

Abstract
Similar to astrocytes at CNS synapses, perisynaptic Schwann cells (PSCs) surround nerve terminals at the neuromuscular junction (NMJ). These special teloglial cells are sensitive to neurotransmitters and upregulate glial fibrillary acidic protein (GFAP) when deprived of synaptic activity. We found that activation of muscarinic acetylcholine receptors (mAChRs) at PSCs, but not purinergic (ATP and adenosine) or peptidergic [substance P (SP) and calcitonin gene-related peptide (CGRP)] receptors, prevented this upregulation. When applied onto single PSCs, muscarine evoked Ca2+ responses that fatigued but prevented upregulation of this glial cytoskeletal protein. Application of ATP onto single PSCs evoked Ca2+signals that showed little fatigue, and GFAP upregulation occurred. Thus, Ca2+ signals alone cannot prevent GFAP upregulation in the PSCs. After blockade of cholinergic receptors by gallamine, neuronal activity was not effective in maintaining low GFAP levels in the perisynaptic glia. Last, immunohistochemistry disclosed mAChRs on PSCs and nearby fibroblasts. Thus, acetylcholine secreted by the nerve terminal acts on the PSCs via mAChRs to regulate GFAP. Cytoskeletal changes may influence perisynaptic glial functions, including growth, remodeling, and modulation of the synapse.
Keywords: nerve–glia signaling, nerve terminal, glial cell, transmitter, synapse, neuromuscular junction, terminal Schwann cell, GFAP, muscarine, muscarinic acetylcholine receptors, Ca2+, cytoskeleton, plasticity
CNS synapses are surrounded by astrocytes (Kuffler and Nicholls, 1966; S̆pac̆ek, 1971), and these glial cells are implicated in diverse functions (for review, seeBarres, 1991). Glia express ion channels, peptide, and neurotransmitter receptors (for review, see Wilkin and Cholewinski, 1988; Barres et al., 1990; Sontheimer, 1994) and respond to neuronal activity and transmitters (MacVicar et al., 1989; Cornell-Bell et al., 1990; Dani et al., 1992; for review, see Smith, 1992; Finkbeiner, 1993). Glia guide neuronal migration (Hatten, 1990), buffer extracellular ions (Orkand et al., 1966; Walz, 1989), and take up released transmitters (for review, see Hertz, 1979; Nicholls and Attwell, 1990). Astrocytes store transmitters and glycogen as an energy reserve for subsequent use by nerve terminals (for review, see Pfrieger and Barres, 1996; Tsacopoulos and Magistretti, 1996), synthesize, store, and release neuroactive compounds (Martin, 1992; Giulian, 1993), and can modulate neuronal activity (Nedergaard, 1994; Parpura et al., 1994; Araque et al., 1998). Thus, glia have significant roles at synapses.
Alterations in the glial environment induce changes in their morphology and phenotype (for review, see Shao and McCarthy, 1994). Neuronal degeneration, injury, disease, and other insults induce a glial reaction (for review, see Eng and DeArmond, 1982; Norenberg, 1994) characterized by cytoplasmic hypertrophy and increased glial fibrillary acidic protein (GFAP) (Eng et al., 1971). GFAP, a major glial intermediate filament protein that constitutes the cytoskeleton, is implicated in many functions, including morphology, adhesion, and signaling pathways (for review, see Galou et al., 1997; Rutka et al., 1997). Altered nerve activity affects GFAP in astrocytes (Steward et al., 1991; Canady and Rubel, 1992; Canady et al., 1994) and in the specialized perisynaptic Schwann cells (PSCs) (Georgiou et al., 1994) that wrap nerve terminals at the neuromuscular junction (NMJ) [Georgiou and Charlton (1999); for review, see Balice-Gordon (1996)and Son et al. (1996)]. PSCs upregulate GFAP after blockade of synaptic activity by nerve transection (Georgiou et al., 1994); electrical stimulation of transected nerves prevents this reaction, but not when transmitter release is prevented by blockade of presynaptic Ca2+ channels. Thus, events occurring subsequent to presynaptic Ca2+ influx serve to maintain low GFAP levels. However, it is not known whether GFAP regulation by synaptic activity occurs via specific classic neurotransmitters or other messengers.
Nerve terminals at NMJs release transmitters such as acetylcholine (ACh) and ATP from small vesicles and release peptide transmitters such as calcitonin gene-related peptide (CGRP) and substance P (SP) from larger vesicles (Matteoli et al., 1988, 1990). Moreover, PSCs respond to activation of various receptors, including muscarinic acetylcholine receptors (mAChRs) (Jahromi et al., 1992; Robitaille et al., 1997), purinoceptors (Robitaille, 1995), and substance P/neurokinin-1 (NK-1) receptors (Bourque and Robitaille, 1998). PSCs also possess voltage-dependent L-type Ca2+ channels (Robitaille et al., 1996).
We investigated whether neurotransmitters are involved in regulation of PSC GFAP. We found that activation of mAChRs, but not purinergic or peptidergic receptors, prevented GFAP upregulation. Blockade of mAChRs prevented nerve activity-dependent maintenance of low GFAP. We also demonstrated by immunohistochemistry that mAChRs are localized on PSCs.
MATERIALS AND METHODS
Animals and experimental treatment. Rana pipiens frogs (4–6 cm body length; supplied from either Connecticut Valley Biological Supply, Southampton, MA, or Wards Scientific, St. Catherines, Ontario, Canada) were housed at 15°C in running water. We double-pithed frogs and performed experiments at room temperature on pairs of nerve-transected, cutaneous pectoris muscles (Dreyer and Peper, 1974). Preparations were maintained in normal frog Ringer’s saline (FRS; 120 mm NaCl, 2.0 mm KCl, 1.0 mm NaHCO3, 1.8 mmCaCl2, 5.0 mm HEPES, pH adjusted to 7.2).
Excised muscles were pinned down in Petri dishes coated with Sylgard (Dow Corning, Midland, MI) and incubated in FRS. Each contralateral muscle was supplemented with either (+)-muscarine chloride (muscarine), ATP (disodium salt), adenosine, SP (all from Research Biochemicals, Natick, MA), synthetic human α-CGRP (Sigma, St. Louis, MO), or frog CGRP generously provided by Dr. Alain Fournier (Université du Québec). Gallamine triethiodide was from Sigma.
Cutaneous pectoris muscles were denervated as reported previously (Robitaille et al., 1996, 1997; Georgiou and Charlton, 1999). Frogs were anesthetized by injecting 0.3 mg/gm frog body weight of MS-222 (3-aminobenzoic acid ethyl ester, methane sulfonate salt; Sigma) dissolved in FRS into a dorsal lymphatic sac. Two 1 cm incisions were made on the ventral side of the frog, along the skin close to the point where the nerve reaches each muscle. At one muscle, a 0.5 cm portion of the motor nerve was removed without damaging the vasculature. The skin was sutured using silk thread and sealed using Histoacryl blue (B. Braun Melsungen AG, Melsungen, Germany). Frogs were maintained in individual cages containing water for 14 d. Contralateral muscles, with intact motor nerves, served as controls.
PSC Ca2+ imaging experiments. Muscle preparations were incubated for 90 min with the Ca2+indicator fluo-3 AM (10 μm; Molecular Probes, Eugene, OR) in FRS containing a final concentration of 1% (v/v) dimethyl sulfoxide (Sigma) and 0.02% (w/v) Pluronic F-127 (Molecular Probes) to facilitate solubilization of the dye. A site of PSC(s) on the dorsal side of the muscle was selected and imaged for fluo-3 fluorescence (F) using a Bio-Rad (Hercules, CA) 600 confocal laser scanning microscope (argon ion laser attenuated to 1% of the full intensity). A 40× water immersion objective (0.55 NA; Nikon) was used. The 488 nm laser line was used to excite fluo-3, and a low-pass filter with cutoff at 515 nm was used to detect the emitted F. Images were stored digitally, and subsequently, F at the soma of PSCs was calculated using CFOCAL, a program for PC analysis of Bio-Rad images written by T. A. Goldthorpe and modified by B. S. Jahromi. Change in F was expressed as %ΔF/F = [100 × (Fresponse −Fresting)/Fresting].
Muscles were perfused in FRS containing the chelator tetrakis (2-pyridylmethyl), ethylenediamine (20 μm; Molecular Probes) to reduce heavy metal binding to fluo-3 (Jahromi et al., 1992). By the time muscles were excised (15 min), incubated in dye (90 min), and perfused (15 min), ∼2 hr had elapsed. At this point, an electrode (2–3 μm tip diameter) containing a transmitter was positioned parallel to the long axis of a muscle fiber, close to a group of PSC nuclei (∼100 μm away). A short pulse (200 msec) of air pressure was applied to cause ejection (puff). Transmitters were dissolved in the perfusion solution and included either muscarine (40 nm) or ATP (50 μm). Pressure application of perfusion solution alone did not induce Ca2+ signals. At the end of experiments (∼6 hr after excision), synapses were identified using peanut agglutinin (PNA) coupled to tetramethylrhodamine isothiocyanate (PNA-TRITC; 10 μg/ml FRS; Sigma) applied for 10 min and fixed.
Staining and immunohistochemistry. Muscles were fixed using 3% paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA, 19034) in cold (5°C) FRS for 10 min and rinsed in FRS for at least 30 min. PNA-TRITC was applied for 10 min, unless already applied as in Ca2+ imaging experiments.
Immunostaining for mAChRs was performed using an IgM pan-muscarinic antibody (2 μg of clone M35) obtained initially from Dr. E. A. Van der Zee and subsequently purchased from Argene Inc. (North Massapequa, NY); antibodies from the latter source were in ascites fluid and specially ordered without NaN3. Secondary antibodies were FITC-labeled goat anti-mouse IgM (2 μg; Sigma). M35 antibody is ineffective on SDS-PAGE Western blots (André et al., 1984), suggesting that the M35 binding site is sensitive to this detection technique.
In preparations processed for GFAP expression, we used an IgG mouse anti-GFAP antibody (1 μg of clone G-A-5; Boehringer Mannheim, Laval, Québec, Canada). This antibody does not cross-react with other intermediate filaments such as vimentin or desmin but is specific for the C terminus of GFAP (Debus et al., 1983). We have determined previously that the G-A-5 clone is a specific probe for GFAP in frog by performing brain immunoblots (Georgiou et al., 1994, their Fig. 9). Secondary antibody was obtained from Sigma (2 μg of FITC-labeled goat anti-mouse IgG, Fc specific).
During the staining procedure, muscles were incubated in pairs and moved between five well plates each containing 0.2 ml “staining solution” consisting of 10% (w/v) bovine serum albumin (Sigma) and 0.3% Triton X-100 (Boehringer Mannheim) in FRS. (1) Preparations were “blocked” in this solution for 30 min, (2) incubated with primary antibody for 60 min, (3) rinsed for 60 min, (4) incubated with secondary antibody for 30 min, and (5) rinsed twice for 30 min. No fluorescence was observed when the primary antibodies were omitted.
Preparations were mounted onto glass slides in Slowfade antifade reagent (Molecular Probes) under a coverslip. Images were collected under immersion oil with a 40× lens (1.30 NA; Nikon), using the dual wavelength mode of the confocal microscope (argon ion laser light attenuated to 1% of full power). Excitation wavelength in this case was 514 nm; one photomultiplier tube detects green emitted light (selected using 514–550 nm bandpass filter), and the other photomultiplier tube detects red emitted light (selected using low-pass filter with 590 nm cutoff). Photomultiplier gain was always set to maximum, and black level was kept constant within a muscle pair.
Evaluation of GFAP and statistics. We used the same protocol as before (Georgiou et al., 1994), in which PNA-labeled PSCs at synapses on the dorsal side of the muscle were located and assayed for GFAP. PSCs were considered positive for GFAP when at least half their process lengths were labeled. All values shown are means ± SEM. Paired Student’s t tests, at a confidence level of 95% (α = 0.05), were used to evaluate statistical significance. In Ca2+ imaging experiments, we compared the proportion of GFAP-expressing PSCs in a zone including one muscle fiber on either side of the transmitter puff site with that for PSCs on synapses elsewhere on the same preparation and farther than one muscle fiber away from the puff site.
RESULTS
We have shown previously that at nerve–muscle synapses, PSCs normally express low levels of GFAP. Under control conditions, only ∼11% of PSCs are GFAP positive, but once nerve activity is blocked for 6 hr, either by nerve transection, TTX injection, or blockade of presynaptic Ca2+ channels, 81–91% of PSCs express GFAP (Georgiou et al., 1994). Increases in GFAP can be blocked by translation and transcription inhibitors. In this study, we first used pairs of muscles removed from animals and incubated in FRS for 6 hr and tested whether the application of various agents could prevent GFAP upregulation. The location of PSCs was identified routinely by labeling preparations with fluorescent PNA, a lectin that binds to glycosylated residues in the extracellular matrix of frog nerve terminals and effectively outlines the synapse (Ko, 1987). PNA is a reliable indicator of PSC location, and the outline formed around PSCs and their soma allows for their selective identification and corresponding assessment of GFAP immunoreactivity (Georgiou et al., 1994). Typical images from control muscles excised and incubated in FRS for 6 hr appear in Figure 1A (each set contains an image of PNA staining in the top panel and a simultaneously acquired image of GFAP in the bottom panel). In the excised neuromuscular preparations, other nonmuscle/neuronal cells, such as myelinating Schwann cells surrounding motor nerves (data not shown) and fibroblast-like cells, often contained GFAP. The perisynaptic fibroblasts are particularly interesting in that they had GFAP-containing processes that contacted synapses (Fig. 1A, asterisks). We refer to these cells as fibroblasts, because unlike PSCs, they express vimentin (data not shown) and appear similar to perisynaptic fibroblasts described by others (Connor and McMahan, 1987; Gatchalian et al., 1989;Weis et al., 1991). The function of these fibroblasts will be the subject of future study. The remainder of this report concerns only PSCs.
Fig. 1.

Muscarine prevents GFAP upregulation. Shown are simultaneously acquired confocal images from muscles excised and incubated for 6 hr in FRS, then double-labeled using PNA-TRITC (top panels) and anti-GFAP, revealed with goat anti-mouse FITC (bottom panels). Scale bars, 20 μm.A, Two sets of images from separate muscles, each showing three PSCs outlined by PNA (arrows), that express GFAP. The fields also contain GFAP-labeled perisynaptic fibroblasts (asterisks) that have processes contacting the synapse. B, The contralateral muscles toA were incubated in 20 μm muscarine. Note that in the top panel, the two PSCs outlined by PNA (arrows) had no corresponding expression of GFAP. At the synapses shown in the bottom panels, one of two PSCs in the field has upregulated GFAP along most of its entire length (arrowheads). In contrast, the other PSC has little GFAP, mostly in the form of periodic dots, and does not meet the criterion of GFAP positive.
Muscarine prevents GFAP upregulation
The frog neuromuscular synapse is cholinergic, and hence we considered ACh to be a prime candidate for regulating GFAP in PSCs. Because PSCs respond to muscarinic agonists (Jahromi et al., 1992;Robitaille et al., 1997), we tested the hypothesis that activation of mAChRs can prevent GFAP upregulation in PSCs. Excised nerve–muscle preparations were incubated for 6 hr in muscarine, then were fixed and probed for GFAP. Compared with control FRS-treated muscles, incubation in 20 μm muscarine reduced the proportion of PSCs expressing GFAP (significantly different; p = 0.006) (Table 1). The images in Figure1B are from the respective contralateral muscle (same frogs) of those in Figure 1A; they each show two PSCs outlined by PNA (arrows point to somata) but contain no corresponding GFAP in three of the four cells, and this was typical of muscarine-treated preparations. The results indicate that after muscle excision, muscarine will reduce GFAP upregulation in PSCs.
Table 1.
Effect of transmitters on upregulation of GFAP at PSCs, 6 hr after muscle excision
| Transmitter | N | Treated muscles | t test | Contralateral controls | ||
|---|---|---|---|---|---|---|
| % GFAP+ PSCs (± SEM) | n | % GFAP+ PSCs (± SEM) | n | |||
| Muscarine, 20 μm | 5 | 44 (±9) | 132 | p = 0.0061-a | 91 (±4) | 113 |
| ATP, 50 μm | 3 | 86 (±9) | 56 | p = 0.7 | 89 (±6) | 66 |
| Adenosine, 50 μm | 3 | 92 (±5) | 70 | p = 0.7 | 90 (±5) | 58 |
| SP, 10 μm | 3 | 80 (±6) | 56 | p = 0.5 | 86 (±6) | 78 |
| CGRP, 1 μm | 4 | 79 (±12) | 104 | p = 0.3 | 93 (±7) | 101 |
The proportion of PSCs positive (+) for GFAP under various treatments is tabulated. Muscles were incubated in transmitter for 6 hr. Contralateral muscles had vehicle treatment and served as GFAP+ controls. SEM, number of muscles examined (N), and number of cells surveyed (n) are presented.
Statistical differences determined by paired Student’s t tests between treated and control muscles.
Transmitter specificity for regulation of GFAP
In addition to ACh, small clear synaptic vesicles store and release the transmitter ATP, which is hydrolyzed extracellularly to adenosine (Zimmerman, 1994). In astrocytes and also in PSCs, application of ATP induces Ca2+ signals (Jahromi et al., 1992; Salter and Hicks, 1994). PSCs are equipped with three purinoceptors: one receptor prefers adenosine (A1), whereas two additional receptors are selective for ATP (P2x, P2y), and each can be distinguished pharmacologically (Robitaille, 1995). To test whether purine transmitters can prevent GFAP upregulation, we incubated excised preparations with either 50 μm ATP or adenosine for 6 hr.
Compared with contralateral controls, we found no significant differences in the proportion of PSCs expressing GFAP for either ATP-treated (Table 1) or adenosine-treated (Table 1) preparations. Figure 2 shows a typical image from a neuromuscular preparation after it was treated with ATP for 6 hr, in which several (seven) PSCs outlined by PNA have expressed GFAP. Similar patterns of GFAP expression were observed in adenosine-treated preparations (images not shown; see Table 1). Thus, purinoceptor activation cannot prevent the upregulation of GFAP that occurs at PSCs after nerve transection and muscle excision.
Fig. 2.
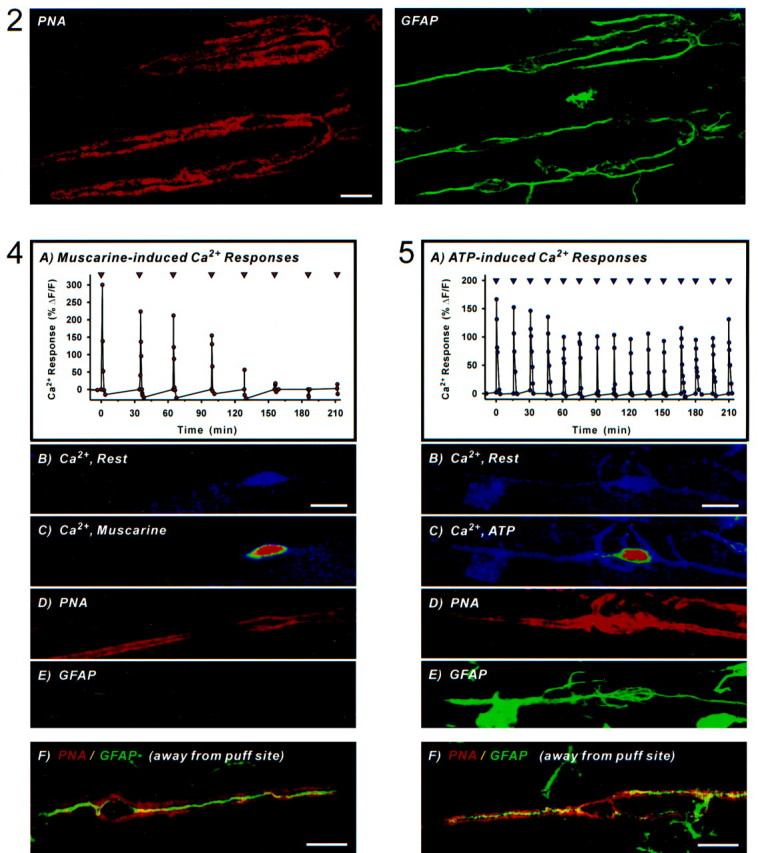
ATP fails to prevent GFAP upregulation. Shown are simultaneously acquired confocal images from muscle treated for 6 hr in 50 μm ATP and subsequently labeled using PNA-TRITC (left; appears in red) and anti-GFAP revealed with goat anti-mouse FITC (right; shown ingreen). Note that several PSCs outlined by PNA have expressed GFAP. Scale bar, 20 μm.
Nerve terminals in frog also contain large dense-cored vesicles that contain the peptide transmitters CGRP and SP (Matteoli et al., 1988,1990). Moreover, PSCs are known to have NK-1 receptors, which when activated by SP induce release of Ca2+ from intracellular stores (Bourque and Robitaille, 1998). CGRP affects ATP-evoked Ca2+ signals in PSCs, and thus it is likely that PSCs are equipped with CGRP receptors (our unpublished observations). Using the same protocol as above, we addressed the hypothesis that neuropeptides participate in the maintenance of low GFAP levels.
We found no significant differences in the proportion of PSCs expressing GFAP for either SP-treated (10 μm; Table 1) or human CGRP-treated (1 μm; Table 1) preparations, compared with their respective contralateral controls. Frog CGRP (1 μm) also did not affect GFAP upregulation, but like human CGRP it did attenuate ATP-evoked Ca2+ signals in PSCs (∼50% reduction; data not shown). The lack of effect of SP and CGRP suggests that these peptides alone do not suffice in keeping GFAP at a low level.
Transmitter-induced Ca2+ signals and regulation of GFAP.
We next asked whether Ca2+ signals evoked by application of cholinergic or purinergic agonists (Jahromi et al., 1992; Georgiou et al., 1994; Robitaille, 1995) might be involved in GFAP regulation. To investigate the role of Ca2+, we released transmitters onto individual PSCs and correlated the presence of Ca2+ signals with regulation of GFAP.
We excised muscles, loaded cells with the Ca2+indicator fluo-3 AM, and began perfusion (see Materials and Methods). We then pressure-released periodically via microelectrode small volumes (puff) of transmitters onto PSCs. At 6 hr after muscle excision, synaptic sites were labeled with PNA, and muscles were fixed. We then probed for GFAP and compared expression in PSCs located at thepuff site (synapses located on the muscle fiber where transmitter was applied, including synapses from the two immediately adjacent muscle fibers), with that for PSCs situated at synapseselsewhere on the same preparation (more than one muscle fiber away from the puff site).
When ATP (50 μm) was applied every 15 min onto identified PSCs, we observed Ca2+ responses that were largely nonfatiguing and persisted throughout the duration of the applications (4–5 hr). In contrast, muscarine (40 nm) induced Ca2+ signals that fatigued with each successive application (every 30 min). Figure 3 shows the average of normalized Ca2+ responses for each successive application of ATP and muscarine. We continued to puff each agonist onto the PSCs for 6 hr from the beginning of each experiment.
Fig. 3.

Ca2+ responses evoked by ATP differ from those evoked by muscarine. Averaged peak Ca2+ responses evoked by consecutive local application of either ATP or muscarine (see inset). Results are pooled from experiments summarized in Table 2 (also see Figs. 4, 5). Note the attenuation of Ca2+ responses that occurred with repetitive application of muscarine. In contrast, ATP, which was applied twice as frequently (15 vs 30 min intervals), evoked Ca2+ responses that were relatively resistant to fatigue. Note that the first application of transmitter occurred between 1.5 and 2 hr after muscle excision (see Materials and Methods), and this corresponds to time = 0 min in the plot.
Release of muscarine onto single cells reduced GFAP upregulation for PSCs at the puff site (Table 2) when compared with PSCs at synaptic sites elsewhere on the same muscles. An example from an experiment in which muscarine was applied at 30 min intervals is shown in Figure 4. The first five applications of muscarine induced Ca2+ signals in this experiment (Figs. 4A–C). After 6 hr, synapses were labeled using PNA (Fig. 4D) and fixed and stained for GFAP. The same synapse was located again, and the PSC was found to be devoid of GFAP (Fig. 4E). In contrast, the double-labeled image in Figure 4Fshows a GFAP-positive (appears in green) PSC from the same muscle but located at a synapse (revealed by PNA, shown inred) away from the muscarine puff site. This demonstrates that activation of mAChRs can prevent GFAP upregulation.
Table 2.
Ca2+ imaging of PSCs and effect of local transmitter application on upregulation of GFAP
| Transmitter | N | Transmitter puff site | t test | Elsewhere (same muscle) | ||
|---|---|---|---|---|---|---|
| % GFAP+ PSCs (± SEM) | n | % GFAP+ PSCs (± SEM) | n | |||
| Muscarine, 40 nm | 6 | 28 (±18) | 34 | p = 0.0092-a | 72 (±10) | 103 |
| ATP, 50 μm | 5 | 90 (±10) | 20 | p = 0.2 | 84 (±9) | 67 |
The proportion of PSCs positive (+) for GFAP under various treatments is tabulated. After loading cells with the Ca2+ indicator fluo-3, transmitter was released onto a site of PSCs (puff site) via micropipette. Puff site includes PSCs within one muscle fiber, whereas “elsewhere” column refers to cells surveyed on the same muscle, but more than one muscle fiber away from the puff site. SEM, number of muscles examined (N), and number of cells surveyed (n) are presented.
Statistical differences determined by paired Student’s t tests.
Fig. 4.
Local muscarine application prevents GFAP upregulation. A, Plot of normalized Ca2+ responses in a PSC from an experiment in which 40 nm muscarine was applied locally via microelectrode every 30 min (▿). Time = 0 min refers to the first application of muscarine, and this corresponds to ∼2 hr after muscle excision.B, Confocal image of the same PSC loaded with fluo-3 AM showing resting levels of Ca2+ fluorescence. Scale bar, 20 μm. C, Ca2+ fluorescence at the peak of the first response evoked by muscarine. PNA was later applied to outline PSCs, and at 6 hr after muscle excision the preparation was fixed, permeabilized (fluo-3 was therefore washed out), and double-labeled for GFAP. D, Pseudocolor image of the same synapse labeled with PNA-TRITC (red).E, Image of the same synapse probed with anti-GFAP revealed with goat anti-mouse FITC (green). Note the lack of GFAP at locations corresponding within the PNA outline.F, Merged image from a synapse located away from the muscarine puff site showing a PSC outlined by PNA (appears inred) that upregulated GFAP (shown ingreen). Overlap of the two colors appearsyellow. Scale bar, 20 μm.
When ATP was released onto single PSCs in the same manner as above (at 30 min intervals), GFAP upregulation could not be prevented (data not shown). We then applied ATP twice as frequently as muscarine (every 15 min) and still found similar proportions of GFAP-positive PSCs at the ATP puff site and at synapses elsewhere on the same muscle (Table 2). An example from an experiment in which microelectrode application of ATP onto a single PSC evoked Ca2+ signals is shown in Figure 5A–C; at the end of the 6 hr experiment, the PSC was outlined using PNA (Fig.5D) and later found to express GFAP (Fig.5E). PSCs from the same muscle but located at synapses away from the ATP puff site also contained GFAP, as exemplified by the double-labeled image of Figure 5F. The ability to detect Ca2+ transients for the duration of this and other ATP experiments (Fig. 3) allows us to conclude that changes in intracellular Ca2+ alone are not sufficient to prevent upregulation of GFAP.
Fig. 5.
Local ATP application cannot prevent GFAP upregulation. A, Plot of normalized Ca2+ fluorescence in a PSC from an experiment in which 50 μm ATP was applied locally via microelectrode every 15 min (▿). Time = 0 min refers to the first application of ATP, and this corresponds to ∼ 2 hr after muscle excision.B, Confocal image of the PSC loaded with fluo-3 AM showing resting levels of Ca2+ fluorescence. Scale bar, 20 μm. C, Ca2+ fluorescence at the peak of the first response evoked by ATP. PNA was later applied to outline PSCs, and at 6 hr after muscle excision the preparation was fixed, permeabilized (fluo-3 was therefore washed out), and double-labeled for GFAP. D, Pseudocolor image of the same synapse labeled with PNA-TRITC (red).E, Image of the same synapse probed with anti-GFAP revealed with goat anti-mouse FITC (green). Note that the PSC expressed GFAP. F, Merged image from a synapse located on the same muscle but away from the ATP puff site. A PSC outlined by PNA (shown in red) has upregulated GFAP (appears in green). Overlap of the two colors appearsyellow. Scale bar, 20 μm.
Cholinergic antagonist blocks nerve-evoked maintenance of low GFAP
If release of ACh at nerve terminals maintains low levels of GFAP by activation of mAChRs, blockade of these receptors should affect this regulation. ACh and muscarine as low as 1 nm cause release of Ca2+ from PSC internal stores (Jahromi et al., 1992) via mAChRs that are insensitive to classic muscarinic antagonists but can be blocked by the allosteric cholinergic antagonist gallamine (Robitaille et al., 1997). Gallamine blocks both muscarinic and nicotinic receptors, but the latter is of no importance because nerve stimulation maintains low GFAP levels even after their blockade (Georgiou et al., 1994).
To test the hypothesis that mAChRs are involved in GFAP regulation, we stimulated nerve-transected muscle preparations in vivoafter first isolating each muscle by applying a perimeter Vaseline dam (Georgiou et al., 1994). Muscles received 5 Hz motor nerve stimulation, and FRS with or without 10 μm gallamine was applied by drip application for 6 hr, at which point preparations were fixed and assayed for GFAP immunoreactivity.
We found that the proportion of PSCs expressing GFAP was greater in gallamine-treated (76 ± 6% of 94 cells) versus vehicle-treated (39 ± 7% of 86 cells) preparations (significantly different by paired Student’s t test, p = 0.01;N, n = 3.94 and 3.86, respectively). The results indicate that blockade of cholinergic receptors is sufficient to induce upregulation of GFAP in PSCs.
Localization of mAChRs
To confirm that PSCs have mAChRs with some similarity to those in mammals, we attempted to localize mAChRs with immunohistochemistry. We used the monoclonal antibody M35 (André et al., 1984), an IgM raised against affinity-purified calf brain mAChRs. M35 does not discriminate between mAChRs and detects human m1–m5 receptors, presumably by binding to an extracellular epitope preserved on all mAChR subtypes (Carsi-Gabrenas et al., 1997).
We found punctate weak mAChR immunoreactivity on PSCs and also fibroblast-like cells in the absence of a permeabilizing agent (data not shown). Staining was considerably greater after permeabilizing with Triton X-100. It is not clear why immunoreactivity increased after permeabilization, but this may indicate that the antibody is hindered from binding to many antigenic sites or that most of the receptors are not normally available on the surface of PSCs. The mAChR staining shown in each of the four images of Figure 6 thus represents receptors on the PSC surface as well as those already internalized or awaiting transport to the membrane. We cannot exclude the possibility that the underlying nerve terminals also express mAChRs. However, it is clear that PSCs express mAChRs because immunoreactivity was seen in the cytoplasm surrounding the PSC nucleus (Fig. 6A), which has an expanded shape and protrudes 5–10 μm from the nerve terminal. Furthermore, when muscles were denervated for 2 weeks to allow degeneration of nerve terminals, PSCs still displayed mAChR staining (Fig. 6B). The data imply that cholinergic substances activate mAChRs localized directly on PSCs.
Fig. 6.

Expression of mAChRs at PSCs and fibroblasts. Shown are four pairs of simultaneously acquired confocal images from neuromuscular junctions identified using PNA-TRITC (top panels) and labeled for mAChRs using clone M35 (pan-mAChR) antibodies revealed with FITC-goat anti-mouse IgM (bottom panels). Scale bars, 20 μm. A, Images revealing synapses from two separate normal (quickly excised and fixed) preparations. PSCs were immunoreactive as judged by staining at the level of PSC cell bodies (arrows); fluorescence elsewhere within the PNA outline corresponds to PSC and possibly also nerve terminal staining. Fibroblast-like cells also had mAChR staining.B, Images from two separate muscle preparations previously denervated (2 weeks). PSCs and fibroblasts continued to express mAChRs.
Because M35 exhibits agonist-like properties (Leiber et al., 1984), we wondered whether M35 could induce Ca2+ signals in PSCs. Microelectrode application of M35 (200 nm) on preparations previously loaded with the Ca2+indicator fluo-3 induced a Ca2+ transient, similar in time course to that observed in PSCs exposed to transmitters such as muscarine or ATP. Similar results occurred in freshly excised muscles as well as in PSCs from denervated muscles. The images in Figure7A show a PSC from a denervated preparation that had been loaded with the Ca2+indicator fluo-3 and that subsequently responded to M35. The plotted time course of the PSC responses to various challenges (spaced ∼5 min apart) appears in Figure 7B–G. Control experiments using ascites fluid containing nonspecific IgM did not evoke a Ca2+ response (Fig. 7B). Application of M35, however, induced Ca2+ transients that attenuated in amplitude (and duration) with repeated applications (Fig.7C–E). Furthermore, subsequent addition of muscarine did not induce a Ca2+ response, suggesting that M35 either induced desensitization of the muscarine-evoked Ca2+ signal pathway (Fig. 4A) or possibly prevented muscarine binding. In the same preparation, when muscarine was puffed from the same microelectrode onto other PSCs that had not received M35, this induced Ca2+ responses (data not shown). Although muscarinic Ca2+ responses were prevented by previous application of M35, the internal Ca2+ stores were likely not depleted because ATP could still evoke a signal (Fig. 7G). These observations support the notion that M35 is capable of activating receptors with characteristics similar to those activated by muscarine and suggest that the antibody likely detects mAChRs in frog.
Fig. 7.

Muscarinic receptor antibody induces Ca2+ transients in PSCs. Results from an experiment on a PSC from a denervated muscle preparation (A) and plots of normalized Ca2+ fluorescence in response to successive (5 min apart) microelectrode application of various agents (B–G). A, Confocal image of a PSC loaded with fluo-3 AM showing fluorescence attributable to resting levels of Ca2+ and fluorescence after application of M35 antibody. Scale bar, 20 μm. B, Application of ascites fluid containing nonspecific IgM did not evoke a Ca2+ response. C–E, Successive additions of M35 pan-muscarinic antibody (200 nm) induced a Ca2+ signal that became attenuated in amplitude.F, Subsequent addition of muscarine (40 nm) failed to evoke a Ca2+ response. G, The PSC was still responsive to ATP (50 μm).
DISCUSSION
The results demonstrate that ACh, operating on PSC mAChRs, can prevent upregulation of GFAP. This reveals that in addition to the known role for neurotransmitters at postsynaptic cells, there are physiological actions on perisynaptic glia. Because the transmitters ATP, adenosine, SP, and CGRP, which are also released or present at this synapse and can also interact with PSCs, failed to prevent GFAP upregulation, this suggests that specific receptor-mediated second messenger pathways are involved. We also found that when endogenous ACh fails to activate mAChRs on PSCs, upregulation of GFAP occurs.
Cholinergic antagonist allows GFAP upregulation
Gallamine application allowed upregulation of GFAP to occur in the PSCs even when the motor nerve was stimulated. Although gallamine blocks postsynaptic nicotinic acetylcholine receptors (nAChRs), results from a previous study (Georgiou et al., 1994) argue against the involvement of nAChRs in GFAP regulation. In similar experiments, GFAP upregulation was blocked by nerve stimulation in the presence of nAChR blockers (d-tubocurarine chloride and α-bungarotoxin). This contrasts with results from the present study in which GFAP upregulation could not be blocked in gallamine-treated muscles. The data indicate that a reduction in mAChR activation triggers GFAP upregulation in PSCs.
PSCs express mAChRs
Five mAChRs have been cloned (m1–m5) and interact with effectors via GTP-binding (G-) proteins. In general, m1, m3, and m5 are coupled to phosphoinositide hydrolysis, whereas m2 and m4 receptors are coupled to inhibition of adenylate cyclase (Bonner, 1989; Hulme et al., 1990). General muscarinic antagonists such as atropine are ineffective in blocking PSC Ca2+ responses evoked by muscarinic agonists (Robitaille et al., 1997), indicating that these mAChRs are unlike the five known types. Muscarinic agonists mobilize Ca2+ from PSC internal stores (Jahromi et el., 1992) via a pertussis-toxin-insensitive mechanism (Robitaille et al., 1997), suggesting that there are mAChRs that are not Gi/Go-coupled. In denervated preparations, muscarine-evoked Ca2+ signals are blocked by atropine (Robitaille et al., 1997). Although the significance of this change in mAChR expression, properties, and/or operation is unclear, it is possible that nerve–glial signals are involved.
We used the M35 antibody, which binds all subtypes of mAChRs (Carsi-Gabrenas et al., 1997), to demonstrate that PSCs express receptors under both innervated and denervated situations. Moreover, M35 induced Ca2+ signals in the PSCs (Fig. 7). These results suggest that muscarinic effects on PSCs, including Ca2+ signals and regulation of GFAP, occur by direct activation of PSCs. Elucidation of the subtypes and properties of the PSC mAChRs, including the second messenger effector systems to which they couple, will be important in determining the transduction pathways involved in GFAP regulation.
Muscarinic regulation of GFAP
Muscarine reduces the upregulation of GFAP that occurs after blockade of nerve activity. We have demonstrated previously, using anin vivo assay (Georgiou et al., 1994), that “normally” only 11% (±4%) of PSCs express GFAP (quickly excised and fixed muscle). However, by 6 and 22 hr after transection of the motor nerve, 86% (±9) and 88% (±4%) of the PSCs expressed GFAP, respectively; when the transected nerves were stimulated electrically (0.5 Hz), the proportion of PSCs expressing GFAP at these two time points was held at 19% (±6%) and 22% (±5%). The latter values are not significantly different from levels in “normal frogs.”
In excised muscles treated for 6 hr either periodically (puffing) or continuously with muscarine, we found that 28% (±18%) and 44% (±9%) of the PSCs expressed GFAP, respectively. Only the latter value is significantly different from previously established levels of “normal muscles” (p = 0.4 andp = 0.01, respectively; unpaired t test). There are several reasons why continuous muscarine application in situ did not maintain GFAP expression as low as previously established “normal” levels.
Repeated activation of PSC mAChRs results in successively smaller Ca2+ signals (Fig. 4) (also see Jahromi et al., 1992). This desensitization phenomenon may be more pronounced with continuous muscarine application, limiting activation of effectors that serve to prevent GFAP upregulation. The experimental protocols thus may not reproduce optimally the temporal and/or spatial pattern of mAChR activation that occurs in vivo. The specific patterns of receptor–effector activation and subsequent second messenger alterations are likely to affect PSC physiology (Finkbeiner, 1993; Toma et al., 1997; Dolmetsch et al., 1998; Mons et al., 1998).
In contrast to the previous study, we conducted experiments on isolatedexcised muscles, and compared PSCs either withinpairs of muscles (drug application to experimental but not control muscle) or between PSCs from single muscles alone (local puff application). These approaches reduce the spread of drugs to contralateral muscles or adjacent synapses, respectively. Moreover, errors attributable to variations between animals are minimized. However, in excised muscles circulation is lost, and potentially important humoral factors are not available and other cellularly released factors may accumulate. It remains to be seen whether such factors contribute to GFAP regulation. Another consideration is that excision of muscles requires between 15 and 30 min, and in Ca2+-imaging experiments, nearly 2 hr elapse before experiments are underway. During this time, GFAP upregulation is occurring, and it is possible that subsequent muscarine application will not reduce the amount of pre-existing GFAP to control levels. It will be interesting to see whether PSCs that have expressed GFAP can be subsequently induced to reduce their GFAP content by applying nerve stimulation or muscarine.
Are Ca2+ signals involved in GFAP regulation?
Four observations lead to the conclusion that changes in bulk cytoplasmic Ca2+ alone are likely insufficient to prevent GFAP upregulation. First, the electrical stimulation paradigm used on nerve transected preparations (0.5 Hz) (Georgiou et al., 1994) does not evoke detectable Ca2+ signals in PSCs; generation of a Ca2+ transient requires a much higher rate and duration of stimulation [>20 Hz for 5 sec, our unpublished observations; also see Jahromi et al. (1992)]. Second, direct application of ATP onto single PSCs evoked Ca2+ signals, but unlike muscarine could not prevent GFAP upregulation. Indeed, we applied ATP both at the same and twice the frequency used for muscarine (30 and 15 min intervals), but neither treatment blocked GFAP upregulation. Third, several noncholinergic transmitters, including ATP, adenosine, and SP, which all induce Ca2+ signals in PSCs, were used in this study, but these failed to prevent GFAP upregulation. Furthermore, the Ca2+ mobilizations in each of these cases are similar to that evoked by muscarine in that they activate intracellular PSC stores via G-proteins (Jahromi et al., 1992; Robitaille, 1995;Bourque and Robitaille, 1998). PSCs appear to release Ca2+ from a single type of store that is sensitive to thapsigargin [our unpublished observations; also see Castonguay and Robitaille (1998)]; thus, it is unlikely that Ca2+signals linked to ATP, adenosine, or SP receptors cannot regulate GFAP because they originate from a different source than muscarinic Ca2+ signals. Fourth, repetitive muscarine application induced Ca2+ signals that attenuated in amplitude and were nearly abolished by the fifth application and over the time course of each experiment represented a total Ca2+ load that was smaller than that produced by ATP. In summary, although a cooperative role for Ca2+ cannot be ruled out, it appears that Ca2+ alone does not suffice in maintaining low GFAP levels. Instead, other second messengers in addition to Ca2+ that are generated on activation of PSC mAChRs are likely to be important.
Role of GFAP and functions of PSCs
GFAP is an intermediate filament protein implicated in control of glial shape, motility, and process extension (Duffy et al., 1982; Weinstein et al., 1991; for review, see Lazarides, 1982; Galou et al., 1997; Rodnight et al., 1997). Some of these roles may relate to the fact that PSCs sprout processes after denervation that retract on reinnervation (Reynolds and Woolf, 1992). Son and Thompson (1995) found that when a nerve terminal is denervated, the surrounding PSCs extend processes to nearby intact synapses and then guide nerve sprouts back to the denervated synapse. This extraordinary function may require GFAP, which we have found in denervated PSCs and their newly formed processes (our unpublished observations). GFAP is also believed to be important in many other aspects of glial function, such as cell adhesion and signaling pathways to the nucleus (for review, see Galou et al., 1997; Rutka et al., 1997).
Synaptic environment and PSCs
Expression of proteins and operations of PSCs are dependent on the state of the neuromuscular synapse. Denervation induces PSCs to express the low-affinity nerve growth factor receptor (Reynolds and Woolf, 1992) GAP-43 (Woolf et al., 1992) and to synthesize and release ACh (Birks et al., 1960; Bevan et al., 1973). Interestingly, application of botulinum toxin, which induces paralysis by blocking neurotransmitter release, is a sufficient stimulus for extension of PSC and neuronal processes at the NMJ (Son and Thompson, 1995; Caroni et al., 1997). Cessation of neurotransmitter release is a likely trigger for many of these and other changes in PSCs. Because evidence of PSC feedback onto nerve terminals does exist (Robitaille, 1998), modifications in perisynaptic glial operation may serve to direct the formation, maintenance, and modulation of synapses.
Footnotes
This work was supported by a Medical Research Council of Canada Group Grant “Nerve Cells and Synapses” to M.P.C. and by the Neuroscience Network. J.G. was supported by a Neuroscience Network Studentship. We thank Dr. Eddy A. Van der Zee for his generous gift of M35 antibody and Dr. Alain Fournier (Université du Québec, Institut National de la Recherche Scientifique–Santé, Point Claire, Québec, Canada) for kindly supplying frog CGRP.
Correspondence should be addressed to Milton P. Charlton, Department of Physiology, Medical Sciences Building 3232, University of Toronto, 1 King’s College Circle, Toronto, Ontario, Canada M5S 1A8. E-mail:milton@spine.synaptic.med.utoronto.ca
REFERENCES
- 1.André C, Guillet JG, De Backer J-P, Vanderheyden P, Hoebeke J, Strosberg AD. Monoclonal antibodies against the native or denatured forms of muscarinic acetylcholine receptors. EMBO J. 1984;3:17–21. doi: 10.1002/j.1460-2075.1984.tb01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balice-Gordon RJ. Schwann cells: dynamic roles at the neuromuscular junction. Curr Biol. 1996;6:1054–1056. doi: 10.1016/s0960-9822(02)70662-3. [DOI] [PubMed] [Google Scholar]
- 4.Barres BA. New roles for glia. J Neurosci. 1991;11:3685–3694. doi: 10.1523/JNEUROSCI.11-12-03685.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barres BA, Chun LLY, Corey DP. Ion channels in vertebrate glia. Annu Rev Neurosci. 1990;13:441–474. doi: 10.1146/annurev.ne.13.030190.002301. [DOI] [PubMed] [Google Scholar]
- 6.Bevan S, Miledi R, Grampp W. Induced transmitter release from Schwann cells and its suppression by actinomycin D. Nature New Biol. 1973;241:85–86. doi: 10.1038/newbio241085a0. [DOI] [PubMed] [Google Scholar]
- 7.Birks R, Katz B, Miledi R. Physiological and structural changes at the amphibian myoneural junction in the course of nerve degeneration. J Physiol (Lond) 1960;150:145–168. doi: 10.1113/jphysiol.1960.sp006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonner TI. The molecular basis of muscarinic receptor diversity. Trends Neurosci. 1989;12:148–151. doi: 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- 9.Bourque MJ, Robitaille R. Endogenous peptidergic modulation of perisynaptic Schwann cells at the frog neuromuscular junction. J Physiol (Lond) 1998;512:197–209. doi: 10.1111/j.1469-7793.1998.197bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canady KS, Rubel EW. Rapid and reversible astrocytic reaction to afferent activity blockade in chick cochlear nucleus. J Neurosci. 1992;12:1001–1009. doi: 10.1523/JNEUROSCI.12-03-01001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canady KS, Olavarria JF, Rubel EW. Reduced retinal activity increases GFAP immunoreactivity in rat lateral geniculate nucleus. Brain Res. 1994;663:206–214. doi: 10.1016/0006-8993(94)91265-3. [DOI] [PubMed] [Google Scholar]
- 12.Caroni P, Aigner L, Schneider C. Intrinsic neuronal determinants locally regulate extrasynaptic and synaptic growth at the adult neuromuscular junction. J Cell Biol. 1997;136:679–692. doi: 10.1083/jcb.136.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carsi-Gabrenas JM, Van der Zee EA, Luiten PGM, Potter LT. Non-selectivity of the monoclonal antibody M35 for subtypes of muscarinic acetylcholine receptors. Brain Res Bull. 1997;44:25–31. doi: 10.1016/s0361-9230(96)00422-4. [DOI] [PubMed] [Google Scholar]
- 14.Castonguay A, Robitaille R. Elevation of Ca2+ in perisynaptic Schwann cells modulates synaptic transmission at the frog neuromuscular junction. Soc Neurosci Abstr. 1998;24:824. [Google Scholar]
- 15.Connor EA, McMahan UJ. Cell accumulation in the junctional region of denervated muscle. J Cell Biol. 1987;104:109–120. doi: 10.1083/jcb.104.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 17.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 18.Debus E, Weber K, Osborn M. Monoclonal antibodies specific for glial fibrillary acidic (GFA) protein and for each of the neurofilament triplet polypeptides. Differentiation. 1983;25:193–203. doi: 10.1111/j.1432-0436.1984.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 19.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 20.Dreyer F, Peper K. A monolayer preparation of innervated skeletal muscle fibers of the M. cutaneus pectoris of the frog. Pflügers Arch. 1974;348:257–262. doi: 10.1007/BF00587416. [DOI] [PubMed] [Google Scholar]
- 21.Duffy PE, Huang Y-Y, Rapport MM. The relationship of glial fibrillary acidic protein to the shape, motility, and differentiation of human astrocytoma cells. Exp Cell Res. 1982;139:145–157. doi: 10.1016/0014-4827(82)90328-7. [DOI] [PubMed] [Google Scholar]
- 22.Eng LF, DeArmond SJ. Immunocytochemical studies of astrocytes in normal development and disease. Adv Cell Neurobiol. 1982;3:145–171. [Google Scholar]
- 23.Eng LF, Vanderhaeghen JJ, Bignami A, Gerstl B. An acidic protein isolated from fibrous astrocytes. Brain Res. 1971;28:351–354. doi: 10.1016/0006-8993(71)90668-8. [DOI] [PubMed] [Google Scholar]
- 24.Finkbeiner SM. Glial calcium. Glia. 1993;9:83–104. doi: 10.1002/glia.440090202. [DOI] [PubMed] [Google Scholar]
- 25.Galou M, Gao J, Humber J, Mericskay M, Li Z, Paulin D, Vicart P. The importance of intermediate filaments in the adaptation of tissues to mechanical stress: evidence from gene knockout studies. Biol Cell. 1997;89:85–97. [PubMed] [Google Scholar]
- 26.Gatchalian CL, Schachner M, Sanes JR. Fibroblasts that proliferate near denervated synaptic sites in skeletal muscle synthesize the adhesive molecules tenascin(J1), N-CAM, fibronectin, and a heparin sulfate proteoglycan. J Cell Biol. 1989;108:1873–1890. doi: 10.1083/jcb.108.5.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georgiou J, Charlton MP (1999) Non-myelin forming perisynaptic Schwann cells express protein zero and myelin-associated glycoprotein. Glia, in press. [DOI] [PubMed]
- 28.Georgiou J, Robitaille R, Trimble WS, Charlton MP. Synaptic regulation of glial protein expression in vivo. Neuron. 1994;12:443–455. doi: 10.1016/0896-6273(94)90284-4. [DOI] [PubMed] [Google Scholar]
- 29.Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia. 1993;7:102–110. doi: 10.1002/glia.440070116. [DOI] [PubMed] [Google Scholar]
- 30.Hatten ME. Riding the glial monorail: a common mechanism for glial-guided neuronal migration in different regions of the developing mammalian brain. Trends Neurosci. 1990;13:179–184. doi: 10.1016/0166-2236(90)90044-b. [DOI] [PubMed] [Google Scholar]
- 31.Hertz L. Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol. 1979;13:277–323. doi: 10.1016/0301-0082(79)90018-2. [DOI] [PubMed] [Google Scholar]
- 32.Hulme EC, Birdshall NJM, Buckley NJ. Muscarinic receptor subtypes. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 33.Jahromi BS, Robitaille R, Charlton MP. Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron. 1992;8:1069–1077. doi: 10.1016/0896-6273(92)90128-z. [DOI] [PubMed] [Google Scholar]
- 34.Ko C-P. A lectin, peanut agglutinin, as a probe for the extracellular matrix in living neuromuscular junctions. J Neurocytol. 1987;16:567–576. doi: 10.1007/BF01668509. [DOI] [PubMed] [Google Scholar]
- 35.Kuffler SW, Nicholls JG. The physiology of neuroglial cells. Ergeb Physiol. 1966;57:1–90. [PubMed] [Google Scholar]
- 36.Lazarides E. Intermediate filaments: a chemically heterogeneous, developmentally regulated class of proteins. Annu Rev Biochem. 1982;51:219–250. doi: 10.1146/annurev.bi.51.070182.001251. [DOI] [PubMed] [Google Scholar]
- 37.Leiber D, Harbon S, Guillet JG, André C, Strosberg AD. Monoclonal antibodies to purified muscarinic receptor display agonist-like activity. Proc Natl Acad Sci USA. 1984;81:4331–4334. doi: 10.1073/pnas.81.14.4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacVicar BA, Tse FWY, Crichton SA, Kettenmann H. GABA-activated Cl- channels in astrocytes of hippocampal slices. J Neurosci. 1989;9:3577–3583. doi: 10.1523/JNEUROSCI.09-10-03577.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin DL. Synthesis and release of neuroactive substances by glial cells. Glia. 1992;5:81–94. doi: 10.1002/glia.440050202. [DOI] [PubMed] [Google Scholar]
- 40.Matteoli M, Haimann C, Torri-Tarelli F, Polak JM, Ceccarelli B, De Camilli P. Differential effect of alpha-latrotoxin on exocytosis from small synaptic vesicles and from large dense-core vesicles containing calcitonin gene-related peptide at the frog neuromuscular junction. Proc Natl Acad Sci USA. 1988;85:7366–7370. doi: 10.1073/pnas.85.19.7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matteoli M, Haimann C, De Camilli P. Substance P-like immunoreactivity at the frog neuromuscular junction. Neuroscience. 1990;37:271–275. doi: 10.1016/0306-4522(90)90213-n. [DOI] [PubMed] [Google Scholar]
- 42.Mons N, Decorte L, Jaffard R, Cooper DM. Ca2+-sensitive adenylyl cyclases, key integrators of cellular signalling. Life Sci. 1998;62:17–18. doi: 10.1016/s0024-3205(98)00122-2. [DOI] [PubMed] [Google Scholar]
- 43.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 44.Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 45.Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol. 1994;53:213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- 46.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 47.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 48.Pfrieger FW, Barres BA. New views on synapse-glia interactions. Curr Opin Neurobiol. 1996;6:615–621. doi: 10.1016/s0959-4388(96)80093-6. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds ML, Woolf CJ. Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol. 1992;21:50–66. doi: 10.1007/BF01206897. [DOI] [PubMed] [Google Scholar]
- 50.Robitaille R. Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1995;15:7121–7131. doi: 10.1523/JNEUROSCI.15-11-07121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 1998;21:847–855. doi: 10.1016/s0896-6273(00)80600-5. [DOI] [PubMed] [Google Scholar]
- 52.Robitaille R, Bourque M-J, Vandaele S. Localization of L-type Ca2+ channels at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci. 1996;16:148–158. doi: 10.1523/JNEUROSCI.16-01-00148.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robitaille R, Jahromi BS, Charlton MP. Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J Physiol (Lond) 1997;504:337–347. doi: 10.1111/j.1469-7793.1997.337be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodnight R, Gonçalves CA, Wofchuk ST, Leal R. Control of the phosphorylation of the astrocyte marker glial fibrillary acidic protein (GFAP) in the immature rat hippocampus by glutamate and calcium ions: possible key factor in astrocytic plasticity. Braz J Med Biol Res. 1997;30:325–338. doi: 10.1590/s0100-879x1997000300005. [DOI] [PubMed] [Google Scholar]
- 55.Rutka JT, Murakami M, Dirks PB, Hubbard SL, Becker LE, Fukuyama K, Jung S, Tsugu A, Matsuzawa K. Role of glial filaments in cells and tumors of glial origin: a review. J Neurosurg. 1997;87:420–430. doi: 10.3171/jns.1997.87.3.0420. [DOI] [PubMed] [Google Scholar]
- 56.Salter MW, Hicks JL. ATP-evoked increases in intracellular calcium in neurons and glia from the dorsal spinal cord. J Neurosci. 1994;14:1563–1575. doi: 10.1523/JNEUROSCI.14-03-01563.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shao Y, McCarthy KD. Plasticity of astrocytes. Glia. 1994;11:147–155. doi: 10.1002/glia.440110209. [DOI] [PubMed] [Google Scholar]
- 58.Smith SJ. Do astrocytes process neural information? Prog Brain Res. 1992;94:119–136. doi: 10.1016/s0079-6123(08)61744-6. [DOI] [PubMed] [Google Scholar]
- 59.Son Y-J, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995;14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- 60.Son Y-J, Trachtenberg JT, Thompson WJ. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996;19:280–285. doi: 10.1016/S0166-2236(96)10032-1. [DOI] [PubMed] [Google Scholar]
- 61.Sontheimer H. Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–172. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- 62.S̆pac̆ek J. Three-dimensional reconstructions of astroglia and oligodendroglia cells. Z Zellforsch. 1971;112:430–442. [PubMed] [Google Scholar]
- 63.Steward O, Torre ER, Tomasulo R, Lothman E. Neuronal activ-ity up-regulates astroglial gene expression. Proc Natl Acad Sci USA. 1991;88:6819–6823. doi: 10.1073/pnas.88.15.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toma JG, Rogers D, Senger DL, Campenot RB, Miller FD. Spatial regulation of neuronal gene expression in response to nerve growth factor. Dev Biol. 1997;184:1–9. doi: 10.1006/dbio.1997.8515. [DOI] [PubMed] [Google Scholar]
- 65.Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci. 1996;16:877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walz W. Role of glial cells in the regulation of the brain ion microenvironment. Prog Neurobiol. 1989;13:277–323. doi: 10.1016/0301-0082(89)90005-1. [DOI] [PubMed] [Google Scholar]
- 67.Weinstein DE, Shelanski ML, Liem RKH. Suppression by anti- sense mRNA demonstrates a requirement for the glial fibrillary acidic protein in the formation of stable astrocytic processes in response to neurons. J Cell Biol. 1991;112:1205–1213. doi: 10.1083/jcb.112.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weis J, Fine SM, David C, Savarirayan S, Sanes JR. Integration site-dependent expression of a transgene reveals specialized features of cells associated with neuromuscular junctions. J Cell Biol. 1991;113:1385–1397. doi: 10.1083/jcb.113.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilkin GP, Cholewinski A. Peptide receptors on astrocytes. In: Kimelberg HK, editor. Glial cell receptors. Raven; New York: 1988. pp. 223–241. [Google Scholar]
- 70.Woolf CJ, Reynolds ML, Chong MS, Emson P, Irwin N, Benowitz LI. Denervation of the motor endplate results in the rapid expression by terminal Schwann cells of the growth-associated protein GAP-43. J Neurosci. 1992;12:3999–4010. doi: 10.1523/JNEUROSCI.12-10-03999.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zimmerman H. Signalling via ATP in the nervous system. Trends Neurosci. 1994;17:420–426. doi: 10.1016/0166-2236(94)90016-7. [DOI] [PubMed] [Google Scholar]