

Abstract
We have previously shown that NMDA receptor activation during status epilepticus (SE) is required to produce epilepsy in in vitro and in vivo models. As in human symptomatic epilepsy, the epilepsy in these models is permanent, suggesting that the pathological activation of NMDA receptors causes permanent plasticity changes in the brain. Ca2+influx through NMDA receptors is known to transiently activate a key transcription factor, serum response factor (SRF). Thus, we investigated whether this factor, in terms of its expression and ability to bind to the consensus serum response element, was altered long term in the pilocarpine model of epilepsy. In hippocampal nuclear extracts, SRF binding to DNA was significantly increased over saline-injected control rats at 24 hr and at 8 weeks after the onset of SE. This increase was shown to be the result of significantly elevated levels of SRF. DNA binding was also persistently increased in the cortical, but not in the cerebellar, extracts. Hippocampal expression of SRF was localized to neurons using immunohistochemistry. NMDA receptor activation during SE was required for these changes to take place, and the spontaneous seizures seen in epileptic rats did not appear to be responsible for the increase in SRF. The results demonstrate that SRF is persistently elevated after SE in the pilocarpine model of epilepsy and support the theory that long-term gene changes in this model occur and are associated with the long-lasting plasticity changes that are initiated during epileptogenesis.
Keywords: epilepsy, serum response factor, neuronal plasticity, seizure, status epilepticus, hippocampus, SRF
Temporal lobe epilepsy is a common form of epilepsy characterized by spontaneous recurrent seizures that are often intractable to treatment (Meldrum, 1983; Lothman et al., 1991; McNamara, 1994). The process of inducing epilepsy in normal brain tissue is called epileptogenesis (McNamara, 1994). A variety of traumatic insults to the human brain can result in epilepsy. Status epilepticus (SE), defined as 30 min or more of continuous or repetitive seizures without regaining consciousness, can cause epilepsy in humans (Lothman and Bertram, 1993). SE can also give rise to spontaneous recurrent seizures in in vitro (Rafiq et al., 1993; Sombati and DeLorenzo, 1995) and in vivo (Mello et al., 1993) models of epilepsy. Thus, acute SE must trigger some long-term change(s) in neuronal plasticity that results in spontaneous recurrent seizures. It has been proposed that long-term changes in gene expression may underlie epileptogenesis (DeLorenzo, 1991; DeLorenzo and Morris, 1999). Persistent changes in gene expression in epileptogenesis are likely to require long-term changes in transcriptional regulation.
Epilepsy and long-term potentiation (LTP) are both models of altered neuronal plasticity and excitability (for review, see Martinez and Derrick, 1996; Suzuki, 1996). LTP requires Ca2+ entry through NMDA receptors (Malenka, 1991) and new protein synthesis (late-phase) and is correlated with expression of many of the immediate early genes (IEGs) (Kaczmarek, 1992; Suzuki, 1996). Ca2+entry through activated NMDA receptors rapidly induces the IEGc-fos by using the mitogen-activated protein kinase cascade (Xia et al., 1996). This occurs via activation of serum response factor (SRF), which, when bound to the serum response element (SRE) on the c-fos promoter, will dramatically enhance the basal level of c-fos expression. If epileptogenesis has similarities with LTP, it would be expected that SE could induce Ca2+-dependent changes in neuronal excitability that gives rise to spontaneous recurrent seizures. During epileptogenesis, Ca2+ influx through NMDA channels is required for epilepsy in the in vitrohippocampal culture (Sombati and DeLorenzo, 1995; DeLorenzo et al., 1998), in the kindled in vitro hippocampal slice (Stasheff et al., 1989), and in the in vivo pilocarpine rat (Rice and DeLorenzo, 1998) models of epilepsy. Based on these observations, whatever neuroplastic alterations occur during SE that are responsible for epileptogenesis should be NMDA–Ca2+-dependent and may involve SRF as a downstream effector. Epilepsy-dependent alterations in SRF could play a key role in changing the expression of many genes, including many of the IEGs.
To evaluate the effect of epileptogenesis on SRF, we chose to use the rat pilocarpine model of epilepsy (Turski et al., 1983; Mello et al., 1993). These epileptic rats have a very similar pathology to humans with epilepsy by displaying necrosis of neurons within the CA1, CA3, and dentate gyrus regions of the hippocampus, mossy fiber sprouting, and lasting partial–complex spontaneous seizures (Mello et al., 1993). For these reasons, we have examined the effect of SE and the resultant epilepsy on SRF and have found that its DNA-binding activity and expression are persistently increased in this in vivo model of epilepsy.
MATERIALS AND METHODS
Induction of epilepsy in rats. All animal treatments used were approved and in accordance with the Institutional Animal Care and Use Committee guidelines. The pilocarpine model of epilepsy was used to induce spontaneous recurrent seizures after SE in rats by established procedures (Mello et al., 1993; Rice and DeLorenzo, 1998). SE was induced in adult male Sprague Dawley rats (∼200 gm) by intraperitoneally injected pilocarpine (350 mg/kg; Sigma, St. Louis, MO). Control animals were injected with saline. To control the peripheral effects of pilocarpine, all animals were given methyl-scopolamine (1 mg/kg, i.p.; Sigma) 30 min before treatment. One hour after the onset of SE, diazepam (4 mg/kg, i.p.; Sigma) was used to terminate the seizures. Additional doses of diazepam were given at 3 and 5 hr after SE onset as necessary. Control rats were treated with an equal number of diazepam doses. Pilocarpine animals were used for long-term experiments only if they were observed by video monitoring to have spontaneous recurrent seizures within 40 d of injection. In some animals, NMDA receptor activation was blocked by treatment with MK-801 (4 mg/kg, i.p.; Research Biochemicals, Natick, MA) 20 min before pilocarpine or saline injections (Ormandy et al., 1989; Rice and DeLorenzo, 1998).
In a set of pilocarpine-treated rats, spontaneous recurrent seizures were inhibited with the anticonvulsant phenytoin (50 mg/kg, i.p., twice daily; Elkins-Sinn, Cherry Hill, NJ) starting the day after pilocarpine injection. Sets of pilocarpine-treated and saline-treated (control) rats were injected twice daily for comparison. Phenytoin-treated and nonphenytoin-treated epileptic rats were video monitored 8 hr/d until they were killed to determine percent change in seizure frequency. These animals were killed 4 weeks after pilocarpine treatment.
Electroconvulsive shock induced seizures. Tonic–clonic seizures were induced in adult male Sprague Dawley rats (∼250 gm) via maximal electroshock using corneal electrodes (150 mA, 39 V, 0.2 sec, 60 Hz) (Porter et al., 1984). Control animals were treated with the same handling conditions, except that they were not exposed to electroconvulsive shock (ECS). ECS animals were killed at 3, 12, and 24 hr after treatment, and nuclear extracts were prepared (see below).
Preparation of nuclear- and cytosolic-enriched extracts and crude homogenates. Rats were killed at various time points after treatments, and nuclear-enriched fractions and cytoplasmic-enriched fractions were prepared from hippocampi, cortices, and cerebellums at 4°C by the procedure of Moore et al. (1996) with modifications as follows. Brain tissues were quickly rinsed in PBS and then homogenized by 14 strokes in a Dounce homogenizer (A pestle) in 1.5 ml of tissue-homogenizing solution (15 mm HEPES, pH 7.9, 0.25 m sucrose, 60 mmKCl, 10 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mmphenylmethylsulfonyl fluoride [PMSF] [Sigma], and phosphatase inhibitors [600 nm Okadaic acid (Calbiochem, La Jolla, CA); 5 nm cypermethrin (Calbiochem); and 2 mm NaF]). Cells were pelleted at 2000 ×g for 10 min, resuspended in 1.5 ml cell lysis solution (10 mm HEPES, pH 7.9, 1.5 mmMgCl2, 10 mm KCl, 1 mm PMSF, and phosphatase inhibitors) and homogenized by seven strokes with a B pestle in a Dounce homogenizer. Nuclei were pelleted at 4000 × g for 10 min. The supernatant was saved as the cytoplasmic-enriched fraction, and the nuclei were resuspended in 0.2 ml (hippocampi) and 0.4 ml (cortices and cerebellums) of nuclei lysis solution (10 mmHEPES, pH 7.9, 1.5 mm MgCl2, 1 mm EDTA, 0.8 m NaCl, 25% glycerol, and phosphatase inhibitors). The nuclei suspension was gently rocked for 30 min to release soluble nuclear factors, followed by centrifugation at 14,000 × g for 30 min to remove membrane debris and DNA.
Crude hippocampal homogenates were prepared from phenytoin- and saline-treated rats. Hippocampi were homogenized in 50 mmTris, pH 7.5, 6 mm EGTA, 320 mm sucrose, 1 mm DTT, and 0.3 mm PMSF, followed by removal of cellular debris by centrifugation at 14,000 × g for 20 min. Protein concentrations in nuclear-enriched fractions, cytoplasmic-enriched fractions, and crude homogenates were determined by the Bradford assay (Bradford, 1976) (Bio-Rad, Hercules, CA). Extracts were stored at −80°C for up to 1 year before use.
Electrophoretic mobility-shift assays. The level of specific SRF binding to SRE was assessed using electrophoretic mobility-shift assays (EMSAs). The SRE consensus (Attar and Gilman, 1992) double-stranded oligonucleotide (5′-GGATGTCCATATTAGGACATCT-3′; Santa Cruz Biotechnology, Santa Cruz, CA) was end-labeled with [γ-32P]ATP (>4000 Ci/mm; ICN Biochemicals, Costa Mesa, CA) using T4 polynucleotide kinase (10 U; Life Technologies, Gaithersburg, MD). Extracts (3 μg of nuclear-enriched fraction, 8 μg of cytoplasmic-enriched fraction, or 6 μg of crude homogenate) were incubated for 10 min at room temperature in a 15 μl volume containing 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mmMgCl2, 0.5 mm DTT, 0.5 mm EDTA, 4% glycerol, and 0.05 mg/ml poly(dI-dC) · poly(dI-dC) (Amersham Pharmacia Biotech, Piscataway, NJ). Labeled SRE (∼0.028 pmol or 3 × 104 cpm) was then added and incubated for an additional 20 min before electrophoresis. EMSAs were also performed on cytoplasmic-enriched fractions in the same manner, except that 6 μg of cytoplasmic-enriched fraction was used, and additional glycerol was added (1 μl of 50%). Protein complexes were separated on 4% nondenaturing polyacrylamide gels in 0.5× TBE (45 mm Tris-borate and 1 mmEDTA), dried, and either viewed by autoradiography or quantitated by phosphor imaging (Molecular Dynamics, Sunnyvale, CA).
To determine protein–SRE complex specificity, a 50-fold molar excess of unlabeled SRE or nonspecific oligonucleotide was added at the start of the assay. Supershifts were performed after the extract was allowed to interact with the oligonucleotide for 20 min by the addition of 2 μg of an anti-SRF rabbit polyclonal antibody (sc-335; Santa Cruz Biotechnology) and then incubated at 4°C for 1 hr before electrophoresis (Sato-Bigbee et al., 1994). The specificity of antibody binding was determined by preincubating the anti-SRF antibody for 1 hr at room temperature with 1 μg of SRF immunizing peptide (sc-335P; Santa Cruz Biotechnology).
Normalization of EMSA data with internal standards. Each EMSA reaction was performed using the standard amounts (in micrograms) of protein specified above. However, to control for any selective differences between epileptic and control rats in protein levels or in the EMSAs, we used internal standards. The first standard was α and β tubulin visualized by SDS-PAGE of 20 μg of each nuclear-enriched fraction, followed by Coomassie brilliant blue staining and densitometry (Molecular Dynamics) of the prominent tubulin bands (Stryer, 1981). The second standard used EMSA data from a specific complex formed using a labeled Oct1 (5′-TGTCGAATGCAAATCACTAGAA-3′; Promega, Madison, WI) oligonucleotide and the same nuclear-enriched fractions. Using these internal standards, we compared SRF binding normalized by protein levels with the data normalized by tubulin levels and Oct1 binding.
Western blotting. Nuclear-enriched fraction proteins (20 μg/well) were separated by SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were blocked in PBS containing 0.05% Tween 20 and 3% Bio-Rad blocking reagent for 1 hr. Fresh blocking solution was added with diluted primary antibody and incubated at room temperature for 1.5 hr. Polyclonal rabbit anti-SRF antibody (0.5 μg/ml; Santa Cruz Biotechnology) was used to quantitate SRF protein and polyclonal rabbit anti-N-terminal SRF [1:10,000 dilution, kindly donated by Dr. Michael E. Greenburg, Harvard Medical School, Boston, MA (Misra et al., 1991)] was used to verify specificity of the Santa Cruz Biotechnology antibody. Specificity of anti-SRF antibody binding was also tested by preincubating the Santa Cruz Biotechnology antibody for 1 hr with a 10-fold weight excess of immunizing peptide. The membranes were then washed three times with PBS and blocked again for 30 min, followed by incubation for 45 min with goat anti-rabbit IgG antibody conjugated to horseradish peroxidase (0.2 μg/ml in blocking solution; Santa Cruz Biotechnology). The blots were washed five times with PBS, and bound secondary antibody was detected by enhanced chemiluminescence (Pierce, Rockford, IL) and exposure to x-ray film. Films were quantitated by densitometry (Molecular Dynamics).
Immunohistochemistry for hippocampal cellular distribution of SRF. Eight weeks after injection, pilocarpine- and saline-treated rats were anesthetized and perfused first with saline, followed by 4% paraformaldehyde in sodium phosphate buffer, pH 7.0 (Churn et al., 1992). The perfused brains were removed and placed in 10% formalin. After 1 or 2 d, 2 mm coronal sections were dehydrated and paraffin-embedded. Ten micrometer slices were placed on gelatin-coated slides, rehydrated, and blocked with 3% blocking reagent (Bio-Rad) in PBS for 1 hr at room temperature. Slices were incubated overnight at 4°C with 0.5 μg/ml polyclonal rabbit anti-SRF antibody (Santa Cruz Biotechnology) in blocking reagent and then washed three times with PBS–Tween 20 (0.05%). Biotin-conjugated goat anti-rabbit polyclonal secondary antibody (5 μl/ml; Vector Laboratories, Burlingame, CA) in blocking solution was added to the slices for 30 min at room temperature and washed as before. SRF immunoreactivity was detected using the Vector Elite ABC and 3′3′ diaminobenzidine (DAB) substrate kits for peroxidase. For cytoplasmic staining, slides of adjacent slices were rehydrated and stained for 5 min in 0.1% cresyl violet acetate, washed in H2O, and then dehydrated. Antibody and cresyl violet-stained slides were imaged using Nikon Imaging System (Nikon, Garden City, NY).
Statistical analysis. Significant changes in SRF DNA binding and protein levels were determined using the two-tailed Student’st test.
RESULTS
Long-term and acute effects of SE-induced epilepsy on SRF binding
Pilocarpine was used to induce SE and subsequent spontaneous recurrent seizures in male rats (Mello et al., 1993; Rice and DeLorenzo, 1998). Only rats that developed generalized tonic–clonic, stage 3–5 spontaneous recurrent seizures were used in these studies (Racine, 1972). Eight weeks after the initial SE, rats that developed spontaneous recurrent seizures and their saline-treated controls were killed, nuclear- and cytoplasmic-enriched extracts were prepared from hippocampi, and EMSAs were conducted in vitro to study SRF binding to its 32P-labeled consensus DNA element, SRE.
Figure 1 shows the electrophoretic mobility shift pattern for SRE using hippocampal nuclear-enriched fractions from control and epileptic rats 8 weeks after treatment. Only one specific shifted band was obtained for SRE using a pilocarpine-treated rat nuclear-enriched fraction as determined by competition with both a 50-fold excess of unlabeled SRE and an unrelated oligonucleotide. Because several different transcription factors are known to specifically interact with SRE (Johansen and Prywes, 1995), a supershift experiment was performed using an anti-SRF antibody (Fig. 1). This antibody was found to be able to retard the mobility of the specific band by interacting with the SRF–SRE complex in both the control and epileptic nuclear-enriched fractions. When a small amount of SRF peptide was preincubated with the antibody, the supershift was diminished, indicating that the antibody was specifically recognizing SRF in the SRF–SRE complex rather than some other SRE-binding transcription factor. This demonstrates that the specific shifted SRE band seen in Figure 1 was composed at least of an SRF–SRE complex using both control and epileptic hippocampal nuclear-enriched fractions.
Fig. 1.

SRF in control and epileptic (Pilo) rat hippocampal nuclear-enriched extracts specifically interacts with SRE in EMSAs. In a standard binding reaction (lanes 1, 2), nuclear-enriched extracts (3 μg) were incubated with 32P-labeled SRE consensus oligonucleotide probe before native PAGE and autoradiography. Free SRE probe ran at the bottom of the gel, and protein–DNA complexes were retarded in the gel. To determine specificity of protein–DNA complexes visualized in the gel, the pilocarpine reaction was compared with reactions to which a 50-fold excess of either unlabeled SRE consensus oligonucleotide (+SRE, lane 3) or an unrelated oligonucleotide (+nonSRE, lane 4) was added. One specific complex (SRE–SRF) and two nonspecific bands (NS) are shown. The specific complex was determined to contain SRF protein using the supershift assay (lanes 5–9). Control and pilocarpine reactions (lanes 5, 7, respectively) were compared with reactions to which an anti-SRF antibody was added (+Ab, lanes 6, 8). The specific complex was further retarded by the anti-SRF antibody (Ab/SRF/SRE). Anti-SRF antibody specificity was tested by the addition of an excess of immunizing peptide (+Ab+P, lane 9).
Figure 2 shows the comparison of epileptic and control hippocampal nuclear-enriched fraction binding to SRE. At 8 weeks, the nonspecific shifted bands were not affected by the pilocarpine treatment. However, the specific shifted band for SRF was increased long term by the pilocarpine treatment (Fig.2A). After quantitation of the 8 week epileptic and control animals by phosphor imaging, a statistically significant increase of 44.6% in SRF binding was observed (Fig.2B). No significant change was observed in SRF binding using the cytoplasmic-enriched fractions from these same animals (8% increase over control; Student’s t test;p = 0.21), indicating that the increase in SRF binding in the nuclear-enriched fractions was not caused by an increased translocation of SRF from the cytoplasm to the nucleus with a resulting decrease in cytoplasmic levels of SRF.
Fig. 2.
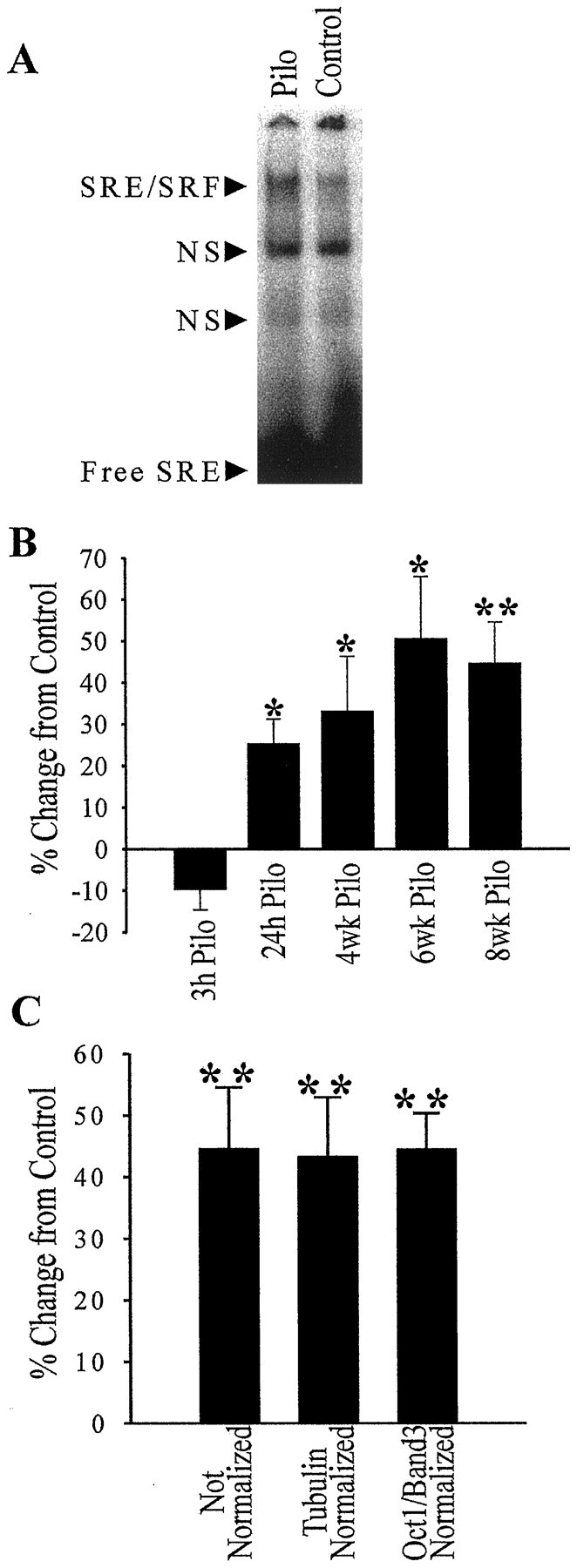
Specific binding of SRF protein to its SRE DNA element is induced during epileptogenesis. EMSAs were conducted in the same manner as in the control reactions in Figure 1, except that the gels were visualized and quantitated by phosphor imaging. A representative EMSA using 8 week post-treatment hippocampal nuclear-enriched fractions from pilocarpine-treated (Pilo) and saline-treated (Control) rats is shown (A). Quantitation of these gels and determination of percent change in SRE binding over paired control nuclear-enriched fractions for five time points (B) revealed a significant increase in SRF binding acutely at 24 hr (n = 6) and at 4 (n = 4), 6 (n = 4), and 8 (n = 6) weeks. The 8 week pilocarpine EMSA data that was not normalized was compared with the data (C) that was normalized by either tubulin protein levels in these nuclear-enriched fractions or EMSA data for transcription factor binding that did not show any changes (Oct1 band three). Error bars represent SEM; *p < 0.05; **p < 0.01; Student’s ttest.
To control for the possibility that variations in the levels of nuclear protein expression may have occurred in epileptic rats or that some differences occurred in the preparation of nuclear-enriched fractions from these animals, hippocampal binding data were normalized against two internal standards (Fig. 2C) in addition to being standardized by protein loading levels. Visualized SDS-polyacrylamide gels, quantitated by densitometry and containing these nuclear-enriched fractions, showed no discernable differences in the banding pattern between pilocarpine-treated and control rat nuclear-enriched fractions. These results control for changes in the levels of the highly expressed proteins in the nucleus as a result of the treatment with pilocarpine. α and β tubulin are highly expressed in the nucleus and were used as one of the internal standards. The second source of internal standards used a specific shifted band for another transcription factor consensus sequence, Oct1. When the SRE binding data were normalized using tubulin protein levels or the data from the Oct1 band three gel shift, the resulting percent increase in SRE binding seen in Figure2C in the epileptic rats (43.4% increase from control, tubulin; 44.6% from Oct1 band three; p < 0.01; Student’s t test) was not different from the SRE binding data that was normalized only by protein loading levels (44.6% increase from control; p < 0.01). This suggests that normalizing data from the nuclear-enriched fractions by protein concentration was sufficient to control for the minor variations that occur in the preparation of the nuclear-enriched fractions.
To determine whether SRF binding was altered at time points earlier than 8 weeks after the SE episode, pilocarpine- and saline-treated (control) rats were killed 3 and 24 hr acutely and at 4 and 6 weeks after the onset of SE, and hippocampal nuclear-enriched fractions were obtained. Figure 2B shows the quantitated data for SRF binding at these time points. At 3 hr after SE, there was no significant change in SRF binding to SRE. However, by 24 hr after SE, there was a significant increase in SRF binding in the pilocarpine animals (25.3% increase over control). This statistically significant increase was maintained at 4 and 6 weeks after SE (33.2 and 50.6% increase from control, respectively).
Alterations in SRF expression in the pilocarpine epilepsy model
The increased binding of SRF to the SRE consensus oligonucleotide could be attributable to an increased steady-state level of SRF in the hippocampus or to post-translational modification of preexisting SRF. To investigate these possibilities, Western blots of nuclear-enriched fractions from epileptic and control hippocampi were probed with anti-SRF antibodies, visualized on film by chemiluminescence, and quantitated by densitometry (Fig. 3). The Santa Cruz Biotechnology anti-SRF antibody detected a major band at 68 kDa that had the same mobility as the band detected by a different polyclonal anti-SRF, kindly provided by Dr. Greenburg’s laboratory (Misra et al., 1991), demonstrating its specificity (data not shown). Two other minor bands were also present: one at 60 kDa and another at 135 kDa (data not shown). The 60 kDa band probably represented a newly synthesized form of SRF, which may lack some of the post-translational modifications of the mature form (Misra et al., 1991). Because SRF does dimerize, the 135 kDa band could be an SRF dimer that was incompletely dissociated by SDS and β-mercaptoethanol before electrophoresis. Binding of the Santa Cruz Biotechnology antibody to all three bands was completely abolished when the antibody was preincubated with the immunizing peptide (data not shown). Using the Santa Cruz Biotechnology antibody to assay the level of the mature 68 kDa SRF protein at 24 hr after SE, there was a statistically significant 68.6% increase in SRF protein in the pilocarpine nuclear-enriched fractions ( p < 0.01 Student’st test) (Fig. 3). At 8 weeks after SE, there was also a significant 66.6% increase in SRF protein in pilocarpine hippocampal nuclei (p < 0.05). Similar results were obtained when these data were normalized to internal standards with either tubulin (68.3% increase over control; p < 0.01) or microtubule-associated protein (71.0% increase over control;p < 0.01), confirming the significant increase in SRF in the epileptic animals. Thus, the induction of epilepsy in the pilocarpine model produced a long-lasting increase in the DNA binding and protein expression of SRF.
Fig. 3.

SRF protein expression is upregulated by SE. Representative Western blots using pilocarpine- and saline-treated rat nuclear-enriched fractions obtained 24 hr (A;n = 10) and 8 weeks (B;n = 4) after injection. SRF immunoreactive bands were visualized using an anti-SRF antibody, a peroxidase-conjugated secondary antibody, and enhanced chemiluminescence. The mature form of SRF is shown at 68 kDa, and the immature, newly synthesized form is at 60 kDa. Quantitation of the mature SRF band at both time points by densitometry (C) revealed a significant increase in SRF protein. Error bars represent SEM; *p < 0.05; **p < 0.01; Student’s ttest.
Long-term changes in SRF binding in cortex and cerebellum in epileptic rats
In the pilocarpine model of epilepsy, seizures are presumed to originate in the hippocampus (Mello et al., 1993). However, other brain regions are likely to play roles in seizure generalization in epilepsy. We investigated whether SRF binding was altered long term in the epileptic state in cortical tissue in which seizure propagation is necessary for the animals to display generalized tonic–clonic seizures. At 8 weeks after SE, there was a significant increase in SRF DNA binding of 14.1% over control rats using cortical nuclear-enriched fractions (Fig. 4). This increase was not as large as the 44.6% increase seen in the hippocampal nuclear-enriched fractions but was statistically significant (p < 0.05; Student’s t test). We also investigated SRF binding in the cerebellum, which does not manifest spontaneous recurrent seizures or play a role in seizure propagation. No change in SRF binding to DNA was seen using cerebellar nuclear-enriched fractions (−0.1% change from control;p = 0.50) (Fig. 4). Therefore, there was a region-specific correlation between areas of the brain that manifest spontaneous recurrent seizures (epilepsy) and altered SRF function.
Fig. 4.
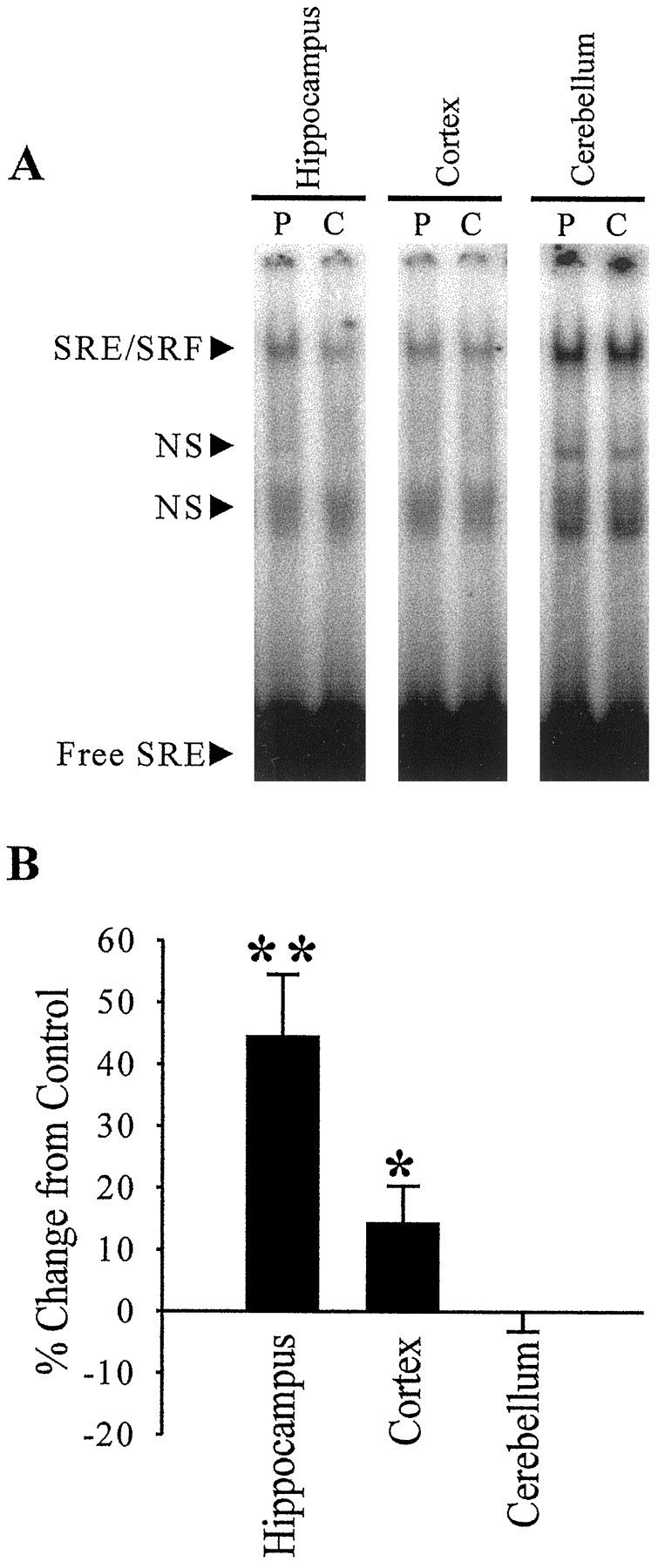
Long-term alterations in specific binding of SRF to its SRE DNA element in nuclear-enriched fractions from specific brain regions of epileptic animals. Hippocampal (n= 6), cortical (n = 6), and cerebellar (n = 5) nuclear-enriched fractions were prepared from pilocarpine-treated epileptic (P) and saline-treated control (C) rats 8 weeks after treatment. EMSAs were performed as in Figure 2. A representative EMSA using these nuclear-enriched fractions is shown (A). Quantitation of these gels and determination of percent change in SRE binding over paired control nuclear-enriched fractions for three brain regions (B) revealed a significant increase in SRF binding in the hippocampus and cortex. Error bars represent SEM; *p < 0.05; **p < 0.01; Student’s ttest.
The effect of ECS-induced seizures on SRF binding
To evaluate whether the elevation in SRF protein binding to its DNA element in the pilocarpine animals was caused by isolated seizures that occurred intermittently in the epileptic animals, the effect of individual seizures on elevating SRF was investigated. If SRF expression was regulated by individual seizures, then this change should also result after a vigorous tonic–clonic seizure induced by ECS. Maximal ECS was used to induce a tonic–clonic seizure in naive rats. ECS-induced seizures were more severe than spontaneous recurrent seizures in the pilocarpine rats as evaluated by video monitoring andRacine’s (1972) rating scale. These seizures lasted ∼1 min and were followed by a postictal period, after which the rats returned to an apparently normal, alert state. Control rats were handled identically, except that they did not receive ECS. Groups of ECS rats were killed 3, 12, and 24 hr after treatment, and nuclear-enriched fractions were prepared and compared with control rats using EMSAs with the SRE DNA probe. There was no significant difference in SRE binding between control and ECS rats at all time points (Fig.5A). These results indicate that individual seizures that were greater in severity than the spontaneous recurrent seizures in the epileptic pilocarpine animals were not able to elevate SRF binding.
Fig. 5.
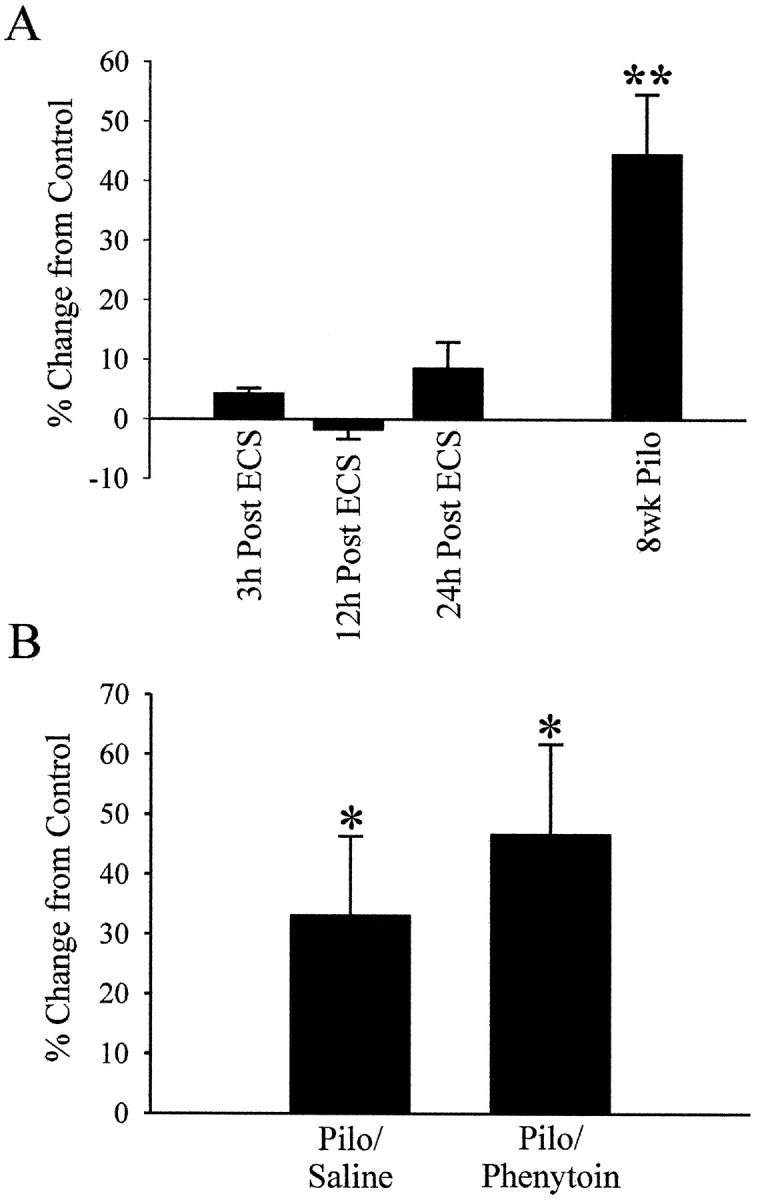
Intense ECS-induced seizures do not significantly alter SRF binding to its DNA element SRE, and anti-convulsant reduction of seizure frequency in epileptic rats does not diminish the increase in SRF binding. A, Groups of rats were treated with ECS (see Materials and Methods) and killed 3, 12, and 24 hr after ECS (n = 5, n = 5, andn = 6, respectively). Nuclear-enriched fractions were prepared from animals and control rats that did not receive ECS and compared using EMSAs conducted in the same manner as in Figure 2. Data are represented in the bar graph as a percent change in SRE binding from control nuclear-enriched fractions. Also shown is the significant percent increase from control seen in nuclear-enriched fractions obtained from epileptic rats 8 weeks after pilocarpine treatment (n = 6) (Fig. 2). B, Seizure frequency was reduced by 53% in a set of epileptic rats by twice daily injections of phenytoin (Pilo/Phenytoin;n = 4) compared with epileptic rats that received saline injections (Pilo/Saline; n = 4). Hippocampal homogenates were prepared 4 weeks after pilocarpine injection, and EMSAs were conducted as in Figure 2. Thebars represent the percent change from saline injected control rats. Error bars represent SEM; *p < 0.05; **p < 0.01; Student’s ttest.
The effect of anticonvulsant drug treatment of epileptic animals on SRF binding
Another approach to determining whether recurrent seizures in the pilocarpine model of epilepsy were playing a role in elevating SRF DNA binding was to treat the epileptic rats with chronic anticonvulsant drug therapy to reduce seizure frequency. If increased SRF expression was related to the recurrent seizures in the epileptic animals, then anticonvulsant treatment and reduction of seizure frequency should decrease SRF binding in these animals. The anticonvulsant drug phenytoin has been shown to significantly decrease seizure frequency in epilepsy patients (Schmidt, 1982) and in pilocarpine-treated animals (Leite and Cavalheiro, 1995). To evaluate this possibility, a group of pilocarpine-treated rats was injected twice daily with phenytoin beginning the day after SE until they were killed. The seizure frequency was reduced by 53% in these animals compared with control pilocarpine-treated animals based on video monitoring. Animals were killed 4 weeks after pilocarpine injection, and hippocampal homogenates were prepared. EMSAs were used to compare SRF binding between homogenates from epileptic phenytoin-treated and nonepileptic control rats that had received saline injections twice daily instead of phenytoin. A significant increase in SRF binding (46.7% from control;p < 0.05; Student’s t test) (Fig.5B) was observed in the phenytoin-treated epileptic animals. The increase was not reduced compared with epileptic animals, which received saline injection instead of phenytoin (33.2% increase from control; p < 0.05) (Fig. 5B).These results demonstrate that elevated SRF binding did not diminish with anticonvulsant-decreased recurrent seizure frequency, indicating that seizure activity in the pilocarpine animals was not likely responsible for the elevated SRF levels.
The role of NMDA receptor activation during SE on increased SRF binding
We have found that NMDA receptor activation during SE is required for epileptogenesis in the pilocarpine epilepsy model (Rice and DeLorenzo, 1998) and in the hippocampal culture model of epilepsy (Sombati and DeLorenzo, 1995; DeLorenzo et al., 1998). When NMDA receptors were blocked by the injection of an NMDA receptor inhibitor, MK-801, before pilocarpine injection, prolonged electrographic SE still occurred, but spontaneous recurrent seizures did not develop (Rice and DeLorenzo, 1998). If the elevation in SRF plays a role in epileptogenesis in this model, then the increase in SRF, like epileptogenesis, should require NMDA receptor activation during SE.
To investigate this possibility, a group of rats was injected with MK-801 before pilocarpine or saline injections. Nuclear-enriched fractions were prepared 8 weeks later, and SRF binding was compared between four groups by EMSAs (pilocarpine, pilocarpine plus MK-801, saline plus MK-801, and saline). Figure6 shows that MK-801 effectively blocked the increased SRF binding in the pilocarpine rats under the same conditions that blocked epileptogenesis (Rice and DeLorenzo, 1998). Epileptic pilocarpine rats had an increase in SRF binding of 45.2% (p < 0.01; Student’s t test) (data not shown in Fig. 6) over “nonepileptic” pilocarpine plus MK-801 rats. In addition, the SRF binding for the pilocarpine plus MK-801 animals (−4.4% of control; p = 0.68) was the same as the binding seen in the saline-treated control animals (Fig. 6).
Fig. 6.

Upregulation of specific SRF DNA binding at 8 weeks after SE was prevented by NMDA channel inhibition during SE. MK-801, an NMDA channel antagonist, was administered before pilocarpine or saline injection. Nuclear-enriched fractions prepared 8 weeks after treatment from four groups of rats [Pilo/Saline(n = 6) (Fig. 2), Pilo/MK-801(n = 4), Saline/MK-801(n = 4), and Saline/Saline(n = 6)]. Electrophoretic mobility-shift assays were conducted and quantitated as described in Figure 2. Data are represented as a percent change in SRE binding from control animals (Saline/MK-801; n = 4), and controls (Saline/Saline). Error bars represent SEM; **p < 0.01; Student’s ttest.
SRF immunohistochemistry in rat hippocampus
All of the above experiments were performed using nuclear and cytoplasmic fractions from whole homogenized hippocampi. The hippocampus, in addition to having several types of neurons, also contains many types of non-neuronal cells, including glia and fibroblasts. All of these cell types could potentially express SRF and be responsible for the altered expression of SRF in epileptic rats. To evaluate the cellular distribution of SRF in the rat hippocampus, immunohistochemistry, using the same anti-SRF antibody used in the above studies, was performed on paraffin-embedded coronal sections obtained 8 weeks after pilocarpine or saline injections. Adjacent sections were stained with cresyl violet. Figure7 shows the SRF and cresyl violet hippocampal and CA1 regional staining pattern in a saline- and a pilocarpine-treated adult rat. The majority of the SRF immunoreactivity was enriched in the nuclei of neurons in the dentate gyrus, CA1, CA2, and CA3 regions of the hippocampus. Intense staining was present in the CA1 and CA2 and in the dentate gyrus, with less staining in the CA 3 region of the hippocampus. Staining in other hippocampal cell types was not apparent. These results demonstrate that the majority of SRF antibody reactivity was highly enriched in the pyramidal neuron cell layer in the hippocampus and primarily associated with neurons in both control and epileptic animals.
Fig. 7.
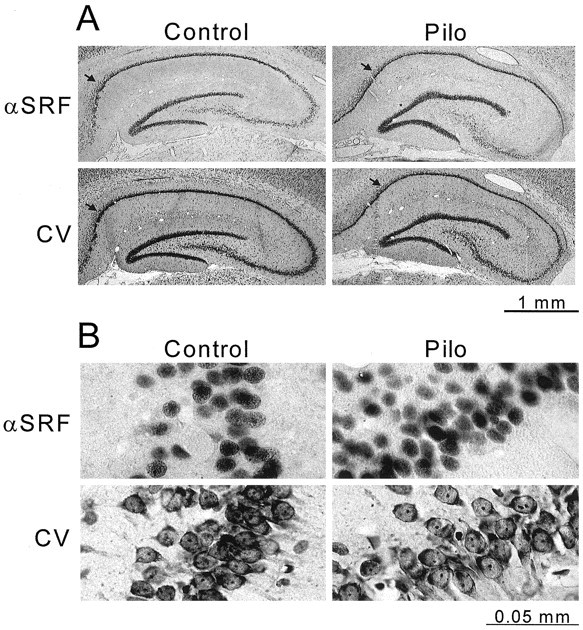
SRF immunoreactivity is localized to the nuclei of the neuronal cell regions of the epileptic (Pilo) and control rat hippocampus. Adjacent coronal sections were sliced from a paraformaldehyde-fixed, paraffin-embedded brain of control saline-injected (left panels) and pilocarpine-injected (right panels) rats at 8 weeks after treatment. SRF immunoreactivity (αSRF) was visualized using a DAB substrate kit (see Materials and Methods). Cell bodies in adjacent slices were stained with cresyl violet (CV). Images of cresyl violet- and SRF-stained slides were captured using a digital camera. Low-magnification views of control and epileptic hippocampi are shown in A. Higher magnification views are presented below in B of the CA1 hippocampal region, designated by arrows in A. Scale bars below A and B indicate actual size.
Although immunohistochemical determinations are not reliable for quantitation, we attempted to evaluate the number of positively stained neurons in epileptic and control slices of the CA1 hippocampal region. To do this, the number of anti-SRF stained nuclei were counted in an area of the CA1 region, and cell loss and variation was controlled for by counting cresyl violet-stained cells in the same area in an adjacent section. This qualitative measurement revealed an increase in anti-SRF staining in the epileptic animals. This increase in the number of SRF-positive neurons did coincide with the long-term increased SRF expression assessed by Western blotting (Fig. 3). Although it is difficult to use immunohistochemical studies for quantitative analysis, these results suggest that the long-term differences in SRF expression in this model of epilepsy are occurring in the neuronal nuclei of the hippocampus and not in non-neuronal cell types.
DISCUSSION
Epilepsy represents a permanent change in neuronal function mediated by lasting changes in neuronal plasticity. The possible role of altered genetic expression in mediating symptomatic epilepsy represents a molecular mechanism that could account for long-lasting changes in neuronal function in response to environmental influences (DeLorenzo, 1991; DeLorenzo and Morris, 1999). If changes in genetic expression underlie epilepsy, long-lasting alterations in transcriptional regulation should accompany epileptogenesis. The results of this study indicate that epilepsy induced by SE in the pilocarpine model is associated with a long-lasting increase in the binding of the transcription factor SRF to its DNA consensus sequence SRE. The increase in DNA binding was present in both hippocampal and cortical nuclear-enriched fractions but not in cerebellar nuclear-enriched fractions. The hippocampus and cortex both play roles in seizure generation and propagation (Mello et al., 1993). This increase in binding seen using the hippocampal nuclear-enriched fractions from epileptic rats was shown to be caused by an elevated level of SRF protein. The increase in SRF binding was not seen 3 hr after the onset of SE but was seen by 24 hr and 8 weeks later, suggesting that the change in SRF expression takes time to develop and parallels the development of spontaneous recurrent seizures in this model of epilepsy (Mello et al., 1993).
Using the in vivo rat pilocarpine and in the in vitro 0 Mg2+ primary hippocampal culture models of epilepsy and the NMDA receptor antagonists MK-801 and 2-amino-5-phosphonovaleric acid, we have found that NMDA receptor activation during SE is required for epileptogenesis (Sombati and DeLorenzo, 1995; DeLorenzo et al., 1998; Rice and DeLorenzo, 1998). In this work, blockage of NMDA receptor activation during pilocarpine-induced SE completely blocked the long-term increase in SRF binding. Thus, a long-lasting increase in SRF expression and DNA binding occurs in association with the persistent plasticity changes that underlie epilepsy in this model. To our knowledge, this is the first reported instance of a long-term alteration in a transcription factor coinciding with epileptogenesis. The data suggest that long-term changes in epilepsy may be mediated by persistent changes in genetic expression.
Seizures are known to rapidly and transiently induce the expression ofzif/268, NGFI-A and the members of fos andjun families (Saffen et al., 1988; Morgan and Curran, 1991;Robertson, 1992; Nahm and Noebels, 1998). SE in the pilocarpine model has been shown to transiently induce fos expression (Elmer et al., 1997; Penschuck et al., 1997). Recently, Herdegen et al. (1997)reported that the expression of SRF, which is considered to be a “late” IEG (Norman et al., 1988; Misra et al., 1991), was elevated at 2 hr after intraperitoneally kainic acid-induced seizures. However, SRF expression returned to normal levels within 5 hr and remained so for 24 hr after kainic acid injection. The kainic acid in this model does not cause epileptogenesis and does not manifest chronic elevations in SRF (Herdegen et al., 1997). In this work, using the pilocarpine model of epilepsy, SRF binding was significantly elevated at 24 hr and at 4, 6, and 8 weeks after the onset of pilocarpine-induced SE. Therefore, the ability to produce prolonged increases in SRF expression was associated with epileptogenesis.
It is possible that the recurrent seizures that occur in this model may be playing some role in the elevation of SRF. However, ECS did not cause an increase in SRF binding after acute, intense seizures. These data demonstrate that seizures do not elevate SRF. Furthermore, reduction of the recurrent seizure frequency by 53% with anticonvulsants in the pilocarpine model did not block the increase in SRF binding. It is still possible that some minimal seizure frequency threshold that still occurs in the phenytoin-treated epileptic rats could play a role in the increased SRF expression. However, inducing seizures by intraperitoneally kainic acid or ECS is not sufficient for a prolonged increase in SRF. These data indicate that repeated seizures are not causing the elevated SRF binding. Thus, the long-term elevation in SRF observed in association with epilepsy in the pilocarpine model of epilepsy are likely the result of a long-lasting plasticity change induced during epileptogenesis.
The level of SRF protein was upregulated 24 hr after pilocarpine-induced seizures and maintained for a prolonged period of time. Evidence indicates that this upregulation requires NMDA receptor activation during epileptogenesis. For most proteins, the level of protein present in the cell is controlled at the transcriptional level (Lewin, 1987). The expression of SRF has been shown to be transiently induced, in a manner similar to the IEGs, by mitogenic agents in the absence of new protein synthesis (Norman et al., 1988; Misra et al., 1991), putting SRF in a class of delayed IEGs. The half-life of the SRF protein has been determined to be ∼12 hr (Misra et al., 1991). SRF is autoregulated by the binding of activated SRF to SREs on its own promoter (Spencer and Misra, 1996). SRF is known to be activated by phosphorylation caused by NMDA receptor activation and Ca2+ entry into primary hippocampal neurons (Bading et al., 1993; Xia et al., 1996). A plausible mechanism for the initial induction of SRF expression after SE in the pilocarpine model would be that Ca2+ entry through NMDA channels activates SRF protein already present in the nucleus, which in turn stimulates expression of more SRF. Can this mechanism also account for the long-term elevation in SRF? Using an in vitro primary hippocampal culture model of epilepsy (Sombati and DeLorenzo, 1995), we have found that the Ca2+ equilibrium was perturbed in the epileptic state after SE (DeLorenzo et al., 1998). The [Ca2+]i in the “epileptic” pyramidal neurons was elevated to ∼300 nm compared with the sham-treated neurons, which had a [Ca2+]i of 165 nm. [Ca2+]i may also be elevated long term in the in vivo model, allowing for continual increased SRF expression and binding to SRE. Another possible mechanism to account for the continual elevated SRF expression in the epileptic animals would be that SRF is initially pathologically upregulated within 24 hr of the SE. This abnormal amount of SRF may itself, through a positive feedback loop, cause the continual synthesis of more SRF by binding to the SREs on the SRF promoter (Spencer and Misra, 1996). This mechanism would not be dependent on Ca2+ entry or NMDA receptor activation after the initial SE was over.
These results lead to the important question as to what role, if any, does elevated SRF expression have in epilepsy and hyperexcitability. Preliminary data indicate that the AP-1 complex binding is elevated long term in the pilocarpine epilepsy model (Morris et al., 1997). SRF is known to be a positive regulator of the expression of the fos genes (Prywes et al., 1988; Lazo et al., 1992). Elevated SRF could be responsible for the prolonged expression of FosB seen in these models and, perhaps, may play a role in the long-term regulation of other IEGs. Pathological alterations in the expression of IEGs would likely affect the expression of many other genes in hippocampal neurons, including membrane receptors and channels, which could play a direct role in hyperexcitability. SRF might also act directly on the promoters for neuronal-specific genes. SRF is known to play a key role in myogenesis by functionally interacting with promoters of many muscle-specific genes, including the α-actins (Johansen and Prywes, 1995). Long-term changes in the expression of mRNAs for specific isoform of the GABAA receptor have been identified in the pilocarpine model (Rice et al., 1996). The possible role of SRF in the regulation of the expression of the GABAA receptor genes is not known. The role of SRF in the direct control of neuronal-specific genes has not been characterized, and its role in neural development and differentiation is unknown.
Other mechanisms for SRF elevations seen in association with epilepsy may account for a role of this transcription factor in the maintenance of the epileptic state. The pathological overexpression of SRF may also act to repress transcription of a number of genes. High amounts of SRF were found to inhibit or “squelch” its own activity and the activity of other transcription factors, including cAMP-response element-binding protein (CREB) in in vitro transcription assays (Prywes and Zhu, 1992). Squelching occurs when an overexpressed transcription factor binds up a common coactivator so that it is no longer available to act as an intermediary between transcription factors and core transcription complexes (Prywes and Zhu, 1992). Apparently, SRF and CREB share the same coactivator. Unfortunately, transcriptional squelching is difficult to study in vivo, and we do not know whether enough SRF is expressed in the epileptic rats to cause this phenomenon. However, Mello et al. (1996) reported that c-fos was not elevated or was very weakly elevated in epileptic pilocarpine rats after spontaneous recurrent seizures or a second episode of SE. This would indicate that the transcriptional machinery has been chronically altered in these animals so that a seizure that would normally induce c-fos expression is no longer able to do so. Further studies on the role of altered gene expression in modulating long-term plasticity changes as seen in epilepsy are necessary to establish the specific gene changes that lead to hyperexcitability changes.
Footnotes
This work was supported National Institutes of Health Training Grant T32 NS07288 to T.A.M. and A.C.R., a Research Training grant from the American Epilepsy Society with support from the Milken Family Medical Foundation to T.A.M., National Institutes of Health Grants RO1-NS23350 and PO1-NS25630 to R.J.D., the Nathan and Sophie Gumenick Neuroscience Research Fund, and the Milton L. Markel Alzheimer’s Disease Research Fund. The gift of the SRF antibody by Dr. Michael E. Greenburg is greatly appreciated. The phenytoin studies were conducted with the help of Dr. Lisa D. Kochan. We also thank Drs. Shubhro Pal and Severn B. Churn for suggestions during the course of this research effort and for critical reading of this manuscript.
Correspondence should be addressed to Dr. Robert J. DeLorenzo, P.O. Box 980599, Department of Neurology, Medical College of Virginia, Virginia Commonwealth University, Richmond, VA 23298-0599.
REFERENCES
- 1.Attar RM, Gilman MZ. Expression cloning of a novel zinc finger protein that binds to the c-fos serum response element. Mol Cell Biol. 1992;12:2432–2443. doi: 10.1128/mcb.12.5.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 3.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 4.Churn SB, Yaghmai A, Povlishock J, Rafiq A, DeLorenzo RJ. Global forebrain ischemia results in decreased immunoreactivity of calcium/calmodulin-dependent protein kinase II. J Cereb Blood Flow Metab. 1992;12:784–793. doi: 10.1038/jcbfm.1992.109. [DOI] [PubMed] [Google Scholar]
- 5.DeLorenzo RJ. The challenging genetics of epilepsy. Epilepsy Res Suppl. 1991;4:3–17. [PubMed] [Google Scholar]
- 6.DeLorenzo RJ, Morris TA. Long-term modulation of genetic expression in epilepsy and other neurological diseases. The Neuroscientist. 1999;5:86–99. [Google Scholar]
- 7.DeLorenzo RJ, Pal S, Sombati S. Prolonged activation of the NMDA receptor-Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture. Proc Natl Acad Sci USA. 1998;95:14482–14487. doi: 10.1073/pnas.95.24.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elmer E, Kokaia M, Ernfors P, Ferencz I, Kokaia Z, Lindvall O. Suppressed kindling epileptogenesis and perturbed BDNF and TrkB gene regulation in NT-3 mutant mice. Exp Neurol. 1997;145:93–103. doi: 10.1006/exnr.1997.6478. [DOI] [PubMed] [Google Scholar]
- 9.Herdegen T, Blume A, Buschmann T, Georgakopoulos E, Winter C, Schmid W, Hsieh TF, Zimmermann M, Gass P. Expression of activating transcription factor-2, serum response factor and cAMP/Ca response element binding protein in the adult rat brain following generalized seizures, nerve fibre lesion and ultraviolet irradiation. Neuroscience. 1997;81:199–212. doi: 10.1016/s0306-4522(97)00170-x. [DOI] [PubMed] [Google Scholar]
- 10.Johansen FE, Prywes R. Serum response factor: transcriptional regulation of genes induced by growth factors and differentiation. Biochim Biophys Acta. 1995;1242:1–10. doi: 10.1016/0304-419x(94)00014-s. [DOI] [PubMed] [Google Scholar]
- 11.Kaczmarek L. Expression of c-fos and other genes encoding transcription factors in long-term potentiation. Behav Neural Biol. 1992;57:263–266. doi: 10.1016/0163-1047(92)90276-a. [DOI] [PubMed] [Google Scholar]
- 12.Lazo PS, Dorfman K, Noguchi T, Mattei MG, Bravo R. Structure and mapping of the fosB gene. FosB downregulates the activity of the fosB promoter. Nucleic Acids Res. 1992;20:343–350. doi: 10.1093/nar/20.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leite JP, Cavalheiro EA. Effects of conventional antiepileptic drugs in a model of spontaneous recurrent seizures in rats. Epilepsy Res. 1995;20:93–104. doi: 10.1016/0920-1211(94)00070-d. [DOI] [PubMed] [Google Scholar]
- 14.Lewin B. Genes, Ed 3, p 183. Wiley; New York: 1987. [Google Scholar]
- 15.Lothman EW, Bertram EH. Epileptogenic effects of status epilepticus. Epilepsia [Suppl 34] 1993;1:S59–S70. doi: 10.1111/j.1528-1157.1993.tb05907.x. [DOI] [PubMed] [Google Scholar]
- 16.Lothman EW, Bertram EH, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- 17.Malenka RC. The role of postsynaptic calcium in the induction of long-term potentiation. Mol Neurobiol. 1991;5:289–295. doi: 10.1007/BF02935552. [DOI] [PubMed] [Google Scholar]
- 18.Martinez JLJ, Derrick BE. Long-term potentiation and learning. Annu Rev Psychol. 1996;47:173–203. doi: 10.1146/annurev.psych.47.1.173. [DOI] [PubMed] [Google Scholar]
- 19.McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meldrum BS. Metabolic factors during prolonged seizures and their relation to nerve cell death. Adv Neurol. 1983;34:261–275. [PubMed] [Google Scholar]
- 21.Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia. 1993;34:985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 22.Mello LE, Kohman CM, Tan AM, Cavalheiro EA, Finch DM. Lack of Fos-like immunoreactivity after spontaneous seizures or reinduction of status epilepticus by pilocarpine in rats. Neurosci Lett. 1996;208:133–137. doi: 10.1016/0304-3940(96)12562-3. [DOI] [PubMed] [Google Scholar]
- 23.Misra RP, Rivera VM, Wang JM, Fan PD, Greenberg ME. The serum response factor is extensively modified by phosphorylation following its synthesis in serum-stimulated fibroblasts. Mol Cell Biol. 1991;11:4545–4554. doi: 10.1128/mcb.11.9.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore AN, Waxham MN, Dash PK. Neuronal activity increases the phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in rat hippocampus and cortex. J Biol Chem. 1996;271:14214–14220. doi: 10.1074/jbc.271.24.14214. [DOI] [PubMed] [Google Scholar]
- 25.Morgan JI, Curran T. Proto-oncogene transcription factors and epilepsy. Trends Pharmacol Sci. 1991;12:343–349. doi: 10.1016/0165-6147(91)90594-i. [DOI] [PubMed] [Google Scholar]
- 26.Morris TA, Rice AC, DeLorenzo RJ. Status epilepticus (SE) results in long-term changes in transcription factor binding and expression in a in vivo model of epilepsy. Epilepsia [Abstr] 1997;38:124. [Google Scholar]
- 27.Nahm WK, Noebels JL. Nonobligate role of early or sustained expression of immediate-early gene proteins c-fos, c-jun, and Zif/268 in hippocampal mossy fiber sprouting. J Neurosci. 1998;18:9245–9255. doi: 10.1523/JNEUROSCI.18-22-09245.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norman C, Runswick M, Pollock R, Treisman R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 1988;55:989–1003. doi: 10.1016/0092-8674(88)90244-9. [DOI] [PubMed] [Google Scholar]
- 29.Ormandy GC, Jope RS, Snead OC. Anticonvulsant actions of MK-801 on the lithium-pilocarpine model of status epilepticus in rats. Exp Neurol. 1989;106:172–180. doi: 10.1016/0014-4886(89)90091-5. [DOI] [PubMed] [Google Scholar]
- 30.Penschuck S, Luscher B, Fritschy JM, Crestani F. Activation of the GABA(A)-receptor Δ-subunit gene promoter following pentylenetetrazole-induced seizures in transgenic mice. Brain Res Mol Brain Res. 1997;51:212–219. doi: 10.1016/s0169-328x(97)00243-x. [DOI] [PubMed] [Google Scholar]
- 31.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferferg HJ, Scoville B, White BG. Antiepileptic drug development program. Cleve Clin Q. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 32.Prywes R, Zhu H. In vitro squelching of activated transcription by serum response factor: evidence for a common coactivator used by multiple transcriptional activators. Nucleic Acids Res. 1992;20:513–520. doi: 10.1093/nar/20.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prywes R, Dutta A, Cromlish JA, Roeder RG. Phosphorylation of serum response factor, a factor that binds to the serum response element of the c-FOS enhancer. Proc Natl Acad Sci USA. 1988;85:7206–7210. doi: 10.1073/pnas.85.19.7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 35.Rafiq A, DeLorenzo RJ, Coulter DA. Generation and propagation of epileptiform discharges in a combined entorhinal cortex/hippocampal slice. J Neurophysiol. 1993;70:1962–1974. doi: 10.1152/jn.1993.70.5.1962. [DOI] [PubMed] [Google Scholar]
- 36.Rice A, Rafiq A, Shapiro SM, Jakoi ER, Coulter DA, DeLorenzo RJ. Long-lasting reduction of inhibitory function and γ-aminobutyric acid type A receptor subunit mRNA expression in a model of temporal lobe epilepsy. Proc Natl Acad Sci USA. 1996;93:9665–9669. doi: 10.1073/pnas.93.18.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998;782:240–247. doi: 10.1016/s0006-8993(97)01285-7. [DOI] [PubMed] [Google Scholar]
- 38.Robertson HA. Immediate-early genes, neuronal plasticity, and memory. Biochem Cell Biol. 1992;70:729–737. doi: 10.1139/o92-112. [DOI] [PubMed] [Google Scholar]
- 39.Saffen DW, Cole AJ, Worley PF, Christy BA, Ryder K, Baraban JM. Convulsant-induced increase in transcription factor messenger RNAs in rat brain. Proc Natl Acad Sci USA. 1988;85:7795–7799. doi: 10.1073/pnas.85.20.7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato-Bigbee C, Chan EL, Yu RK. Oligodendroglial cyclic AMP response element-binding protein: a member of the CREB family of transcription factors. J Neurosci Res. 1994;38:621–628. doi: 10.1002/jnr.490380604. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt D. The influence of antiepileptic drugs on the electroencephalogram: a review of controlled clinical studies. Electroencephalogr Clin Neurophysiol [Suppl] 1982;36:453–466. [PubMed] [Google Scholar]
- 42.Sombati S, DeLorenzo RJ. Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol. 1995;73:1706–1711. doi: 10.1152/jn.1995.73.4.1706. [DOI] [PubMed] [Google Scholar]
- 43.Spencer JA, Misra RP. Expression of the serum response factor gene is regulated by serum response factor binding sites. J Biol Chem. 1996;271:16535–16543. doi: 10.1074/jbc.271.28.16535. [DOI] [PubMed] [Google Scholar]
- 44.Stasheff SF, Anderson WW, Clark S, Wilson WA. NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science. 1989;245:648–651. doi: 10.1126/science.2569762. [DOI] [PubMed] [Google Scholar]
- 45.Stryer L. Biochemistry, Ed 2, pp 832–833. Freeman; San Francisco: 1981. [Google Scholar]
- 46.Suzuki T. Messengers from the synapses to the nucleus (MSNs) that establish late phase of long-term potentiation (LTP) and long-term memory. Neurosci Res. 1996;25:1–6. doi: 10.1016/0168-0102(96)01023-1. [DOI] [PubMed] [Google Scholar]
- 47.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–335. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]