tumor growth and invasiveness by affecting ECM remodeling through producing MMPs [25,26]. Several studies have also indicated the vital role of CAFs in OSCC development and metastasis [27,28]. Other biological processes such as the collagen metabolic process were also enriched, which might be due to the production of MMPs [29]. Similarly, the top 3 cellular components were associated with ECM. ECM is a highly dynamic structure, continuously performing the remodeling process, including ECM components deposition, degradation, or other modification [26]. These results of biological process and cellular component ontology highlight the pivotal effect of abnormal ECM dynamics on OSCC occurrence and progression. For the molecular function ontology, the enriched categories of structural constituent of muscle, actin binding, heparin binding, and growth factor binding may be relevant to CAFs as well. Fibroblasts can be activated by various growth factors secreted by cancer or immune cells, such as transforming growth factor-beta1 (TGF-β1), hepatocyte growth factor (HGF), and fibroblast growth factor (FGF) [30]. Once activated, fibroblasts undergo phenotypic transformation and turn into CAFs, which can express characteristic markers α-smooth muscle actin (α-SMA), indicating the myofibroblast phenotype and strong contractility [30,31]. In turn, CAFs can synthesize a series of growth factors, cytokines, and chemokines, including TGF-β, HGF, MMPs, VEGF, MCT-4, and IL-6, all of which are critical to inducing the deposition of ECM, promoting epithelial-mesenchymal transition (EMT), and ultimately resulting in tumor initiation and metastasis [32,33]. Consistent with GO analysis, KEGG analysis showed ECM-receptor interaction and focal adhesion were the 2 most significantly enrichment categories. The enrichment of ECM-receptor interaction and focal adhesion signaling pathway may be related to expression of the integrin (ITG) gene [34,35]. Integrins are heterodimeric cell surface receptors that participate in specific interactions between cells and ECM, as well as in cell adhesion [36]. Focal adhesion is a cell–substrate adhesion structure mediated by integrin, with functions including fixing the ends of actin filaments, promoting strong attachments to substrates, and playing a functional role as an integrin signaling platform [37].
Module analysis for the PPI network showed that most of the top 10 genes in module 1 were collagen-related genes. Collagens are one of the most important components of ECM, and increasing evidence suggests collagen affects tumor growth and metastasis through multiple mechanisms [38]. Some studies have explored the mechanism of collagen XVI and XVII facilitating proliferation and invasion of OSCC cells [39,40]. Additionally, the overexpression of some collagen-related genes is also possibly due to the net result of overproduction and degradation.
In this study, we integrated 3 parts of results and combined them with survival analysis to identify 7 genes (PLAU, SERPINE1, LAMC2, TGFBI, ITGA5, FSCN1, and HLF) as hub genes. PLAU (plasminogen activator, urokinase) and SERPINE1 (plasminogen activator inhibitor-1) are both part of the plasminogen activation system. LAMC2, TGFBI, and ITGA5 are ECM protein-related gene. The remaining 2 genes (FSCN1 and HLF) are novel molecules that have received little research interest to date.
PLAU encodes the urokinase-type plasmin activator (uPA), a serine protease which converts inactive plasminogen to plasmin by binding to its receptor (uPAR). uPA has been shown to be involved in tissue remodeling and migration under physiological conditions and tumorigenesis [41,42]. uPA/uPAR plays critical roles in promoting extracellular proteolysis, regulation of cell/ECM interactions, and cell migration, all of which are related to the malignant progression of various tumors [43]. Elevated expression of uPA was observed in OSCC tissues, which was correlated with increased invasiveness of OSCC [44].
SERPINE1, also known as plasminogen activator inhibitor-1 (PAI-1), is the principal inhibitor of tissue plasminogen activator (tPA) and uPA. However, higher expression of SERPINE1 has been described as a poor prognostic marker in several cancers. Especially, SERPINE1 had been validated as a biological marker for treatment regimen selection in patients with node-negative breast cancer [45]. Bajou et al. observed that absence of SERPINE1 expression in mice had the effect of inhibiting malignant cell invasion and angiogenesis after transplantation of malignant keratinocytes, whereas invasion and vascularization recurred when mice were injected with adenovirus carrying hunman SERPINE1 [46]. These observations indicate that SERPINE1 has a multifunctional role in promoting tumor development, invasion, and metastasis, independent of the ability to function as a protease inhibitor. Recent findings have suggested that SERPINE1 can regulate apoptosis and make the tumor cells detach from vitronectin and the ECM by displacing vitronectin from the uPA receptor-vitronectin interaction, thus enhancing tumor cell migration and metastasis [47,48].
LAMC2 is a protein-coding gene that encodes the gamma chain isoform laminin and gamma 2, which form laminin 332 combined with alpha 3 and beta 3 chains. Laminin 332 can drive tumorigenesis and enhances tumor invasion via interactions with collagen VII and integrin receptors alpha6beta4, as well as activation of PI3K and RAC1 [49]. Notably, LAMC2 appears to be preferentially expressed in invading malignant cells in many human cancers [50]. LAMC2 is also one of the 4 signatures accurately predicting lymph node metastasis of OSCC [51].
Integrin-α5 (ITGA5) is a member of the intergrins family, which has been demonstrated to regulate various complex biological events such as differentiation, development, cell adhesion, and control of cancer growth and progression. Integrin α5, which is associates with integrin β1 to form a fibronectin receptor, has been shown to exert a pivotal role in certain cancers such as non-small cell cancer, esophageal squamous cell carcinoma, and breast cancer [52–54]. Integrin α5β1 can promote cancer cell migration and invasion through activating the focal adhesion kinase (FAK) and Src [55]. In addition, Claudia et al. found that integrin α5β1 enhances cancer cell invasiveness by facilitating the generation of contractile forces [56]. However, its expression pattern and function in OSCC are still elusive.
TGFBI, also called βig-h3, encodes transforming growth factor-beta-induced protein, which is an extracellular matrix protein implicated in physiological and pathological processes, including development of corneal dystrophy and tumor formation. TGFBI has dual functions in tumor progression, acting as a tumor promoter or tumor suppressor depending on the tumor microenvironment [57]. TGFBI has been reported to promote metastasis of colon cancer through facilitating tumor cell extravasation [58]. Laura et al. found that TGFBI is necessary for proliferation and survival of melanoma cells as well as metastatic outgrowth [59]. Li et al. demonstrated the role of TGFBI as a tumor suppressor in breast cancer and mesothelioma [60]. Several studies have shown that TGFBI is upregulated in OSCC tissues [61,62], and, compared with normal mouth mucosa, its expression is increased in precancerosis and, more significantly, in OSCC [62]. Our results indicate that TGFBI might have a positive regulatory effect, and higher expression of TGFBI in OSCC tissues suggests a poor prognosis.
FSCN1, known as fascin actin-bundling protein 1, exerts a critical function in regulating cell migration, cell motility, and cell-to-cell interactions [63]. FSCN1 is usually absent in normal epithelial tissues but is overexpressed in many human carcinomas, suggesting aggressive, metastatic carcinomas and poor prognosis [64–66]. FSCN1 was found to participate in EMT and promote invasive filopodia formation, which confers increased motility and metastatic properties to cancerous cells [67]. Given the lack of studies showing involvement of FSCN1 in OSCC, further studies are required.
HLF (hepatic leukemia factor) is a member of the proline and acid-rich (PAR) bZIP transcription factor family, which can form homodimers or heterodimers among each other and regulates transcriptional activity [68]. Chromosomal translocations fuse portions of HLF with the E2A gene to form E2A-HLF, a chimeric transcription factor created by the t(17;19) gene, which contributes to leukemogenesis through its potential to inhibit apoptosis [69,70]. Moreover, HLF can increase miR-132 expression through the HLF binding site BS1 of miR-132 promoter, suppressing the proliferation of glioma cells, metastasis, and radioresistance via inhibiting a downstream factor TTK protein kinase [71]. Intriguingly, in our study, the expression of HLF in OSCC tissues was downregulated compared to normal tissues, and patients with low expression of the HLF gene had shorter overall survival compared to patients with high expression. This result indicates HLF might have potential value in OSCC treatment. Unfortunately, there has been no study discussing the regulating role of HLF in OSCC, and further studies are needed.
Zhang et al. recently used WGCNA to identify 10 hub genes (MMP1, TNFRSF12A, PLAU, FSCN1, PDPN, KRT78, EVPL, GGT6, SMIM5, and CYSRT1) that are associated with OSCC carcinogenesis and undertook survival analysis to validate the prognostic value of these genes [16]. Among these 10 hub genes, 4 genes (PLAU, FSCN1, MMP1, and PDPN) were involved in our analysis. PLAU and FSCN1 were also identified as hub genes in our study, which might have diagnostic and prognostic perspectives for OSCC patients. However, in our study, MMP1 and PDPN were identified as the candidate genes and had no effect on the prognosis of OSCC. These different conclusions regarding MMP1 and PDPN might be due to the difference in data used for survival analysis. The other 6 hub genes (TNFRSF12A, KRT78, EVPL, GGT6, SMIM5, and CYSRT1) identified by Zhang et al. were not included in our 2 parts of results (DEGs of 5 gene microarray data and hub genes in the co-expression network). Several factors might be responsible for this difference between the 2 studies. First, Zhang et al. used a single dataset (GSE30784) to construct the gene co-expression network to identify these 6 genes. In our study, we integrated 5 datasets (GSE30784, GSE13601, GSE37991, GSE31056, and GSE9844) for subsequent analysis. Therefore, these 6 genes might not be statistically significant in our integrative data, and cannot meet our cut-off criteria. Second, Zhang et al. used WGCNA to study the genes related to the transformation of normal mucosa to oral dysplasia and oral dysplasia to carcinoma. However, among the 5 datasets obtained from GEO, only GSE30784 contains normal mucosa, oral dysplasia, and OSCC samples. Thus, we could merely identify hub genes related to the process of normal to OSCC, which might also lead to the different results.
There are several limitations worth noting in our study. First, in a co-expression network, a connection between 2 genes cannot be assumed to correspond with a connection in regulatory or PPI networks. When compared with other biological networks where the edges represent well-defined biological interactions, the edges in a co-expression network might be a limited representation of the correlation of the data. Therefore, experimental or clinical studies are needed to further validate these findings. Second, due to the lack of specific clinical information in these 5 datasets from GEO, we failed to construct a co-expression network to explore the relationship between genes and clinical features. Furthermore, we simply studied the effect of hub genes expression on OSCC patient prognosis, while more clinical parameters such as HPV infection and cancer staging should be included in further analysis. Third, among the 5 datasets obtained from GEO, only GSE30784 consists of normal mucosa, oral dysplasia, and OSCC samples. Thus, we could merely analyze the transformation from normal to carcinoma. To include the dynamic analysis from normal to precancerous lesions and further to OSCC would be much better. Fourth, this study only used TCGA and GEO data, and more data from other databases should be assessed to produce a more comprehensive analysis.
Conclusions
We have identified 7 hub genes (PLAU, SERPINE1, LAMC2, TGFBI, ITGA5, FSCN1, and HLF) by using WGCNA and found they are closely correlated with the initiation and prognosis of OSCC. The GO and KEGG pathway enrichment analysis combined with the hub genes has emphasized the essential role of ECM in OSCC occurrence and progression. Among the 7 genes, ITGA5, FSCN1, and HLF are relatively new biomarkers for OSCC, and few studies about their roles in OSCC are currently available, so this topic needs further experimental verification. These novel biomarkers will greatly contribute to the early diagnosis and prognosis prediction in OSCC.
Supplementary Data
The heatmap of the integrated data before removing batch effects. Each column and row represent one sample and one gene, respectively. Red shows high relative expression and green suggests low relative expression. The cluster tree shows that the samples was divided into 5 categories.
The heatmap of the integrated data after removing batch effects. Each column and row represent one sample and one gene, respectively. Red shows high relative expression and green suggests low relative expression. The cluster tree shows that all the samples of 5 datasets were evenly mixed together.
Supplementary Table 1.
Thirty-four hub genes in the co-expression network.
| Gene names | GS. Cancer | MM. turquoise |
|---|---|---|
| LAMC2* | 0.764 | 0.911 |
| PLAU* | 0.754 | 0.892 |
| MMP1* | 0.788 | 0.891 |
| SERPINE1* | 0.714 | 0.882 |
| PDPN* | 0.717 | 0.879 |
| PTHLH* | 0.721 | 0.874 |
| FSCN1* | 0.707 | 0.873 |
| HOMER3* | 0.729 | 0.873 |
| ITGA3* | 0.674 | 0.867 |
| MMP10* | 0.700 | 0.864 |
| CDH3 | 0.750 | 0.860 |
| MYO1B* | 0.720 | 0.858 |
| PLAUR | 0.699 | 0.852 |
| ACTN1 | 0.640 | 0.846 |
| MSN | 0.737 | 0.846 |
| SHC1 | 0.729 | 0.837 |
| TGFBI* | 0.649 | 0.832 |
| TMSB10 | 0.709 | 0.828 |
| MYH9 | 0.613 | 0.817 |
| SLC3A2 | 0.649 | 0.817 |
| ITGA5* | 0.667 | 0.814 |
| PTK7* | 0.724 | 0.812 |
| CYP27B1* | 0.626 | 0.807 |
| SNAI2* | 0.667 | 0.807 |
| ITGB4 | 0.674 | 0.805 |
| IL24* | 0.653 | 0.802 |
| ITGA6* | 0.653 | 0.801 |
| NCOA1 | −0.671 | −0.801 |
| EPHX2* | −0.705 | −0.816 |
| RRAGD* | −0.702 | −0.834 |
| UBL3* | −0.741 | −0.845 |
| ACADSB* | −0.686 | −0.846 |
| HLF* | −0.747 | −0.888 |
| GPD1L* | −0.733 | −0.896 |
24 candidate genes identified through intersection of three parts of results (DEGs of three gene microarray data, DEGs of TCGA data, hub genes in the co-expression network).
GS – gene significance; MM – module membership.
Survival analysis of the other 17 genes.
Survival analysis of the other 17 genes.
Acknowledgements
The authors gratefully acknowledge the researchers who provided the datasets used in this analysis.
Footnotes
Source of support: This study was supported by the Humanities and Social Sciences Program of the Ministry of Education Planning China (16YJA840014)
Cnflict of interests
None.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Thomson PJ. Perspectives on oral squamous cell carcinoma prevention-proliferation, position, progression and prediction. J Oral Pathol Med. 2018;47(9):803–7. doi: 10.1111/jop.12733. [DOI] [PubMed] [Google Scholar]
- 3.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 4.Zheng CM, Ge MH, Zhang SS, et al. Oral cavity cancer incidence and mortality in China, 2010. J Cancer Res Ther. 2015;11(Suppl 2):C149–54. doi: 10.4103/0973-1482.168176. [DOI] [PubMed] [Google Scholar]
- 5.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4–5):309–16. doi: 10.1016/j.oraloncology.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Smokeless tobacco and some tobacco-specific N-nitrosamines. IARC Monogr Eval Carcinog Risks Hum. 2007;89:1–592. [PMC free article] [PubMed] [Google Scholar]
- 7.Reidy J, McHugh E, Stassen LF. A review of the relationship between alcohol and oral cancer. Surgeon. 2011;9(5):278–83. doi: 10.1016/j.surge.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 8.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Betel-quid and areca-nut chewing and some areca-nut derived nitrosamines. IARC Monogr Eval Carcinog Risks Hum. 2004;85:1–334. [PMC free article] [PubMed] [Google Scholar]
- 9.Dalianis T. Human papillomavirus and oropharyngeal cancer, the epidemics, and significance of additional clinical biomarkers for prediction of response to therapy (Review) Int J Oncol. 2014;44(6):1799–805. doi: 10.3892/ijo.2014.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez I, Seoane J, Varela-Centelles P, et al. Is diagnostic delay related to advanced-stage oral cancer? A meta-analysis. Eur J Oral Sci. 2009;117(5):541–46. doi: 10.1111/j.1600-0722.2009.00672.x. [DOI] [PubMed] [Google Scholar]
- 11.Li G, Li X, Yang M, et al. Prediction of biomarkers of oral squamous cell carcinoma using microarray technology. Sci Rep. 2017;7:42105. doi: 10.1038/srep42105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, Sun S, Zeng X, Cui L. Expression profiles analysis identifies a novel three-mRNA signature to predict overall survival in oral squamous cell carcinoma. Am J Cancer Res. 2018;8(3):450–61. [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 14.Carter SL, Brechbuhler CM, Griffin M, Bond AT. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics. 2004;20(14):2242–50. doi: 10.1093/bioinformatics/bth234. [DOI] [PubMed] [Google Scholar]
- 15.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Feng H, Li Z, et al. Application of weighted gene co-expression network analysis to identify key modules and hub genes in oral squamous cell carcinoma tumorigenesis. Onco Targets Ther. 2018;11:6001–21. doi: 10.2147/OTT.S171791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy – analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 18.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–87. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomson PJ. Perspectives on oral squamous cell carcinoma prevention-proliferation, position, progression and prediction. J Oral Pathol Med. 2018;47(9):803–7. doi: 10.1111/jop.12733. [DOI] [PubMed] [Google Scholar]
- 23.Gandellini P, Andriani F, Merlino G, et al. Complexity in the tumour microenvironment: Cancer associated fibroblast gene expression patterns identify both common and unique features of tumour-stroma crosstalk across cancer types. Semin Cancer Biol. 2015;35:96–106. doi: 10.1016/j.semcancer.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Bagordakis E, Sawazaki-Calone I, Macedo CC, et al. Secretome profiling of oral squamous cell carcinoma-associated fibroblasts reveals organization and disassembly of extracellular matrix and collagen metabolic process signatures. Tumour Biol. 2016;37(7):9045–57. doi: 10.1007/s13277-015-4629-y. [DOI] [PubMed] [Google Scholar]
- 25.Otranto M, Sarrazy V, Bonte F, et al. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6(3):203–19. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12) doi: 10.1101/cshperspect.a005058. pii: a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawashiri S, Tanaka A, Noguchi N, et al. Significance of stromal desmoplasia and myofibroblast appearance at the invasive front in squamous cell carcinoma of the oral cavity. Head Neck. 2009;31(10):1346–53. doi: 10.1002/hed.21097. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Min A, Gao S, Tang Z. Genetic regulation and potentially therapeutic application of cancer-associated fibroblasts in oral cancer. J Oral Pathol Med. 2014;43(5):323–34. doi: 10.1111/jop.12098. [DOI] [PubMed] [Google Scholar]
- 29.Van Doren SR. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015;44–46:224–31. doi: 10.1016/j.matbio.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuzet SE, Gaggioli C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016;365(3):607–19. doi: 10.1007/s00441-016-2467-x. [DOI] [PubMed] [Google Scholar]
- 31.Yamaguchi H, Sakai R. Direct interaction between carcinoma cells and cancer associated fibroblasts for the regulation of cancer invasion. Cancers (Basel) 2015;7(4):2054–62. doi: 10.3390/cancers7040876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’ – more than meets the eye. Trends Mol Med. 2013;19(8):447–53. doi: 10.1016/j.molmed.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60. doi: 10.1016/j.semcancer.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Bohanes P, Yang D, Loupakis F, et al. Integrin genetic variants and stage-specific tumor recurrence in patients with stage II and III colon cancer. Pharmacogenomics J. 2015;15(3):226–34. doi: 10.1038/tpj.2014.66. [DOI] [PubMed] [Google Scholar]
- 35.Thomas GJ, Nystrom ML, Marshall JF. Alphavbeta6 integrin in wound healing and cancer of the oral cavity. J Oral Pathol Med. 2006;35(1):1–10. doi: 10.1111/j.1600-0714.2005.00374.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang HJ, Tao J, Sheng L, et al. Twist2 promotes kidney cancer cell proliferation and invasion by regulating ITGA6 and CD44 expression in the ECM-receptor interaction pathway. Onco Targets Ther. 2016;9:1801–12. doi: 10.2147/OTT.S96535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5(10):816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 38.Chen P, Cescon M, Bonaldo P. Collagen VI in cancer and its biological mechanisms. Trends Mol Med. 2013;19(7):410–17. doi: 10.1016/j.molmed.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Bedal KB, Grassel S, Oefner PJ, et al. Collagen XVI induces expression of MMP9 via modulation of AP-1 transcription factors and facilitates invasion of oral squamous cell carcinoma. PLoS One. 2014;9(1):e86777. doi: 10.1371/journal.pone.0086777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parikka M, Nissinen L, Kainulainen T, et al. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration – a novel role for alphaIIb integrin and tirofiban. Exp Cell Res. 2006;312(8):1431–38. doi: 10.1016/j.yexcr.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Shi Z, Stack MS. Urinary-type plasminogen activator (uPA) and its receptor (uPAR) in squamous cell carcinoma of the oral cavity. Biochem J. 2007;407(2):153–59. doi: 10.1042/BJ20071037. [DOI] [PubMed] [Google Scholar]
- 42.Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: A review. Int J Cancer. 1997;72(1):1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 43.Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: Recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22(2–3):205–22. doi: 10.1023/a:1023099415940. [DOI] [PubMed] [Google Scholar]
- 44.Yoshizawa K, Nozaki S, Kitahara H, et al. Expression of urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor and maspin in oral squamous cell carcinoma: Association with mode of invasion and clinicopathological factors. Oncol Rep. 2011;26(6):1555–60. doi: 10.3892/or.2011.1419. [DOI] [PubMed] [Google Scholar]
- 45.American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer. J Oncol Pract. 2007;3(6):336–39. doi: 10.1200/JOP.0768504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bajou K, Noel A, Gerard RD, et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat Med. 1998;4(8):923–28. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 47.Balsara RD, Ploplis VA. Plasminogen activator inhibitor-1: The double-edged sword in apoptosis. Thromb Haemost. 2008;100(6):1029–36. [PMC free article] [PubMed] [Google Scholar]
- 48.Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160(5):781–91. doi: 10.1083/jcb.200208117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marinkovich MP. Tumour microenvironment: Laminin 332 in squamous-cell carcinoma. Nat Rev Cancer. 2007;7(5):370–80. doi: 10.1038/nrc2089. [DOI] [PubMed] [Google Scholar]
- 50.Pyke C, Romer J, Kallunki P, et al. The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am J Pathol. 1994;145(4):782–91. [PMC free article] [PubMed] [Google Scholar]
- 51.Zanaruddin SN, Saleh A, Yang YH, et al. Four-protein signature accurately predicts lymph node metastasis and survival in oral squamous cell carcinoma. Hum Pathol. 2013;44(3):417–26. doi: 10.1016/j.humpath.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 52.Dingemans AM, van den Boogaart V, Vosse BA, et al. Integrin expression profiling identifies integrin alpha5 and beta1 as prognostic factors in early stage non-small cell lung cancer. Mol Cancer. 2010;9:152. doi: 10.1186/1476-4598-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ju JA, Godet I, Ye IC, et al. Hypoxia selectively enhances integrin alpha5beta1 receptor expression in breast cancer to promote metastasis. Mol Cancer Res. 2017;15(6):723–34. doi: 10.1158/1541-7786.MCR-16-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie JJ, Guo JC, Wu ZY, et al. Integrin alpha5 promotes tumor progression and is an independent unfavorable prognostic factor in esophageal squamous cell carcinoma. Hum Pathol. 2016;48:69–75. doi: 10.1016/j.humpath.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 55.Deng B, Zhang S, Miao Y, et al. Adrenomedullin expression in epithelial ovarian cancers and promotes HO8910 cell migration associated with upregulating integrin alpha5beta1 and phosphorylating FAK and paxillin. J Exp Clin Cancer Res. 2012;31:19. doi: 10.1186/1756-9966-31-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mierke CT, Frey B, Fellner M, et al. Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci. 2011;124(Pt 3):369–83. doi: 10.1242/jcs.071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ozawa D, Yokobori T, Sohda M, et al. TGFBI expression in cancer stromal cells is associated with poor prognosis and hematogenous recurrence in esophageal squamous cell carcinoma. Ann Surg Oncol. 2016;23(1):282–89. doi: 10.1245/s10434-014-4259-4. [DOI] [PubMed] [Google Scholar]
- 58.Ma C, Rong Y, Radiloff DR, et al. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 2008;22(3):308–21. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lauden L, Siewiera J, Boukouaci W, et al. TGF-beta-induced (TGFBI) protein in melanoma: A signature of high metastatic potential. J Invest Dermatol. 2014;134(6):1675–85. doi: 10.1038/jid.2014.20. [DOI] [PubMed] [Google Scholar]
- 60.Li B, Wen G, Zhao Y, et al. The role of TGFBI in mesothelioma and breast cancer: Association with tumor suppression. BMC Cancer. 2012;12:239. doi: 10.1186/1471-2407-12-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tomioka H, Morita K, Hasegawa S, Omura K. Gene expression analysis by cDNA microarray in oral squamous cell carcinoma. J Oral Pathol Med. 2006;35(4):206–11. doi: 10.1111/j.1600-0714.2006.00410.x. [DOI] [PubMed] [Google Scholar]
- 62.He Y, Shao F, Pi W, et al. Largescale transcriptomics analysis suggests over-expression of BGH3, MMP9 and PDIA3 in oral squamous cell carcinoma. PLoS One. 2016;11(1):e0146530. doi: 10.1371/journal.pone.0146530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashimoto Y, Kim DJ, Adams JC. The roles of fascins in health and disease. J Pathol. 2011;224(3):289–300. doi: 10.1002/path.2894. [DOI] [PubMed] [Google Scholar]
- 64.Liu C, Gao H, Cao L, et al. The role of FSCN1 in migration and invasion of pituitary adenomas. Mol Cell Endocrinol. 2016;419:217–24. doi: 10.1016/j.mce.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 65.Hanker LC, Karn T, Holtrich U, et al. Prognostic impact of fascin-1 (FSCN1) in epithelial ovarian cancer. Anticancer Res. 2013;33(2):371–77. [PubMed] [Google Scholar]
- 66.Luo A, Yin Y, Li X, et al. The clinical significance of FSCN1 in non-small cell lung cancer. Biomed Pharmacother. 2015;73:75–79. doi: 10.1016/j.biopha.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 67.Machesky LM, Li A. Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol. 2010;3(3):263–70. doi: 10.4161/cib.3.3.11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiang DM, Sun W, Ning BF, et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut. 2018;67(9):1704–15. doi: 10.1136/gutjnl-2016-313392. [DOI] [PubMed] [Google Scholar]
- 69.Inukai T, Inaba T, Dang J, et al. TEF, an antiapoptotic bZIP transcription factor related to the oncogenic E2A-HLF chimera, inhibits cell growth by down-regulating expression of the common beta chain of cytokine receptors. Blood. 2005;105(11):4437–44. doi: 10.1182/blood-2004-08-2976. [DOI] [PubMed] [Google Scholar]
- 70.Hunger SP, Li S, Fall MZ, et al. The proto-oncogene HLF and the related basic leucine zipper protein TEF display highly similar DNA-binding and transcriptional regulatory properties. Blood. 1996;87(11):4607–17. [PubMed] [Google Scholar]
- 71.Chen S, Wang Y, Ni C, et al. HLF/miR-132/TTK axis regulates cell proliferation, metastasis and radiosensitivity of glioma cells. Biomed Pharmacother. 2016;83:898–904. doi: 10.1016/j.biopha.2016.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The heatmap of the integrated data before removing batch effects. Each column and row represent one sample and one gene, respectively. Red shows high relative expression and green suggests low relative expression. The cluster tree shows that the samples was divided into 5 categories.
The heatmap of the integrated data after removing batch effects. Each column and row represent one sample and one gene, respectively. Red shows high relative expression and green suggests low relative expression. The cluster tree shows that all the samples of 5 datasets were evenly mixed together.
Supplementary Table 1.
Thirty-four hub genes in the co-expression network.
| Gene names | GS. Cancer | MM. turquoise |
|---|---|---|
| LAMC2* | 0.764 | 0.911 |
| PLAU* | 0.754 | 0.892 |
| MMP1* | 0.788 | 0.891 |
| SERPINE1* | 0.714 | 0.882 |
| PDPN* | 0.717 | 0.879 |
| PTHLH* | 0.721 | 0.874 |
| FSCN1* | 0.707 | 0.873 |
| HOMER3* | 0.729 | 0.873 |
| ITGA3* | 0.674 | 0.867 |
| MMP10* | 0.700 | 0.864 |
| CDH3 | 0.750 | 0.860 |
| MYO1B* | 0.720 | 0.858 |
| PLAUR | 0.699 | 0.852 |
| ACTN1 | 0.640 | 0.846 |
| MSN | 0.737 | 0.846 |
| SHC1 | 0.729 | 0.837 |
| TGFBI* | 0.649 | 0.832 |
| TMSB10 | 0.709 | 0.828 |
| MYH9 | 0.613 | 0.817 |
| SLC3A2 | 0.649 | 0.817 |
| ITGA5* | 0.667 | 0.814 |
| PTK7* | 0.724 | 0.812 |
| CYP27B1* | 0.626 | 0.807 |
| SNAI2* | 0.667 | 0.807 |
| ITGB4 | 0.674 | 0.805 |
| IL24* | 0.653 | 0.802 |
| ITGA6* | 0.653 | 0.801 |
| NCOA1 | −0.671 | −0.801 |
| EPHX2* | −0.705 | −0.816 |
| RRAGD* | −0.702 | −0.834 |
| UBL3* | −0.741 | −0.845 |
| ACADSB* | −0.686 | −0.846 |
| HLF* | −0.747 | −0.888 |
| GPD1L* | −0.733 | −0.896 |
24 candidate genes identified through intersection of three parts of results (DEGs of three gene microarray data, DEGs of TCGA data, hub genes in the co-expression network).
GS – gene significance; MM – module membership.
Survival analysis of the other 17 genes.
Survival analysis of the other 17 genes.