

Abstract
We report here a ‘nonspectator’ behavior for an unsupported L-function σ3–P ligand (i.e. P{N[o-NMe-C6H4]2}, 1a) in complex with the cyclopentadienyliron dicarbonyl cation (Fp+). Treatment of 1a•Fp+ with [(Me2N)3S][Me3SiF2] results in fluoride addition to the P-center, giving the isolable crystalline fluorometallophosphorane 1aF•Fp that allows a crystallographic assessment of the variance in the Fe–P bond as a function of P-coordination number. The nonspectator reactivity of 1a•Fp+ is rationalized on the basis of electronic structure arguments and by comparison to trigonal analogue (Me2N)3P•Fp+ (i.e. 1b•Fp+), which is inert to fluoride addition. These observations establish a nonspectator L/X-switching in (σ3–P)–M complexes by reversible access to higher-coordinate phosphorus ligand fragments.
Keywords: coordination modes, hypervalent compounds, ligand effects, ligand reactivity, phosphorous ligands
Graphical Abstract
An L-function trivalent phosphorus ligand accepts fluoride ion by nucleophilic addition to generate a stable, isolable metallophosphorane; the reaction is reversible. Changes to the metal–phosphorus bonding as a function coordination number at P are analyzed crystallographically and computationally.
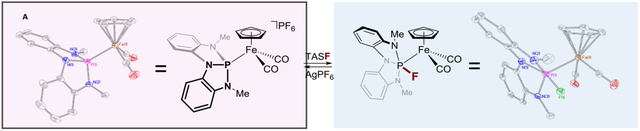
Tricoordinate phosphorus (σ3–P) compounds are archetypal donor ligands in coordination chemistry.1,2,3 Within the Covalent Bond Classification,4,5 σ3–P compounds are designated L-function ligands for transition metals (M) and are overwhelmingly construed as inert, ancillary, spectator ligands within (σ3–P)–M complexes. A rich ‘nonspectator’ reaction chemistry of metal-bound σ3–P compounds, however, belies this prevailing view. Abstraction of a P-substituent from (σ3–P)–M complexes accesses dicoordinate phosphorus ligands (Figure 1a; σ2–P–, phosphide; σ2–P+, phosphenium),6 and the σ2–P+/–/σ3–P interconversion has been the focus of extensive stoichiometric7,8,9,10,11,12,13 and catalytic14 investigation. By complement, addition of an exogenous nucleophile to phosphorus in an L-function (σ3–P)–M complex increases the P-coordination number, resulting in a ‘metallophosphorane’ complex with an X-function (σ4–P)–M formula.15 Literature concerning the addition of a P-substituent to (σ3–P)–M complexes to give higher-coordinate phosphorus congeners is comparatively sparse.16 Verkade has postulated that fluoride addition to PdII-(bis)phosphines induces PdII→Pd0 reduction via initial addition of F– to P.17 Further, Nakazawa and Miyoshi have shown the possibility of nucleophilic substitution of P-substituents in cationic FeII-phosphite complexes, in some cases leading to persistent (σ4–P)–M products.18,19
Figure 1.

Nonspectator modes of reactivity for (σ3–P)–M complexes.
Recently, a κ3-chelate containing a nontrigonal σ3–P center (Figure 1,B) was shown to access directly a (σ4–P)–M metallophosphorane by formal insertion to a Ru–H bond.20 An interpretation of XANES data for B and related compounds A attributed the propensity of the phosphorus center to attain higher coordination to the presence of a low-energy P-based orbital made accessible by the nontrigonal local environment.21 The presence of the low-lying P-centered orbital in A and related compounds raised the prospect of accentuated intermolecular electrophilic reactivity of such nontrigonal σ3–P ligands. We report here the reversible addition of an exogenous nucleophile to the P-center of an unsupported (σ3–P)–M complex C that demonstrates a nonspectator behavior of ligands A. With this study, direct experimental evidence is provided that delineates: (1) the inherent electronic impact on metal-binding arising from nontrigonal distortion of σ3–P ligands without convolution from chelate effects, and (2) the direct crystallographic observation of a nonspectator phosphorus ligand in a higher-coordination state following exogenous nucleophile addition. The ability for nontrigonal σ3–P ligands to reversibly expand local coordination number while remaining σ-bound in the primary ligand sphere of a metal complex forecasts emerging opportunities for functional nonspectator ligands within (σ3–P)–M complexes.22
On the basis of precedent from Martin23 and Nakazawa and Miyoshi,18,19 the cyclopentadienyliron dicarbonyl cation (Fp+) was selected as a coordinatively saturated ‘ancillary metal’24 fragment for study. Iron complexes 1a•Fp+ and 1b•Fp+ were prepared by ligand exchange of [thf•Fp][PF6]25 with P{N[o-NMe-C6H4]2} (1a)26,27 and (Me2N)3P (1b), respectively (Figure 2).
Figure 2.

Synthesis of [R3P•Fp][PF6] complexes, where R3P = 1a or 1b.
According to IR spectroscopy, the CO stretching frequencies of 1a•Fp+ (νasym 2017 cm−1, νsym 2061 cm−1) are higher in energy than those of 1b•Fp+ (νasym 2000 cm−1, νsym 2045 cm−1). This trend tracks qualitatively with the JSe-P coupling constants for phosphorus selenides 1a•Se (JSe-P = 907 Hz) and 1b•Se (JSe-P = 784 Hz), suggesting to a first approximation that 1a is a weaker σ-donor than 1b (see Table 1 for collected metrical data). The 57Fe NMR chemical shifts (obtained indirectly by 2D Fe–P correlation solution NMR experiments due to the low receptivity of the 57Fe nucleus28) for 1a•Fp+ (δ 616 ppm) and 1b•Fp+ (δ 688 ppm) are consistent with this interpretation, based on trends established for related cyclopentadienyliron complexes.29
Table 1.
Collected spectroscopic, structural, and computational data for compounds 1a, 1b, 1a•Fp+, 1b•Fp+, and 1aF•Fp.
| Metric | 31P δ (ppm)a | 1JP-Se (Hz) | 57Fe δ (ppm)b | d(Fe1-P1) (Å) | ν(CO) (cm−1) | FIA (kcal/mol)c | EDA-NOCVd | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Etot | EPauli | Eestat | Esteric | Edisp | Eorb | σ(P→Fe) | π(P←Fe) | |||||||
| 1a | 160.4 | 907e | - | - | - | - | - | - | - | - | - | - | - | - |
| 1b | 122.4 | 784e | - | - | - | - | - | - | - | - | - | - | - | - |
| 1a•Fp+ | 183.5 | - | 616 | 2.1809(4) | 2017, 2061 | 59.3 | −91.9 | 122.7 | −105.0 | 17.7 | −16.1 | −93.6 | −61.7 | −18.1 |
| 1b•Fp+ | 141.4 | - | 688 | 2.2381(5) | 2000, 2045 | 32.9 | −99.8 | 130.0 | −118.4 | 11.6 | −18.4 | −92.9 | −65.8 | −13.2 |
| 1aF•Fp | −3.0 | - | 1013 | 2.3047(9) | 1952, 2007 | - | - | - | - | - | - | - | - | - |
ppm vs. 85% H3PO4.
ppm vs. Fe(CO)5.
Computed (BP86/def2-TZVP(CPCM=CH2Cl2) according to the method in Ref. 32.
EDA-NOCV computational results represent attractive and repulsive energies (kcal/mol) between the Fp+ fragment and phosphorus ligands at the fragment geometry of the complex. The direction of donation is defined to be from phosphorus to iron.
Values from Ref. 25.
Further distinctions between 1a•Fp+ and 1b•Fp+ are manifest in structural analyses based on X-ray diffractometry data obtained with single-crystalline samples (Figure 3). Most evidently, compound 1a•Fp+ features a shorter Fe–P bond length (dFe–P = 2.1809(4) Å) as compared to compound 1b•Fp+ (dFe–P = 2.2381(5) Å). Also, consistent with the aforementioned vibrational data, the average Fe–CCO bond length in 1a•Fp+ (dFe–C = 1.7886(17) Å) is slightly longer than in 1b•Fp+ (dFe–C = 1.7766(19) Å). A further feature of note concerns the dihedral angles φ(N-P-Fe-N); by projection down the P–Fe axis (Figure 3A, right), compound 1a•Fp+ shows a span of dihedral angles Ω(φ) = 28.11(26)°, with a maximum dihedral of φ(N2-P-Fe-N3) = 137.95(13)°. By contrast, compound 1b•Fp+ shows only a span of dihedral angles Ω(φ) = 5.3(3)° and a maximum dihedral of φ(N1-P-Fe-N3) = 121.87(14)°. These metrics illustrate the enhanced nontrigonal local geometry about phosphorus for 1a•Fp+ as compared to 1b•Fp+, consistent with the structural distinctions between the free ligands.26 For reference, the N2–P–N3 bond angle of 1a•Fp+ (116.40(7)⁰) is almost unchanged from that of 1a (115.21(7)⁰), showing that complexation does not significantly perturb the phosphorus triamide framework.
Figure 3.

(A) Left: Thermal ellipsoid plot for 1a•Fp+ rendered at 50% probability level. Hydrogen atoms, noncoordinating PF6– counterion, and a THF solvent molecule are omitted for clarity. Selected metrical data for 1a•Fp+: d(Fe-P): 2.1809(4) Å, d(Fe-(CO)1): 1.7879(17) Å, d(Fe-(CO)2): 1.7893(16) Å, ∠(N1-P-N2): 93.42(6)⁰, ∠(N1-P-N3): 93.04(7)⁰, ∠(N2-P-N3): 116.39(7)⁰. Right: Schematic projection down the P–Fe axis for 1a•Fp+ illustrating dihedral angles φ(N-P-Fe-N). (B) Left: Thermal ellipsoid plot for 1b•Fp+ rendered at 50% probability level. Only one of two molecules in the asymmetric unit is depicted. Hydrogen atoms and a noncoordinating PF6– counterion are omitted for clarity. Selected metrical data for 1b•Fp+: d(Fe-P): 2.2381(5) Å, d(Fe-(CO)1): 1.7739(19) Å, d(Fe-(CO)2): 1.7792(18) Å, ∠(N1-P-N2): 101.59(8) ⁰, ∠(N1-P-N3): 105.03(9)⁰, ∠(N2-P-N3): 107.09(9)⁰. Right: Schematic projection down the P–Fe axis for 1b•Fp+ illustrating dihedral angles φ(N-P-Fe-N). See SI for full details.
In an effort to parse the σ- and π- contributions to the Fe–P bonding interactions in 1a•Fp+ and 1b•Fp+, an energy partitioning into pairwise orbital interactions between σ3–P ligand (1a and 1b, respectively) and Fp+ fragments was undertaken with the Energy Decomposition Analysis – Natural Orbitals for Chemical Valence (EDA-NOCV) method30 as implemented in the ADF modeling program31 at the BP86/def2-TZVP level of density functional theory (Table 1, see SI for full details). Along lines described by Michalak,32 deconvolution of the covalent bonding portion (Eorb) into σ- and π-symmetry components for 1a•Fp+ gives donation σ(P→Fe) = −61.7 kcal/mol (65.9% of Eorb) and back-donation π(P←Fe) = −18.1 kcal/mol (19.3% of Eorb). An illustration of the electron deformation densities for the three principal NOCV interactions of 1a•Fp+ is presented in Figure 4. NOCV deformation density channel Δρ1 depicts depletion of electron density at P (red) and accrual of electron density at Fe (blue) as would be expected for an L-function σ-dative interaction. NOCV deformation density channels Δρ2 and Δρ3 correspond to the backflow of electron density from an Fe dπ orbital into P-based π-acceptor orbitals with two distinct interaction energies (ΔE2orb = −10.7 kcal/mol, ΔE3orb = −7.35 kcal/mol), consistent with the lifting of pπ degeneracy at nontrigonal 1a shown by previous XAS evidence.21 By way of comparison, EDA-NOCV partitioning of the Fe–P bond in 1b•Fp+ gives donation σ(P→Fe) = −65.8 kcal/mol (70.8% of Eorb) and back-donation π(P←Fe) = −13.2 kcal/mol (14.2% of Eorb). This analysis therefore quantifies the relatively weaker σ-donating ability of nontrigonal σ3–P compound 1a as compared to a compositionally related phosphorous triamide 1b evident from spectroscopy (vide supra). Further, a combined consideration of the spectroscopic, structural, and theoretical data suggests a relatively stronger π-accepting ability of 1a vs. 1b.
Figure 4.

Contours of electron deformation density channels Δρ1, Δρ2, and Δρ3 describing the bonding between 1a and the Fp+ metal fragments with corresponding energies and charge estimations obtained from EDA-NOCV method.
To quantify the relative electrophilicity of P-based acceptor orbitals for 1a•Fp+ vs. 1b•Fp+, solvation-corrected fluoride ion affinities (FIAs) were computed at the M06L/def2-TZVP(CPCM:CH2Cl2) level of theory by isodesmic reaction enthalpies according to Christe’s method.33 The FIA for 1a•Fp+ is computed to be significantly larger (–ΔH = 59.3 kcal/mol) than that for 1b•Fp+ (–ΔH = 32.9 kcal/mol). The low absolute values for the FIAs are indicative a modest overall fluoride affinity,34 but the difference Δ(FIA) = 26.4 kcal/mol conforms to the interpretation that P-based electrophilic reactivity should be favored at the nontrigonal complex 1a•Fp+.
The reactivity of 1a•Fp+ and 1b•Fp+ toward fluoride addition was probed experimentally. Treatment of compound 1a•Fp+ with tris(dimethylamino)sulfonium trimethyldifluorosilicate (TASF) in acetonitrile resulted in an immediate change in color from yellow to deep orange (Figure 5a). The formation of a single new phosphorus-containing species was evident by 31P NMR spectroscopy, as indicated by the doublet resonance at δ −3.0 ppm, which displayed large scalar coupling (J = 971 Hz) consistent with the presence of a single fluorine bound to phosphorus via a direct P–F bond. The large upfield shift in 31P NMR chemical shift is consistent with an increased coordination number at phosphorus by fluoride addition, and this inference is confirmed by observation of the complementary coupling in the lone 19F NMR resonance (δ 27.4 ppm, J = 971 Hz, Figure 5b). The product was thus assigned to be fluorometallophosphorane 1aF•Fp, in which a fluoride has been added to the phosphorus of 1a•Fp+ to generate a neutral complex. In solution, compound 1aF•Fp exhibits time-averaged molecular Cs-symmetry with a persistent P–Fe bond;13C NMR spectra demonstrate an equivalence of the CO ligands (one resonance at δ 211 ppm) with well-resolved 2JC–P = 49 Hz and 3JC–F = 5.7 Hz coupling constants. Treatment of 1b•Fp+ to identical fluorinating conditions (TASF, MeCN, rt) does not result in fluorination but instead returns starting materials alongside some decomposition of 1b•Fp+. It is evident that fluoride addition to a higher coordinate phosphorus ligand is enabled by the enhanced electrophilicity of 1a•Fp+ as compared to 1b•Fp+.
Figure 5.

A) Reversible fluorination of 1a•Fp+ and the resulting fluorometallophosphorane 1aF•Fp. B) Solution 31P NMR spectra in CD3CN: (top) spectrum of 1a•Fp+; (middle) spectrum of 1aF•Fp from addition of TASF to 1a•Fp+; (bottom) spectrum of 1a•Fp+ following treatment of 1aF•Fp with AgPF6 and removal of precipitate (AgF). C) Thermal ellipsoid plot rendered at 50% probability level for 1aF•Fp. Hydrogen atoms are removed for clarity. Relevant metrical data for 1aF•Fp: d(Fe-P): 2.3047(9) Å, d(P-F): 1.6687(18) Å, ∠(N1-P-F): 158.11(12)⁰, ∠(N2-P-N3): 134.91(13)⁰, ɸ(C2-Fe-P-F) = 2.63⁰, ɸ(C1-Fe-P-N3) = 8.12⁰. See SI for full details.
The air and moisture sensitive orange 1aF•Fp can be crystallized by slow evaporation of a saturated CH2Cl2 solution at −35° C (Figure 5c). X-ray diffractometry confirms the structural assignment of 1aF•Fp as a metallophosphorane resulting from addition of an exogenous fluoride to σ3–P ligand 1a without further substitution. With respect to the Fe bonding environment, compound 1aF•Fp features an increased Fe–P bond length (dFe–P = 2.3047(9) Å) as compared to 1a•Fp+, as well as a shorter average Fe–CCO bond length (dFe–C = 1.764(3) Å) that coincides with a bathochromic shift of the carbonyl stretching frequencies (νasym 1952 cm−1, νsym 2007 cm−1). With respect to the P bonding environment, metrical parameters give a geometry index of τ = 0.35, indicating a geometry closer to that of a square pyramid than a trigonal bipyramid.35 The addition of fluoride results in an increase in all of the P–N bond lengths by 0.05 Å < ΔdP–N < 0.09 Å as is common for higher-coordinate main group compounds that compensate for their formal ‘hypervalent’ character by distribution of electron density toward the substituents.36 The P–F bond length is quite long (dP–F = 1.6687(18) Å), but falls within the range (1.64(11) Å < dP–F < 1.69(1) Å) observed for the only prior example of a structurally characterized fluorometallophosphorane (i.e. Ir(CO)Cl2(PEt3)2(PF4)) from Holloway.37
Bonding analysis in 1a•Fp+ and 1aF•Fp reveals changes to the nature of the Fe–P σ-interactions as a function of fluoride binding. NBO analysis reports a dative covalent P→Fe σ-interaction for 1a•Fp+ described by an NLMO comprising modest polarization toward the phosphorus (P 56.2%/Fe 38.5%; Figure 6a, left) and involving a P donor NBO with sp1.10 hybridization. The NLMO corresponding to the P–Fe bonding interaction in 1aF•Fp indicates an increased distribution across Fe–P (P 49.8%/Fe 43.9%; Figure 6b, left) with similar phosphorus parentage (sp1.16). Moving from 1a•Fp+ to 1aF•Fp, the Wiberg bond indices decrease (1a•Fp+: WBI = 0.53; 1aF•Fp: WBI = 0.48), in line with the observed increase in bond length from crystallography (ΔdP–Fe = +0.12 Å). For comparison, similar qualitative trends are reported by Gabbai for addition of fluoride to antimony in Pt-Sb bimetallics.38 Here, we invoke a decreased importance of π-backbonding effects in 1aF•Fp to account for this observation; the P-based acceptor orbital is saturated by addition of exogenous fluoride and unavailable for metal bonding.
Figure 6.

Bonding analysis for 1a•Fp+ and 1aF•Fp. (A) Left: NLMO representing P–Fe bond for 1a•Fp+. Right: Contour plot of the Laplacian of the electron-density topology1a•Fp+ in the plane containing the Fe, P, and N atoms. Areas of charge depletion are depicted in red and areas of charge concentration are depicted in blue. Black dots indicate bond critical points. Metrics represent relevant properties at the bond critical points (ρ in e/Å3, ∇2ρ in e/Å5, H/ρ in atomic units). (B) Left: NLMO representing P–Fe bond for 1aF•Fp. Right: Contour plot of the Laplacian of the electron-density topology 1aF•Fp in the plane containing the Fe, P, and F atoms.
Topological analysis of the computed electron density within the Quantum Theory of Atoms in Molecules (QTAIM) framework39 returns bond paths defined by (3, −1) critical points for P–Fe in 1a•Fp+ (Figure 6a, right), and both P–Fe and P–F in 1aF•Fp (Figure 6b, right). No bond paths were located for any F…Fe or N…Fe trajectory, conforming to an η1-formulation of metallophosphorane 1aF•Fp. Qualitatively, P-based valence shell charge concentrations are evident in the Laplacian of the electron density for both 1a•Fp+ and 1aF•Fp along the P–Fe bond path, in line with an L- and X-function ligand classification, respectively. By contrast, the Laplacian distribution for the P–F bond is indicative of a ‘closed-shell interaction’ and a dominant ionic contribution to the P–F bonding in 1aF•Fp.
Consistent with the ionic character of the P–F bonding interaction, treatment of 1aF•Fp with fluoride abstracting reagents leads to removal of the F– ligand and regeneration of 1a•Fp+. Specifically, the addition of 1 equiv of AgPF6 to a CD3CN solution of 1aF•Fp induces the orange solution to become yellow with immediate formation of precipitate. Following filtration, 31P NMR spectroscopy (Figure 5b) confirms full consumption of 1aF•Fp and clean return of compound 1a•Fp+. Evidently, both the nontrigonal phosphorus framework and the P–Fe bond are sufficiently robust as to be retained during the course of the nonspectator L→X→L-switching cycle.
The data reported herein define the spectroscopic, structural, and electronic changes that accrue to phosphorus ligand 1a as it undergoes increase in coordination number upon exogenous fluoride addition. The conversion from L- to X- function roles results in little change to the donor capacity of the phosphorus ligand, but the acceptor capacity is diminished. Further, the reversible nonspectator behavior of tricoordinate phosphorus ligand 1a calls to mind recent developments for higher valent states of Sb ligands from Gabbaï.22 Given this periodic relationship within group 15, the broader implications of nonspectator L/X switching for phosphorus-based ligands in catalysis and sensing warrant further investigation.
Supplementary Material
Acknowledgements
Financial support was provided by NIH NIGMS (R21 GM134240). G.T.C. was supported by an NSF Graduate Research Fellowship. We thank Dr. Clemens Anklin (Bruker) for assistance with 57Fe NMR studies. We thank Drs. Charlene Tsay and Peter Müeller (MIT) for assistance in crystallographic data collection.
References
- [1].Tolman CA, Chem. Rev 1977, 77, 313–348. [Google Scholar]
- [2].Crabtree RH, The Organometallic Chemistry of the Transition Metals. John Wiley and Sons, Inc, 2005, 87–124. [Google Scholar]
- [3].Gillespie JA, Zuidema E, van Leeuwen PWNM, Kamer PCJ, Phosphorus(III) Ligands in Homogenous Catalysis: Design and Synthysis. John Wiley and Sons, Inc, 2012, 1–26. [Google Scholar]
- [4].Green MLH, J. Organomet. Chem 1995, 500, 127–148. [Google Scholar]
- [5].Parkin G, in Comprehensive Organometallic Chemistry III, Volume 1, Chapter 1; Crabtree RH, and Mingos DMP, (Eds), Elsevier, Oxford, 2006. [Google Scholar]
- [6].Rosenberg L, Coord. Chem. Rev 2012, 256, 606–626. [Google Scholar]
- [7].Roddick DM, Santarsiero BD, Bercaw JE, J. Am. Chem. Soc 1985, 107, 4670–4678. [Google Scholar]
- [8].Fryzuk MD, Bhangu K, J. Am. Chem. Soc 1988, 110, 961–963. [Google Scholar]
- [9].Derrah EJ, Pantazis DA, McDonald R, Rosenberg L, Organometallics 2007, 26, 1473–1482. [Google Scholar]
- [10].Hoyle M-AM, Pantazis DA, Burton HM, McDonald R, Rosenberg L, Organometallics 2011, 30, 6458–6465. [Google Scholar]
- [11].Kim Y-E, Oh S, Kim S, Kim O, Kim J, Han SW, Lee Y, J. Am. Chem. Soc 2015, 137, 4280–4283. [DOI] [PubMed] [Google Scholar]
- [12].Oh S, Kim S, Lee D, Gwak J, Lee Y, Inorg. Chem 2016, 55, 12863–12871. [DOI] [PubMed] [Google Scholar]
- [13].Oh S, Lee Y, Organometallics 2016, 35, 1586–1592. [Google Scholar]
- [14].Poitras AM, Knight SE, Bezpalko MW, Foxman BM, Thomas CM, Angew. Chem. Int. Ed 2018, 57, 1497–1500. [DOI] [PubMed] [Google Scholar]
- [15].Goodman J, Macgregor SA, Coord. Chem. Rev 2010, 254, 1295–1306. [Google Scholar]
- [16].Nakazawa H, Kubo K, Miyoshi K, Bull. Chem. Soc. Jpn 2001, 74, 2255–2267. [Google Scholar]
- [17].McLaughlin PA, Verkade JG, Organometallics 1998, 17, 5937–5940. [Google Scholar]
- [18].Kubo K, Nakazawa H, Mizuta T, Miyoshi K, Organometallics 1998, 17, 3522–3531. [Google Scholar]
- [19].Kubo K, Bansho K, Nakazawa H, Miyoshi K, Organometallics 1999, 18, 4311–4316. [Google Scholar]
- [20].Tanushi A, Radosevich A, J. Am. Chem. Soc 2018, 140, 8114–8118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee K, Blake AV, Tanushi A, McCarthy SM, Kim D, Loria SM, Donahue CM, Spielvogel KD, Keith JM, Daly SR, Radosevich AT, Angew. Chem. Int. Ed 2019, 58, 6993–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].For examples of high-valent heavier pnictogen ligands in catalysis and sensing, see:; (a) Jones JS, Gabbaï FP, Acc. Chem. Res 2016, 49, 857–867. [DOI] [PubMed] [Google Scholar]; (b) Jones JS, Wade CR, Gabbaï FP, Angew. Chem. Int. Ed, 2014, 53, 8876–8879. [DOI] [PubMed] [Google Scholar]; (c) You D, Gabbaï FP, J. Am. Chem. Soc 2017, 139, 6843–6846. [DOI] [PubMed] [Google Scholar]; (d) You D, Yang H, Sen S, Gabbaï FP, J. Am. Chem. Soc 2018, 140, 9644–9651. [DOI] [PubMed] [Google Scholar]
- [23].Chopra SK, Martin JC, Heteroat. Chem 1990, 2, 71–79. [Google Scholar]
- [24].Boone MP, Stephan DW, Organometallics, 2014, 33, 387–393. [Google Scholar]
- [25].Catheline D, Astruc D, Organometallics 1984, 3, 1094–1100. [Google Scholar]
- [26].Zhao W, McCarthy SM, Lai TY, Yennawar HP, Radosevich AT, J. Am. Chem. Soc 2014, 136, 17634–17644. [DOI] [PubMed] [Google Scholar]
- [27].Lin Y-C, Hatzakis E, McCarthy SM, Reichl KD, Lai TY, Yennawar HP, Radosevich AT, J. Am. Chem. Soc 2017, 139, 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benn R, Brenneke H, Frings A, Lehmkuhl H, Mehler G, Rufinska A, Wildt T, J. Am. Chem. Soc 1988, 110, 5661–5668. [Google Scholar]
- [29].Mampa RM, Fernandes MA, Carlton L, Organometallics 2014, 33, 3283–3299. [Google Scholar]
- [30].For recent overviews of EDA-NOCV, see:; (a) Frenking G, Bickelhaupt FM, The EDA Perspective of Chemical Bonding In The Chemical Bond; John Wiley & Sons, Ltd, 2014, 121–157. [Google Scholar]; (b) Zhao L, von Hopffgarten M, Andrada DM, Frenking G, WIREs Comput. Mol. Sci 2018, 8, e1345. [Google Scholar]
- [31].ADF2018, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com. [Google Scholar]
- [32].Mitoraj MP, Michalak A, Inorg. Chem 2010, 49, 578–582. [DOI] [PubMed] [Google Scholar]
- [33].Christe KO, Dixon DA, McLemore D, Wilson WW, Sheehy JA, Boatz JA J. Fluorine Chem 2000, 101, 151–153. [Google Scholar]
- [34].Slattery JM, Hussein S, Dalton Trans 2012, 41, 1808–1815. [DOI] [PubMed] [Google Scholar]
- [35].Addison AW, Rao TN, Reedijk J, van Rijn J, C Verschoor G, J. Chem. Soc. Dalt. Trans 1984, 1, 1349–1356. [Google Scholar]
- [36].Crabtree RH, Chem. Soc. Rev 2017, 46, 1720–1729. [DOI] [PubMed] [Google Scholar]
- [37].Blake AJ, Cockman RW, Ebsworth EAV, Henderson SGD, Holloway JH, Pilkington NJ, Rankin DWH, Phosphorus and Sulfur 1987, 30, 143–146. [Google Scholar]
- [38].Jones JS, Wade CR, Gabbaï FP, Organometallics 2015, 34, 2647–2654. [Google Scholar]
- [39].Bader R Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.