

Abstract
Although neurotrophins are best known for their trophic functions, growing evidence suggests that neurotrophins can also be neurotoxic, for instance by enhancing excitotoxic insults. We have shown recently that brain-derived neurotrophic factor (BDNF) limits its neuroprotective action on axotomized rat retinal ganglion cells (RGCs) by upregulating nitric oxide synthase (NOS) activity (Klöcker et al., 1998). The aim of the present study was to investigate this interaction of BDNF and NOS in the lesioned adult rat retina in more detail. We used NOS immunohistochemistry and NADPH-diaphorase (NADPH-d) reaction to characterize morphologically retinal NOS expression and activity. Using reverse transcription-PCR and Western blot analysis, we were able to identify the NOS isoforms being regulated. Six days after optic nerve lesion, we observed an increase in neuronal NOS (NOS-I) mRNA and protein expression in the inner retina. This did not lead to a marked increase in overall retinal NOS activity. Only RGC axons displayed strong de novo NADPH-d reactivity. In contrast, intraocular injection of BDNF resulted in a marked upregulation of NOS activity in NOS-I-immunoreactive structures, leaving the level of NOS-I expression unchanged. In addition, an induction of inducible NOS (NOS-II) was found after BDNF treatment. We identified microglial cells increasing in number and being activated by BDNF, which could serve as the cellular source of NOS-II. In summary, our data suggest that BDNF upregulates retinal NOS activity by both a post-translational regulation of NOS-I activity and an induction of NOS-II. These findings might be useful for developing pharmacological strategies to improve BDNF-mediated neuroprotection.
Keywords: BDNF, nitric oxide synthase, NADPH-diaphorase, microglia, retina, axotomy, neurodegeneration
The nerve growth factor (NGF) gene family, referred to as the neurotrophins, comprises a class of highly related proteins, including NGF itself, brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4/5 (Ibañez, 1994). They have been shown to serve as survival, mitogenic, and differentiation factors in both the developing and adult CNS and PNS (Davies, 1994; Barbacid, 1995; Cellerino and Maffei, 1996). Despite the abundance of in vitro and in vivodata demonstrating neuroprotective properties of neurotrophins (for review, see Snider and Johnson, 1989; Lewin and Barde, 1996), recent in vitro evidence provocatively suggested that neurotrophins under certain circumstances can also be neurotoxic by enhancing excitotoxic insults (Koh et al., 1995; Samdani et al., 1997).
Axonal lesions in the adult mammalian CNS often lead to secondary degeneration and death of the injured neurons. Transection of the optic nerve (ON) in the adult rat, for instance, results in retrograde death of 85% of retinal ganglion cells (RGCs) within 14 d (Villegas-Pérez et al., 1988, 1993). Several neurotrophic factors promote survival of axotomized adult RGCs, BDNF being one of the most effective (Mey and Thanos, 1993; Mansour-Robaey et al., 1994;Peinado-Ramón et al., 1996; Klöcker et al., 1997). However, we have shown recently that exogenously applied BDNF limits its neuroprotective potential on axotomized RGCs by increasing retinal nitric oxide synthase (NOS) activity (Klöcker et al., 1998). Work by Cellerino and collaborators demonstrated that retinal NOS activity is reduced in mice deficient for the bdnf gene, thus suggesting a role also for endogenous BDNF in the regulation of retinal NOS activity (Cellerino et al., 1998b).
We therefore sought out to investigate the interaction of BDNF and NOS in more detail. Up to now, three isoforms of NOS have been characterized and cloned: neuronal NOS (NOS-I or nNOS), inducible NOS (NOS-II or iNOS), and endothelial NOS (NOS-III or eNOS) (for review, see Marletta, 1994; Nathan and Xie, 1994; Griffith and Stuehr, 1995). NOS-I and NOS-III are constitutively expressed, whereas NOS-II is usually not expressed but induced in many cell types by certain immunological stimuli (Nathan and Xie, 1994). In the present study, we first used NOS immunohistochemistry and the NADPH-diaphorase (NADPH-d) reaction (Darius et al., 1995) to describe morphologically the effects of BDNF on retinal NOS expression and activity. Using reverse transcription (RT)-PCR and Western blot analysis, we further addressed the question as to which isoform of NOS is regulated by BDNF. Because BDNF is known to enhance glutamatergic neurotransmission (Carmignoto et al., 1997; Jarvis et al., 1997; Sakai et al., 1997; Suen et al., 1997;Lin et al., 1998) and because NOS-I can be regulated post-translationally by increased intracellular calcium (Nathan and Xie, 1994), we also investigated whether BDNF upregulates NOS-I activity by modifying NMDA receptor activation. To this end, we tested whether the NMDA open-channel blocker memantine could reduce BDNF-induced upregulation of NOS activity.
MATERIALS AND METHODS
Animal surgery. Adult female Dark Agouty rats (150–200 gm; Charles River Wiga, Sulzfeld, Germany) were anesthetized by intraperitoneal injection of chloral hydrate (0.42 gm/kg body weight). The right ON was transected as described previously (Klöcker et al., 1998). Briefly, the orbita was opened saving the supraorbital vein, and the lacrimal gland was subtotally resected. By means of a small retractor, the extraocular muscles were spread, and the ON was exposed after longitudinal incision of the eye retractor muscle and the dura sheath. The ON was transected ∼2 mm from the ocular bulb. After surgery, preservation of the retinal blood supply was checked fundoscopically. To determine RGC densities, cells were retrogradely labeled with the fluorescent tracer Fast Blue (FB) (Dr. Illing Chemie, Gross-Umstadt, Germany). To this end, a small piece of gel foam soaked in 2% aqueous FB was placed at the ocular stump of the ON after transection. For double-labeling experiments (NOS immunohistochemistry and retrograde tracing), we used the fluorescent tracer Fluorogold (FG) (Fluorochrome Inc., Englewood, CO). In this case, animals were anesthetized by diethylether at postnatal day 7, when their superior colliculi offer good surgical access, because they are not yet overgrown by the visual cortex. The skin was incised mediosagitally, and the skull cartilage was opened dorsal to the lambda fissure. FG (5% in normal saline) was then applied to both superior colliculi using a micropipette (Klöcker et al., 1998).
Drug administration. Recombinant human BDNF (Alomone Labs, Jerusalem, Israel) was dissolved in a 1% solution of bovine serum albumin (BSA) in PBS at a concentration of 250 ng/μl. Under diethylether anesthesia, 2 μl of BDNF (500 ng) in BSA–PBS or 2 μl BSA–PBS without BDNF (vehicle) were injected into the vitreous of the right eye by means of a glass microelectrode with a tip diameter of 30 μm, puncturing the eye at the cornea–sclera junction. BDNF–vehicle treatment consisted of a single injection on day 4 after ON transection.
Memantine hydrochloride was purchased from Merz & Co. (Frankfurt, Germany) and was administered intraperitoneally at a dose of 20 mg/kg body weight every 12 hr starting on the day of surgery. The treatment regimens combining memantine and BDNF consisted of the memantine treatment as described above and either a single intraocular injection of 500 ng of BDNF on day 4 after axotomy for the NADPH-d histochemistry or three intraocular injections of 500 ng of BDNF repeated on days 4, 7, and 10 after axotomy for the neuroprotection study (Klöcker et al., 1998).
RGC densities. Fourteen days after ON transection, animals were killed by an overdose of chloral hydrate, and both eyes were removed. The retinas were dissected, flat-mounted on glass slides, and fixed in 4% paraformaldehyde (PFA) in PBS for 20 min. They were examined by fluorescent microscopy (Axiophot 2; Zeiss, Göttingen, Germany) using an UV filter (365/397 nm) for FB and FG fluorescence. RGC densities were determined as described in detail previously (Kermer et al., 1998; Klöcker et al., 1998). Briefly, tracer-labeled RGCs were counted in 12 distinct areas of 62,500 μm2 each (three areas per retinal quadrant at three different retinal eccentricities of , ½, and of the retinal radius). Cell counts were done in duplicate by two investigators.
NADPH-d histochemistry and immunohistochemistry. Six days after ON transection, animals received an overdose of chloral hydrate and were perfused intracardially with 4% PFA in PBS for 10 min. Then, both eyes were dissected and immersion-fixed as eye cups without cornea and lens for additional 20 min in 4% PFA–PBS at 4°C. The retinas were either flat-mounted on glass slides, or cryostat sections were made. For the latter, the eyes were immersed in 30% sucrose in PBS overnight at 4°C, cryoprotected, and then snap-frozen in liquid nitrogen. Sixteen micrometer cryostat sections were made and collected on gelatin-coated slides, air-dried, and stored at −20°C before further processing. NADPH-d histochemistry was performed as described by Huxlin and Bennett (1995). Briefly, the retina whole mounts or sections were incubated for 2–3 hr at room temperature in a solution of 0.5 mg of nitroblue tetrazolium, 2 mg of β-NADPH, and 6 μl of Triton X-100 in 2 ml of PBS (chemicals purchased from Sigma, Deisenhofen, Germany). The development of the staining was checked by repeated microscopic inspections. The histochemical reaction was stopped by washing three times with PBS. Sections were coverslipped in 1:1 glycerol/PBS. BDNF–vehicle-treated and control retinas were always processed in parallel to avoid variability of the histochemical reaction.
For immunohistochemistry, retinal cryostat sections were preincubated in 10% normal goat serum (NGS) in PBS containing 0.03% Triton X-100 (PBST) for 1 hr at room temperature. The sections were incubated at 4°C overnight with either the primary antibody directed against NOS-I (R-20; diluted 1:250 in 2% NGS–PBST; Santa Cruz Biotechnology, Ismaning, Germany) (Heneka et al., 1998) or primary antibodies directed against tissue macrophages (ED-1), the rat equivalent of the human complement receptor CR-3 (Ox-42), and MHC class II antigen (Ox-6) (diluted 1:100 in 2% NGS–PBST; Serotec, Oxford, England). Omission of the primary antibody served as negative control. Immunoreactivity was visualized by incubating the sections with either goat anti-rabbit IgG or goat anti-mouse IgG serum, respectively, both conjugated with Cy-3 (1:250 in a solution of 10% NGS in PBS; Dianova, Hamburg, Germany) for 1 hr at room temperature. Sections were coverslipped in Mowiol (Hoechst, Frankfurt, Germany). For both NADPH-d reaction and immunohistochemistry, retinal sections of at least three different animals per experimental group were examined.
RT-PCR and Western blot experiments. For RT-PCR experiments, retinas were quickly dissected and immediately snap-frozen in liquid nitrogen. Then, total RNA was extracted using Trizol reagent (Life Technologies GmbH, Karlsruhe, Germany) following the manufacturer’s protocol. RT-PCR was performed according to standard protocols. one μg of total RNA was reverse-transcribed using Maloney murine leukemia virus reverse transcriptase (Life Technologies GmbH) in a 20 μl reaction volume. Two microliters were used as a template for PCR. PCR was performed using 0.6 U of Amplitaq polymerase (Perkin-Elmer, Branchburg, NJ) in a 50 μl reaction volume containing 10 mm Tris-HCl, pH 8.3, 50 mmKCl, 0.01% gelatin, 2.5 mm MgCl2, 8 mm dNTP, and 1 μm each of forward and reverse primers. Primer sequences are as follows: NOS-I up, 5′-TTCCGAAGCTTCTGGCAAC-3′; NOS-I down, 5′-GGATGGCTTTGAGGACATC-3′ with annealing temperature of 55°C and 35 amplification cycles run; NOS-II up, 5′-AAGTTTCTTGTGGCAGCAGC-3′; NOS-II down, 5′-CCTCGTGGCTTTGGGCTCCT-3′ with annealing temperature of 54°C and 35 amplification cycles run using the hot-start technique; G3PDH up, 5′-ACCACAGTCCATGCCATCAC-3′; and G3PDH down, 5′-TCCACCACCCTGTTGCTGTA-3′ with annealing temperature of 55°C and 24 amplification cycles run. Five microliters of loading dye were added to the reaction, and 10 μl were analyzed on 1.5% agarose gels containing 0.05% ethidium bromide. We varied the cDNA template concentrations and PCR cycle numbers to control for saturation of the PCR reaction.
For Western blot experiments, retinas were homogenized in lysis buffer containing 50 mm Tris-HCl, 150 mm NaCl, 1% Triton X-100, 0.1 mm PMSF, and 2 μg/ml pepstatin, leupeptin, and aprotinin, pH 8.3. The cell suspension was lysed on ice for 20 min, and cell debris was pelleted at 14,000 ×g for 15 min. The protein concentration of the supernatant was determined using the BCA reagent (Pierce, Rockford, IL). After separation by reducing SDS-PAGE (Ausubel et al., 1987) of the lysates (20 μg of protein per lane), proteins were transferred to a polyvinylidene difluoride membrane and blocked with 5% skim milk in 0.1% Tween 20–PBS (PBS-T). The membranes were incubated with the primary antibodies against NOS-I [1:2000 in 1% skim milk in PBS-T; Transduction Laboratories (Lexington, KY), distributed by Dianova] (Samdani et al., 1997) or NOS-II (M-19; 1:2000 in 1% skim milk in PBS-T; Santa Cruz Biotechnology) (Colville-Nash et al., 1998). After washing in PBS-T, the membranes were incubated with HRP-conjugated secondary antibodies against mouse IgM and rabbit IgG, respectively (1:2000 in PBS-T; Dianova). Labeled proteins were detected using the ECL-plus reagent (Amersham, Arlington Heights, IL) following the supplier’s instructions.
Statistics. Data are given as mean ± SEM. Statistical significance in the neuroprotection study was assessed using one-way ANOVA followed by Duncan’s test.
RESULTS
Effects of ON transection and intraocular BDNF on retinal NADPH-d reactivity
In unlesioned adult rat retinas, NADPH-d staining was mainly found in a subpopulation of amacrine neurons located at the inner margin of the inner nuclear layer (INL). Within this cell population, we could distinguish two different cell types. The most often observed cell type had a large soma, ranging in size from 12 to 16 μm, and was darkly stained for NADPH-d reactivity (type I), whereas the rare second cell type (type II) was smaller, ranging in size from 6 to 10 μm, and was less intensely stained (Fig. 1). In favorable tissue sections, long processes from type I neurons could be followed into sublaminae b and c of the inner plexiform layer (IPL). Type I neurons were concentrated in the central retina, whereas type II neurons did not show a noticeable preference for a certain retinal eccentricity. The ganglion cell layer (GCL) contained a modest number of rather small cells displaying NADPH-d reactivity (Fig.2D). Because of their low staining intensity, they could only be observed after incubation of the retinas for >3 hr in the staining solution. Judged by their morphology, these cells most likely represented displaced amacrine neurons (Darius et al., 1995).
Fig. 1.

Effects of ON transection and BDNF on NOS histochemical activity in the adult rat retina using NADPH-d reactivity as a marker. Radial sections of untreated control retinas (A, C) and retinas 6 d after ON transection without treatment (B) and after a single intraocular injection of 500 ng of BDNF on day 4 after lesion (D). Sections were processed in parallel to allow comparison of NADPH-d reactivity. The arrow in B indicates RGC axons displaying NADPH-d reactivity after ON transection. Thethick arrow in D points to a type I amacrine neuron, and the thin arrow points to a type II amacrine neuron. ONL, Outer nuclear layer. Scale bar, 45 μm.
Fig. 2.

Effects of ON transection and BDNF on NOS histochemical activity in the nerve fiber layer of adult rat retinas using NADPH-d reactivity as a marker. Whole mounts of an untreated control retina (A) and retinas 6 d after ON transection without treatment (B) and after a single intraocular injection of 500 ng of BDNF on day 4 after lesion (C). Whole mounts in A–C were processed in parallel to allow comparison of NADPH-d reactivity. Note the increase in axonal NADPH-d reactivity after ON transection. Magnification: 100×. Close-up into the ganglion cell layer of an untreated control retina (D).Arrows indicate two presumptive displaced amacrine cells. Magnification: 400×.
Six days after ON transection, we observed an increase in retinal NADPH-d staining (Fig. 1B). Interestingly, RGC axons now displayed NOS histochemical activity, although their somas were spared (Fig. 2B,D). Furthermore, the staining intensity of the IPL showing a trilaminar staining grew stronger. Vehicle injection on day 4 after axotomy did not elicit a staining pattern different from ON transection alone (data not shown).
However, a single intraocular injection of 500 ng of BDNF on day 4 after axotomy resulted in an increase of NADPH-d staining, which was much more pronounced than the increase induced by ON lesion itself. Most strikingly, the staining intensity and the number of amacrine neurons in the INL expressing NADPH-d reactivity increased (Fig.1D). In most cases, the arborization of the long type I and short type II cell processes in the IPL were visible in sublaminae a and b, respectively. Although the labeling of RGC axons grew stronger, we still did not find RGC somas being labeled (Fig.2C). The population of NADPH-d-positive displaced amacrine cells in the GCL displayed a little stronger staining without any remarkable change in number.
Quantification of the BDNF effects on NADPH-d-reactive neurons in the INL
We quantified cell densities of NADPH-d-positive amacrine neurons in the INL in flat mount preparations (Fig.3). ON transection alone led to the increase in retinal NADPH-d staining intensity as described above, but the number of NADPH-d-positive amacrine neurons in axotomized retinas did not vary from that in control retinas (Fig.4). However, a single injection of 500 ng of BDNF 4 d after lesion did not only further increase the staining intensity of this subpopulation of amacrine cells but also increased their number. As has been demonstrated in postnatal rats (Cellerino et al., 1998a), this effect was most noticeable in type II amacrine neurons. Their number increased almost fivefold after BDNF injection. The number of type I cells positive for NADPH-d reactivity, on the other hand, increased only marginally by a factor of 1.2. Vehicle injection failed to mimic the effects of BDNF.
Fig. 3.
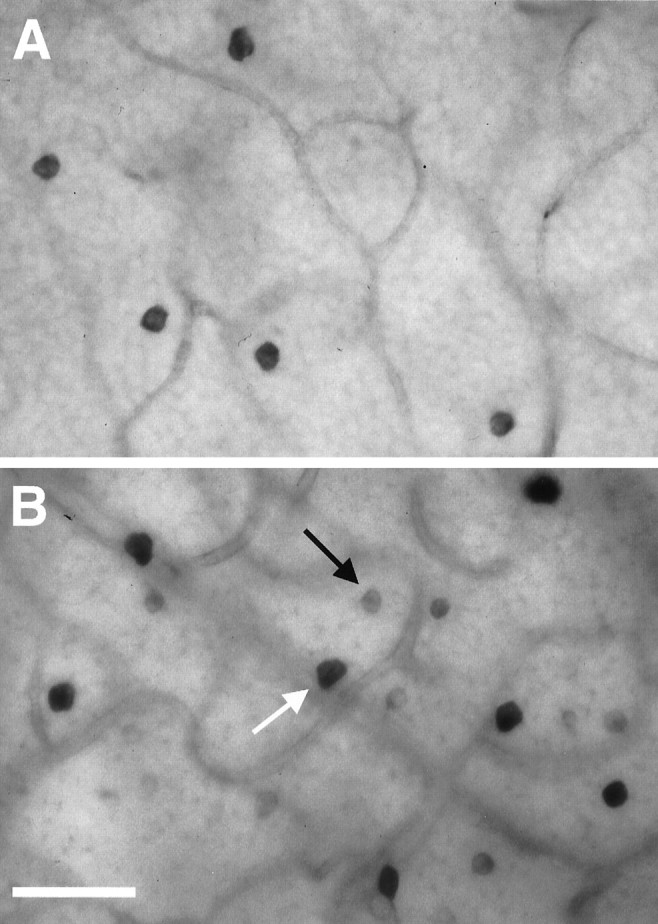
Effect of BDNF on NOS histochemical activity in the INL of adult rat retinas using NADPH-d reactivity as a marker. Whole mounts of an untreated control retina (A) and a retina 6 d after ON transection treated with a single injection of 500 ng of BDNF on day 4 after lesion (B). The retinas were processed in parallel to allow comparison of NADPH-d reactivity. The arrowsindicate type I (white) and type II (black) amacrine neurons in the INL, respectively. Scale bar, 45 μm.
Fig. 4.

A, Quantification of the effect of BDNF on NOS histochemical activity in the INL. Cell counts of NADPH-d-positive type I and type II neurons were performed following the same protocol as described for RGC counts (see Materials and Methods). Numbers are expressed as percentage of the respective contralateral control retinas. Data from three different animals per experimental group were pooled. B, Quantification of the effect of axotomy on NOS immunoreactivity in the INL. Cell counts of NOS-I-immunoreactive type I and type II amacrine neurons in absolute numbers per retinal section. Data from at least five central sections per animal from three animals per experimental group were pooled.
Regulation of retinal NOS mRNA and protein expression after axotomy and intraocular BDNF
To determine whether ON transection and BDNF increased NOS activity in the retina by regulating NOS expression, we performed RT-PCR and Western blot experiments using primers and antibodies specific for the isoforms NOS-I and NOS-II (Fig.5). We found constitutive expression of NOS-I mRNA and protein in control retinas, which markedly increased 6 d after ON lesion. Additional vehicle or BDNF treatment did not further change NOS-I mRNA or protein expression. Low levels of NOS-II mRNA and barely detectable levels of NOS-II protein were also observed in unlesioned control retinas. Axotomy without or with vehicle treatment did not change retinal NOS-II mRNA or protein expression. However, a single intraocular injection of 500 ng of BDNF on day 4 after ON lesion was found to induce both NOS-II mRNA and protein expression in the retina.
Fig. 5.

Regulation of retinal NOS mRNA and protein expression by ON transection and BDNF. A, Representative RT-PCR for NOS-I, NOS-II, and G3PDH of total retinal tissues of untreated controls (CTRL), from retinas 6 d after ON transection without treatment (AXO), and with a single injection of vehicle (VEH) or 500 ng of BDNF (BDNF) on day 4 after lesion.B, Representative Western blot analysis for NOS-I and NOS-II of total retinal tissue of the respective experimental groups. Retinal tissues of four different animals per experimental group were pooled except for NOS-II protein analysis, which displays in each case (VEH and BDNF) test and contralateral control retina of the same animal.
NOS-I immunohistochemistry
In normal control retinas, few type I and type II amacrine neurons in the INL were immunoreactive for NOS-I (Fig.6A). We further found staining of the IPL typically appearing as a trilaminar pattern. NOS-I immunoreactivity was also detected in a subpopulation of cells in the GCL, which was greater than the one staining for NADPH-d reactivity (Fig. 6A, arrow). To distinguish between RGCs and displaced amacrine neurons, we used retrograde fluorescent tracing to unequivocally identify RGCs. As can be derived from Figure6F, subpopulations of both RGCs (arrows) and displaced amacrine neurons (arrowhead) showed NOS-I immunoreactivity. RGC axons, however, were not positive for NOS-I (Fig.6B).
Fig. 6.

Effects of ON transection and BDNF on the expression of NOS-I immunoreactivity in the adult rat retina. Radial sections of an untreated control retina (A) and control ON head (B), of a retina and ON head 6 d after ON transection (C, D), and a retina 6 d after ON transection treated with a single intraocular injection of 500 ng of BDNF (E). We were able to unequivocally identify RGCs that displayed NOS-I immunoreactivity (F), using retrograde fluorescent tracing (inset). See Results for detailed explanation of arrows. ONL, Outer nuclear layer. Scale bars: A, C,E, F, 45 μm. Magnification:B, D, 200×.
Six days after ON transection, retinal NOS-I immunoreactivity not only increased the staining intensity but also the number of particularly type II amacrine neurons in the INL (Figs.4B, 6C). The number of NOS-I-immunoreactive type I amacrine neurons did not change after axotomy. The trilaminar staining pattern of the IPL was still preserved but displayed stronger immunoreactivity. In addition to RGC somas, now their axons were immunoreactive for NOS-I to a significantly greater extent than the cell bodies (Fig. 6C, small arrow). However, immunostaining could only be detected within their intraretinal course up to the optic disk (Fig.6D, arrow). Intraocular injection of vehicle or BDNF after ON transection did not lead to any marked changes in NOS-I immunoreactivity compared with axotomy without treatment (Figs. 4B, 6E).
NMDA antagonism does not prevent the BDNF-induced increase in NADPH-d reactivity
Comparison of the NOS-I immunostainings with the NADPH-d stainings indicated that most of the increase in NOS histochemical activity observed after BDNF treatment could be localized to NOS-I-positive structures. Because we failed to detect an induction of NOS-I mRNA or protein expression by BDNF, however, we assumed that BDNF regulates NOS-I activity post-translationally. In a number of studies, it has been shown that BDNF potentiates glutamatergic neurotransmission, thereby increasing intracellular calcium levels (Jarvis et al., 1997;Sakai et al., 1997). Therefore, we investigated whether BDNF might enhance NOS-I activity via a calcium-dependent mechanism involving NMDA receptors.
To this end, we examined whether simultaneous administration of the NMDA open-channel blocker memantine could reduce the increase in retinal NADPH-d reactivity induced by BDNF. In addition, we addressed the question whether the neuroprotective action of BDNF on axotomized RGCs could be potentiated by simultaneous administration of memantine as shown for a combined treatment with BDNF and the NOS inhibitorl-NAME (Klöcker et al., 1998). We decided to use memantine instead of MK-801, because it can be administered intraperitoneally over a period of 14 d without the side effects of phenylcyclidines on physiological parameters as body temperature and on animal behavior. Systemic application of memantine holds the advantage of avoiding direct pharmacological interactions between the NMDA antagonist and the intraocularly applied BDNF. Moreover, because the block of NMDA receptors by memantine is use-dependent and because neither the delay nor the duration of the BDNF effect on NOS activity could exactly be predicted, we assumed systemic application of the NMDA antagonist repeated twice daily to be superior to single intraocular applications.
As shown in Figure 7, memantine treatment did not affect the upregulation of retinal NADPH-d reactivity by intraocular injection of BDNF. In line with that, we did not observe any potentiation of BDNF-mediated neuroprotection by memantine. ON transection led to the retrograde death of ∼85% RGCs within 14 d as shown in our previous study (density of surviving RGCs, 339 ± 43 RGCs/mm2) (Table1) (Klöcker et al., 1998). Three intraocular injections of 500 ng of BDNF repeated on days 4, 7, and 10 after axotomy resulted in survival of 804 ± 87 RGCs/mm2. Systemic application of 20 mg/kg memantine neither exerted any significant neuroprotective effect when given alone (412 ± 77 RGCs/mm2) nor significantly potentiated the neurotrophic effect of BDNF when given at a combined treatment regimen (919 ± 54 RGCs/mm2). A more detailed analysis of RGC survival with respect to retinal eccentricity suggested a regional difference in survival rates in response to combined BDNF and memantine treatment versus single BDNF treatment. In the peripheral retina, the combined treatment strategy led to higher RGC survival than single BDNF treatment. However, this difference did not reach statistical significance (p > 0.05).
Fig. 7.
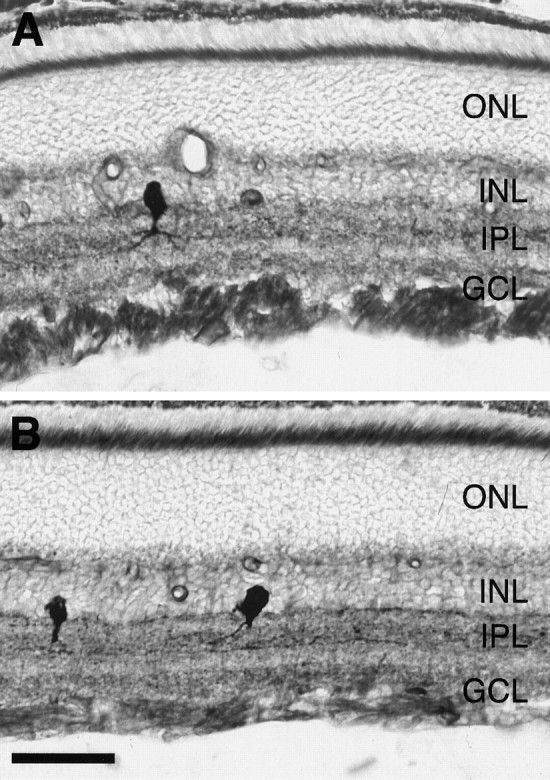
Memantine does not prevent the BDNF-induced increase in retinal NADPH-d reactivity. Radial sections of retinas 6 d after ON transection treated with a single intraocular injection of 500 ng of BDNF on day 4 after lesion (A,B). In B, the intraocular BDNF treatment was combined with systemic administration of memantine at a dose of 20 mg/kg body weight twice daily. Sections were processed in parallel to allow comparison of NADPH-d reactivity. ONL, Outer nuclear layer. Scale bar, 45 μm.
Table 1.
Effects of single and combined treatment with systemic memantine and intraocular BDNF on RGC survival 14 d after optic nerve transection in the adult rat
| Retinal radius | Retinal radius | Retinal radius | Average | |
|---|---|---|---|---|
| Normal control | 2647 ± 95 | 2289 ± 53 | 1180 ± 46 | 2039 ± 56 |
| (n = 11) | ||||
| Axotomy only | 409 ± 50 | 354 ± 56 | 254 ± 49 | 339 ± 43 |
| (n = 6) | ||||
| Vehicle | 587 ± 75 | 394 ± 90 | 255 ± 77 | 412 ± 77 |
| (n = 4) | ||||
| BDNF 500 | 1253 ± 138 | 777 ± 80 | 382 ± 61 | 804 ± 87* |
| (n = 7) | ||||
| Memantine 20 | 400 ± 46 | 321 ± 49 | 208 ± 27 | 310 ± 39 |
| (n = 5) | ||||
| BDNF 500 + | 1249 ± 100 | 975 ± 42 | 535 ± 24 | 919 ± 54* |
| Menamtine 20 | (n = 3) |
Data are given as mean ± SEM RGC densities (cells/mm2) at one-sixth, one-half, and five-sixth, of the retinal radius and averaged over the radius. BDNF 500, Intraocular injections of BDNF at a dose of 500 ng/injection on days 4, 7, and 10 after axotomy. Memantine 20, Intraperitoneal injections of memantine at a dose of 20 mg/kg body weight every 12 hr. For statistical analysis, one-way ANOVA with Duncan’s post hoc test was used (*p < 0.05 compared with axotomy alone).
BDNF activates microglia
Attempts to identify the cellular source of NOS-II upregulation after intraocular BDNF injection using several NOS-II antibodies remained unsuccessful because of unspecific background staining. Because CNS microglia and macrophages have been reported to be responsive to neurotrophins (Elkabes et al., 1996), we asked whether the induction of NOS-II was caused by a BDNF-mediated activation of immune-competent cells known to express NOS-II (Nathan and Xie, 1994).
We performed immunohistochemistry using antibodies directed against monocytes–macrophages (ED-1), complement receptor-3 (Ox-42), and MHC-II antigen (Ox-6) to distinguish between macrophages and microglial cells and to determine their state of activation (Ng and Ling, 1997; Watanabe et al., 1999). Independent of the experimental treatment, ED-1-positive cells were not detected. In contrast, we observed few Ox-42-immunoreactive cells and cell processes in unlesioned control retinas (Fig.8A). They were either located in close proximity of blood vessels or scattered in the outer sublaminae of the IPL with a preference for the central retina. Six days after axotomy without treatment, Ox-42-positive cells not only increased slightly in number but were now also seen in the inner IPL and the GCL (Fig. 8C). Ox-42 expression in vehicle-injected retinas did not differ from only axotomized retinas (Fig.8E). Still most of the Ox-42-positive cells displayed a ramified phenotype as observed in unlesioned controls. A single injection of BDNF markedly enlarged the population of Ox-42-immunopositive cells, particularly in the central retina (Fig.8G). They were distributed in all sublaminae of the IPL, in the GCL, and sometimes even visible in the INL and outer plexiform layer (OPL). Ox-6 immunohistochemistry mainly paralleled Ox-42 immunoreactivity, with the exception that we did not detect any Ox-6-positive cells in control tissue (Fig. 8B). Six days after axotomy without or with additional injection of vehicle, few Ox-6-immunoreactive cells with oval- or round-shaped somas appeared in the inner IPL and GCL (Fig.8D,F). Additional injection of BDNF led to a strong increase in the number of Ox-6-immunoreactive cells, now distributed from the OPL to all sublaminae of the IPL and the GCL. Various phenotypes could be observed, ranging from ramified over rod-shaped to amoeboid morphology (Fig. 8H).
Fig. 8.
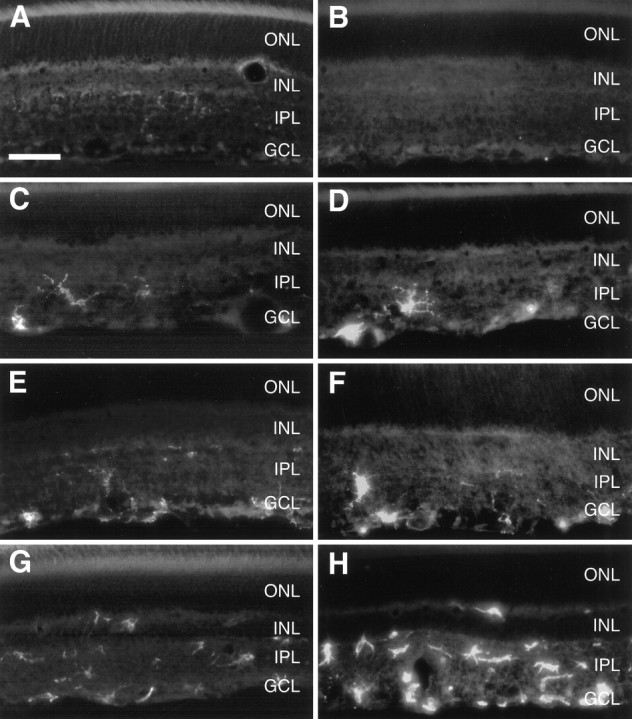
BDNF activates retinal microglia. Complement receptor-3 (Ox-42; left column) and MHC-II antigen (Ox-6; right column) immunoreactivity is shown in radial sections of untreated control retinas (A,B) and of retinas 6 d after ON transection either without treatment (C, D) or with a single injection of vehicle (E, F) or 500 ng of BDNF (G, H) on day 4 after lesion. ONL, Outer nuclear layer. Scale bar, 45 μm.
DISCUSSION
In the present study, we characterized the effects of ON transection and BDNF on retinal NOS expression and activity. Although ON transection does substantially increase retinal NOS-I expression, it leads only to a rather small increase in NOS activity. Additional application of exogenous BDNF, although not further increasing NOS-I expression, leads to a dramatic post-translational upregulation of NOS-I activity. Furthermore, BDNF induces NOS-II expression, most likely by activating retinal microglia.
In unlesioned control retinas, we found NOS-I-immunoreactive amacrine cells in the inner INL and a modest number of NOS-I-immunoreactive displaced amacrine cells and RGCs in the GCL. These observations are in good agreement with previous studies showing a similar distribution of NOS-I immunoreactivity in the rat retina (Yamamoto et al., 1993). The pattern of NADPH-d staining corresponds well with NOS-I immunoreactivity, suggesting that retinal NOS activity is mainly activity of the neuronal isoform. The only two exceptions to this were (1) the photoreceptor segments that were NADPH-d-reactive but not NOS-I-immunoreactive (Koistinaho and Sagar, 1995) and (2) a subpopulation of RGCs that we found to be NOS-I-immunoreactive without showing NADPH-d reactivity. The small population of NADPH-d-positive cells we observed in the GCL were by morphology rather displaced amacrine cells than RGCs. ON transection induced an increase in retinal NOS-I expression as we could demonstrate by RT-PCR and Western blot analysis. Retinal NOS-I immunoreactivity also increased in that we observed a greater number of type II amacrine neurons in the INL and a higher overall staining intensity of labeled cells and the IPL. Besides RGC somas, to an even greater extent, their axons also became immunoreactive. NADPH-d reactivity, however, only increased in the IPL and in RGC axons, still sparing the RGC somas. Thus, there seem to exist tools to specifically target NOS-I activity to certain subcellular locations. The ways how neurons target NOS-I protein to specific subcellular locations are just beginning to be identified. The N-terminal domain of NOS-I, for example, contains a PDZ binding motif that is found in a diverse group of cytoskeletal proteins (Cho et al., 1992). It is known that NOS-I interacts with the postsynaptic density protein PSD-95, which leads to a close association of NOS-I and the NMDA receptor in the postsynapse (Brenman et al., 1996; Brenman and Bredt, 1997). Another example of subcellular targeting of NOS protein has been discovered for the third isoform. NOS-III phosphorylation results in a translocation from the membrane to the cytosol (Michel et al., 1993). In our study, however, we did not only find a higher expression of NOS-I protein in RGC axons, but we also observed a specific targeting of its activity to axons sparing the RGC somas. One could speculate whether a localized expression of the protein inhibitor of neuronal NOS (PIN) preventing dimerization of NOS-I (Jaffrey and Snyder, 1996), suppresses NOS-I activity in RGC somas.
Despite its constitutive expression, it has been repeatedly described that NOS-I protein and its activity can be subject to dramatic upregulation in axotomized neurons in both the CNS and PNS (for review, see Garthwaite and Boulton, 1995). It still remains unclear, however, whether such regulation of NOS is causally involved in neuronal death after axotomy, whether it is just an epiphenomenon, or whether it has even a neuroprotective role (Verge et al., 1992; Yu, 1994; Huxlin and Bennett, 1995; Rossiter et al., 1996). Although NOS expression increases after ON transection, the overall changes in retinal NOS histochemical activity after ON transection that we and others have found are rather small (Huxlin and Bennett, 1995). Therefore, we favor the hypothesis that NO is not a major cause of cell death of axotomized RGCs, which is supported by our previous results showing that the NOS inhibitor l-NAME by itself was ineffective in promoting the survival of axotomized RGCs (Klöcker et al., 1998). It is worth noting that lesion-induced NOS-I protein regulation was not only found in axons of RGCs, which are the cells directly affected by ON transection, but also in neurons of other retinal cell layers that were not lesioned. We have to postulate, therefore, that RGCs communicate retrogradely with their input neurons. The nature of this retrograde messenger system, however, remains speculative.
Intraocular injection of BDNF did not produce the same qualitative changes in the expression of NOS-I mRNA or protein compared with axotomy alone as verified by RT-PCR, Western blot, and immunohistochemistry. However, we observed a strong increase in NADPH-d activity after BDNF treatment, which could be localized to NOS-I-immunoreactive tissue. Most likely, this can be explained by a post-translational regulation of NOS-I activity. Because BDNF can increase intracellular calcium levels by enhancing glutamatergic neurotransmission (Jarvis et al., 1997; Sakai et al., 1997) and because NMDA receptors are expressed in the INL and GCL of the retina (Brandstätter et al., 1994; Hartveit and Veruki, 1997), we tested whether simultaneous application of an NMDA antagonist could reduce the upregulation of retinal NOS activity by BDNF and whether it could improve BDNF neuroprotection on axotomized RGCs as a specific NOS inhibitor had done before (Klöcker et al., 1998). Systemic application of memantine neither changed the upregulation of NOS activity induced by BDNF nor significantly potentiated the neuroprotective effects of BDNF. This is probably not attributable to a dosage problem, because we used the highest dose described in the literature predicted to provide substantial neuroprotection in models of ischemia and glutamate toxicity when given intraperitoneally (Seif el Nasr et al., 1990; Block and Schwarz, 1996; Vorwerk et al., 1996;Lagreze et al., 1998; Osborne, 1999). We can also exclude possible toxic effects of memantine because of high dosage, because both single memantine treatment and the combination of BDNF and memantine did not result in lower, but even slightly higher, RGC rescue compared with controls. These results therefore strongly suggest that enhancement of NMDA receptor-mediated neurotransmission is not the mechanism by which BDNF post-translationally regulates retinal NOS-I activity. Besides modifying NMDA receptors, BDNF could use many other ways to increase intracellular calcium, such as by increasing its release from intracellular stores (Roback et al., 1995; Finkbeiner et al., 1997). Alternatively, a modification of AMPA receptors, also known to be expressed in the INL and GCL and to have rather high calcium conductances (Hamassaki Britto et al., 1993; Rorig and Grantyn, 1993), is conceivable. That single memantine treatment did not result in significant rescue of axotomized RGCs is in contrast to neuroprotective effects of the NMDA antagonist MK-801 on the β subpopulation of RGCs after ON transection in cats (Russelakis Carneiro et al., 1996) but in good agreement with experiments in rats demonstrating even adverse effects of MK-801 on the survival of axotomized RGCs (Schmitt and Sabel, 1996).
In addition to upregulation of NOS-I activity, intraocular application of BDNF induced NOS-II expression as detected by RT-PCR and Western blot experiments. Unfortunately, we were not able to localize the cellular source of NOS-II expression in the retina by immunohistochemistry. However, our data support the hypothesis that BDNF activates immune-competent cells known to express NOS-II (Garthwaite and Boulton, 1995). Although we have never found any macrophages in retinas of any experimental group, we observed few Ox-42-positive microglial cells, even in unlesioned retinas. Six days after ON transection, their number markedly increased, and they translocated into the GCL, now displaying phagocytic activity as revealed by their morphology and Ox-6 immunoreactivity. This observation can easily be explained by the induction of apoptotic cell death among RGCs, which starts at approximately day 4 and reaches a maximum on day 7 after ON lesion (Garcia-Valenzuela et al., 1994;Isenmann et al., 1997). Additional application of BDNF further stimulated the microglial response, which is in good agreement within vitro data showing that CNS microglia produces and responds to neurotrophins (Condorelli et al., 1995; Elkabes et al., 1996; Miwa et al., 1997). Indeed, the observed microglial activation in our model does not have to be a direct response to activation of the BDNF receptor TrkB or the low-affinity neurotrophin receptor p75 but could alternatively be an indirect response to BDNF-induced changes in the retina. Moreover, further studies are needed to actually prove that BDNF upregulates NOS-II expression in microglial cells, because our results do not exclude that retinal neurons normally expressing only NOS-I can additionally upregulate NOS-II in response to BDNF. We detected a basal expression of NOS-II in control tissue, which could be explained by the small population of resting microglial cells we observed also in unlesioned controls. Alternatively, blood cells remaining in retinal vessels during tissue preparation could account for that observation.
In the CNS, NO has highly diverse functions, being involved in neuronal communication, host defense, and vascular regulation (Nathan, 1992;Nathan and Xie, 1994; Schmidt and Walter, 1994; Yun et al., 1996), even exerting neuroprotective effects by inhibiting apoptosis (Melino et al., 1997; Ogura et al., 1997; So et al., 1998) but also being neurotoxic in various CNS disease conditions (Gross and Wolin, 1995;Iadecola, 1997). Besides the redox state of NO, its concentration and the kinetics of its formation often determine the quality of its action. Because NOS-I depends on intracellular calcium, it can produce NO in small and highly regulated bursts well suited for its physiological functions (Garthwaite and Boulton, 1995). NOS-II, however, is independent of calcium and can produce large amounts of NO continuously for long periods of time, which probably add to the cytotoxic effects of NO (Gross and Wolin, 1995). Future studies will have to reveal whether the differential effects of BDNF on the NOS isoforms we described contribute to different extents to the adverse effect limiting BDNF neuroprotection (Klöcker et al., 1998). Then, the development of feasible strategies for isoform-specific inhibition of NOS in vivo could be useful to potentiate BDNF neuroprotection more powerfully and less afflicted with side effects.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 430-B4. We thank S. Thomsen for her skillful technical assistance.
Correspondence should be addressed to Nikolaj Klöcker, Department of Physiology, University of Tübingen, Ob dem Himmelreich 7, 72074 Tübingen, Germany.
REFERENCES
- 1.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. Wiley; New York: 1987. [Google Scholar]
- 2.Barbacid M. Neurotrophic factors and their receptors. Curr Opin Cell Biol. 1995;7:148–155. doi: 10.1016/0955-0674(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 3.Block F, Schwarz M. Memantine reduces functional and morphological consequences induced by global ischemia in rats. Neurosci Lett. 1996;208:41–44. doi: 10.1016/0304-3940(96)12545-3. [DOI] [PubMed] [Google Scholar]
- 4.Brandstatter JH, Hartveit E, Sassoe Pognetto M, Wassle H. Expression of NMDA and high-affinity kainate receptor subunit mRNAs in the adult rat retina. Eur J Neurosci. 1994;6:1100–1112. doi: 10.1111/j.1460-9568.1994.tb00607.x. [DOI] [PubMed] [Google Scholar]
- 5.Brenman JE, Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol. 1997;7:374–378. doi: 10.1016/s0959-4388(97)80065-7. [DOI] [PubMed] [Google Scholar]
- 6.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 7.Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol Lond. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cellerino A, Maffei L. The action of neurotrophins in the development and plasticity of the visual cortex. Prog Neurobiol 49 1996. 53 71[Erratum 50:333]. [DOI] [PubMed] [Google Scholar]
- 9.Cellerino A, Pinzón-Duarte G, Carroll P, Kohler K. Brain-derived neurotrophic factor modulates the development of the dopaminergic network in the rodent retina. J Neurosci. 1998a;18:3351–3362. doi: 10.1523/JNEUROSCI.18-09-03351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cellerino A, Arango-González BA, Kohler K. BDNF controls the development of nitric oxide synthase expression in the rodent retina. Invest Ophthalmol Vis Sci. 1998b;39:S576. [Google Scholar]
- 11.Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 12.Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol. 1998;161:978–984. [PubMed] [Google Scholar]
- 13.Condorelli DF, Salin T, Dell’Albani P, Mudo G, Corsaro M, Timmusk T, Metsis M, Belluardo N. Neurotrophins and their trk receptors in cultured cells of the glial lineage and in white matter of the central nervous system. J Mol Neurosci. 1995;6:237–248. doi: 10.1007/BF02736783. [DOI] [PubMed] [Google Scholar]
- 14.Darius S, Wolf G, Huang PL, Fishman MC. Localization of NADPH-diaphorase/nitric oxide synthase in the rat retina: an electron microscopic study. Brain Res. 1995;690:231–235. doi: 10.1016/0006-8993(95)00559-9. [DOI] [PubMed] [Google Scholar]
- 15.Davies AM. The role of neurotrophins in the developing nervous system. J Neurobiol. 1994;25:1334–1348. doi: 10.1002/neu.480251103. [DOI] [PubMed] [Google Scholar]
- 16.Elkabes S, DiCicco Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z, Sharma SC. Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol. 1994;25:431–438. doi: 10.1002/neu.480250408. [DOI] [PubMed] [Google Scholar]
- 19.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 20.Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 21.Gross SS, Wolin MS. Nitric oxide: pathophysiological mechanisms. Annu Rev Physiol. 1995;57:737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- 22.Hamassaki Britto DE, Hermans Borgmeyer I, Heinemann S, Hughes TE. Expression of glutamate receptor genes in the mammalian retina: the localization of GluR1 through GluR7 mRNAs. J Neurosci. 1993;13:1888–1898. doi: 10.1523/JNEUROSCI.13-05-01888.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartveit E, Veruki ML. AII amacrine cells express functional NMDA receptors. NeuroReport. 1997;8:1219–1223. doi: 10.1097/00001756-199703240-00032. [DOI] [PubMed] [Google Scholar]
- 24.Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T. Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem. 1998;71:88–94. doi: 10.1046/j.1471-4159.1998.71010088.x. [DOI] [PubMed] [Google Scholar]
- 25.Huxlin KR, Bennett MR. NADPH diaphorase expression in the rat retina after axotomy—a supportive role for nitric oxide. Eur J Neurosci. 1995;7:2226–2239. doi: 10.1111/j.1460-9568.1995.tb00644.x. [DOI] [PubMed] [Google Scholar]
- 26.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 27.Ibañez CF. Structure-function relationships in the neurotrophin family. J Neurobiol. 1994;25:1349–1361. doi: 10.1002/neu.480251104. [DOI] [PubMed] [Google Scholar]
- 28.Isenmann S, Wahl C, Krajewski S, Reed JC, Bähr M. Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur J Neurosci. 1997;9:1763–1772. doi: 10.1111/j.1460-9568.1997.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 29.Jaffrey SR, Snyder SH. PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774–777. doi: 10.1126/science.274.5288.774. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- 31.Kermer P, Klöcker N, Labes M, Bähr M. Inhibition of CPP32-like proteases rescues axotomized retinal ganglion cells from secondary cell death in vivo. J Neurosci. 1998;18:4656–4662. doi: 10.1523/JNEUROSCI.18-12-04656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klöcker N, Braunling F, Isenmann S, Bähr M. In vivo neurotrophic effects of GDNF on axotomized retinal ganglion cells. NeuroReport. 1997;8:3439–3442. doi: 10.1097/00001756-199711100-00005. [DOI] [PubMed] [Google Scholar]
- 33.Klöcker N, Cellerino A, Bähr M. Free radical scavenging and inhibition of nitric oxide synthase potentiates the neurotrophic effects of brain-derived neurotrophic factor on axotomized retinal ganglion cells in vivo. J Neurosci. 1998;18:1038–1046. doi: 10.1523/JNEUROSCI.18-03-01038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 35.Koistinaho J, Sagar SM. NADPH-diaphorase-reactive neurones in the retina. Prog Retinal Eye Res. 1995;15:69–87. [Google Scholar]
- 36.Lagreze WA, Knorle R, Bach M, Feuerstein TJ. Memantine is neuroprotective in a rat model of pressure-induced retinal ischemia. Invest Ophthalmol Vis Sci. 1998;39:1063–1066. [PubMed] [Google Scholar]
- 37.Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 38.Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- 39.Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci USA. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 41.Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, Finazzi Agro A. S-Nitrosylation regulates apoptosis. Nature. 1997;388:432–433. doi: 10.1038/41237. [DOI] [PubMed] [Google Scholar]
- 42.Mey J, Thanos S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993;602:304–317. doi: 10.1016/0006-8993(93)90695-j. [DOI] [PubMed] [Google Scholar]
- 43.Michel T, Li GK, Busconi L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1993;90:6252–6256. doi: 10.1073/pnas.90.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miwa T, Furukawa S, Nakajima K, Furukawa Y, Kohsaka S. Lipopolysaccharide enhances synthesis of brain-derived neurotrophic factor in cultured rat microglia. J Neurosci Res. 1997;50:1023–1029. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1023::AID-JNR13>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 45.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 46.Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 47.Ng YK, Ling EA. Induction of major histocompatibility class II antigen on microglial cells in postnatal and adult rats following intraperitoneal injections of lipopolysaccharide. Neurosci Res. 1997;28:111–118. doi: 10.1016/s0168-0102(97)00033-3. [DOI] [PubMed] [Google Scholar]
- 48.Ogura T, Tatemichi M, Esumi H. Nitric oxide inhibits CPP32-like activity under redox regulation. Biochem Biophys Res Commun. 1997;236:365–369. doi: 10.1006/bbrc.1997.6948. [DOI] [PubMed] [Google Scholar]
- 49.Osborne NN. Memantine reduces alterations to the mammalian retina, in situ, induced by ischemia. Vis Neurosci. 1999;16:45–52. doi: 10.1017/s0952523899161017. [DOI] [PubMed] [Google Scholar]
- 50.Peinado-Ramón P, Salvador M, Villegas-Perez MP, Vidal Sanz M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Invest Ophthalmol Vis Sci. 1996;37:489–500. [PubMed] [Google Scholar]
- 51.Roback JD, Marsh HN, Downen M, Palfrey HC, Wainer BH. BDNF-activated signal transduction in rat cortical glial cells. Eur J Neurosci. 1995;7:849–862. doi: 10.1111/j.1460-9568.1995.tb01072.x. [DOI] [PubMed] [Google Scholar]
- 52.Rorig B, Grantyn R. Rat retinal ganglion cells express Ca(2+)-permeable non-NMDA glutamate receptors during the period of histogenetic cell death. Neurosci Lett. 1993;153:32–36. doi: 10.1016/0304-3940(93)90070-2. [DOI] [PubMed] [Google Scholar]
- 53.Rossiter JP, Riopelle RJ, Bisby MA. Axotomy-induced apoptotic cell death of neonatal rat facial motoneurons: time course analysis and relation to NADPH-diaphorase activity. Exp Neurol. 1996;138:33–44. doi: 10.1006/exnr.1996.0044. [DOI] [PubMed] [Google Scholar]
- 54.Russelakis Carneiro M, Silveira LC, Perry VH. Factors affecting the survival of cat retinal ganglion cells after optic nerve injury. J Neurocytol. 1996;25:393–402. doi: 10.1007/BF02284810. [DOI] [PubMed] [Google Scholar]
- 55.Sakai N, Yamada M, Numakawa T, Ogura A, Hatanaka H. BDNF potentiates spontaneous Ca2+ oscillations in cultured hippocampal neurons. Brain Res. 1997;778:318–328. doi: 10.1016/s0006-8993(97)01052-4. [DOI] [PubMed] [Google Scholar]
- 56.Samdani AF, Newcamp C, Resink A, Facchinetti F, Hoffman BE, Dawson VL, Dawson TM. Differential susceptibility to neurotoxicity mediated by neurotrophins and neuronal nitric oxide synthase. J Neurosci. 1997;17:4633–4641. doi: 10.1523/JNEUROSCI.17-12-04633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt HH, Walter U. NO at work. Cell. 1994;78:919–925. doi: 10.1016/0092-8674(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 58.Schmitt U, Sabel BA. MK-801 reduces retinal ganglion cell survival but improves visual performance after controlled optic nerve crush. J Neurotrauma. 1996;13:791–800. doi: 10.1089/neu.1996.13.791. [DOI] [PubMed] [Google Scholar]
- 59.Seif el Nasr M, Peruche B, Rossberg C, Mennel HD, Krieglstein J. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol. 1990;185:19–24. doi: 10.1016/0014-2999(90)90206-l. [DOI] [PubMed] [Google Scholar]
- 60.Snider WD, Johnson EM. Neurotrophic molecules. Ann Neurol. 1989;26:489–506. doi: 10.1002/ana.410260402. [DOI] [PubMed] [Google Scholar]
- 61.So HS, Park RK, Kim MS, Lee SR, Jung BH, Chung SY, Jun CD, Chung HT. Nitric oxide inhibits c-Jun N-terminal kinase 2 (JNK 2) via S-nitrosylation. Biochem Biophys Res Commun. 1998;247:809–813. doi: 10.1006/bbrc.1998.8788. [DOI] [PubMed] [Google Scholar]
- 62.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verge VM, Xu Z, Xu XJ, Wiesenfeld Hallin Z, Hokfelt T. Marked increase in nitric oxide synthase mRNA in rat dorsal root ganglia after peripheral axotomy: in situ hybridization and functional studies. Proc Natl Acad Sci USA. 1992;89:11617–11621. doi: 10.1073/pnas.89.23.11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villegas-Perez MP, Vidal Sanz M, Bray GM, Aguayo AJ. Influences of peripheral nerve grafts on the survival and regrowth of axotomized retinal ganglion cells in adult rats. J Neurosci. 1988;8:265–280. doi: 10.1523/JNEUROSCI.08-01-00265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villegas-Perez MP, Vidal Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993;24:23–36. doi: 10.1002/neu.480240103. [DOI] [PubMed] [Google Scholar]
- 66.Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest Ophthalmol Vis Sci. 1996;37:1618–1624. [PubMed] [Google Scholar]
- 67.Watanabe T, Yamamoto T, Abe Y, Saito N, Kumagai T, Kayama H. Differential activation of microglia after experimental spinal cord injury. J Neurotrauma. 1999;16:255–265. doi: 10.1089/neu.1999.16.255. [DOI] [PubMed] [Google Scholar]
- 68.Yamamoto R, Bredt DS, Snyder SH, Stone RA. The localization of nitric oxide synthase in the rat eye and related cranial ganglia. Neuroscience. 1993;54:189–200. doi: 10.1016/0306-4522(93)90393-t. [DOI] [PubMed] [Google Scholar]
- 69.Yu WH. Nitric oxide synthase in motor neurons after axotomy. J Histochem Cytochem. 1994;42:451–457. doi: 10.1177/42.4.7510317. [DOI] [PubMed] [Google Scholar]
- 70.Yun HY, Dawson VL, Dawson TM. Neurobiology of nitric oxide. Crit Rev Neurobiol. 1996;10:291–316. doi: 10.1615/critrevneurobiol.v10.i3-4.20. [DOI] [PubMed] [Google Scholar]