

Abstract
The N-methyl-d-aspartate (NMDA) receptor antagonist ketamine is associated with rapid but transient antidepressant effects in patients with treatment resistant unipolar depression (TRD). Based on work suggesting that ketamine and lithium may share overlapping mechanisms of action, we tested lithium compared to placebo as a continuation strategy following ketamine in subjects with TRD. Participants who met all eligibility criteria and showed at least an initial partial response to a single intravenous infusion of ketamine 0.5 mg/kg were randomized under double-blind conditions to lithium or matching placebo before receiving an additional three infusions of ketamine. Subsequent to the ketamine treatments, participants remained on lithium or placebo during a double-blind continuation phase. The primary study outcome was depression severity as measured by the Montgomery–Åsberg Depression Rating Scale compared between the two groups at Study Day 28, which occurred ~2 weeks following the final ketamine of four infusions. Forty-seven participants with TRD were enrolled in the study and underwent an initial ketamine infusion, of whom 34 participants were deemed to have at least a partial antidepressant response and were eligible for randomization. Comparison between treatment with daily oral lithium (n = 18) or matching placebo (n = 16) at the primary outcome showed no difference in depression severity between groups (t32 = 0.11, p = 0.91, 95% CI [−7.87, 8.76]). There was no difference between lithium and placebo in continuing the acute antidepressant response to ketamine. The identification of a safe and effective strategy for preventing depression relapse following an acute course of ketamine treatment remains an important goal for future studies.
Subject terms: Translational research, Depression
Introduction
Major Depressive Disorder (MDD) is a disabling medical illness highly prevalent throughout the world [1]. Patients with MDD who fail to respond to at least two antidepressant trials of adequate dose and duration may be classified as suffering from treatment resistant depression (TRD) [2]. The Food and Drug Administration (FDA)-approved antidepressant medications show remission rates of 40–60% in randomized trials [3] and lower remission rates (20–40%) in naturalistic studies [4]. The persistence of depressive symptoms is associated with a worsened quality of life for patients and caregivers, increased risk for suicide, and higher economic costs to society [1, 5]. Despite an urgent need for more efficient and rapidly acting antidepressants, the development of new drugs for MDD in the last 50 years has been almost exclusively based on medications targeting the monoamine system [6].
In this context, the discovery of a rapid antidepressant effect of a sub-anesthetic dose of the N-methyl-d-aspartate (NMDA) receptor antagonist ketamine by Berman et al. [7] triggered considerable enthusiasm within the scientific community. Multiple replications have demonstrated ketamine’s acute antidepressant efficacy [8, 9]; however, these therapeutic effects are relatively transient, lasting about 1 week following a single infusion [10, 11]. The transient nature of the antidepressant response following treatment with intravenous (IV) ketamine has raised concerns regarding the use of ketamine in clinical settings. Considering that the risk of relapse for pharmacotherapy-resistant patients is undoubtedly a matter of primary importance [2], a critical issue in the field is the identification of strategies that will enhance the durability of the initial antidepressant response to ketamine. Repeated infusions of ketamine have been investigated as one such strategy. An open-label study investigating the effects of six ketamine infusions showed that the median time-to-relapse among responders was 18 days (ranging from 4 to >83 days) [12]. However, identifying relapse-prevention strategies with a more feasible route of administration (e.g., oral versus IV), which would improve accessibility for patients, remains a critical issue for the clinical utility of ketamine for TRD.
Lithium has shown clinical efficacy as an adjuvant in patients with unipolar TRD [13], and the inhibition of glycogen synthase kinase (GSK)-3 by lithium has been shown to be critical to its therapeutic efficacy and tolerability [14, 15]. Basic research on the molecular mechanisms underpinning the antidepressant and neuroplastic effects of ketamine implicate both activation of the mammalian target of rapamycin (mTOR) signaling pathway and inhibitory phosphorylation of GSK-3 within the prefrontal cortex (PFC) [16, 17]. Mice with knock-in mutations designed to prevent phosphorylation of GSK-3 show attenuated antidepressant response to ketamine, suggesting that inhibition of GSK-3 is involved in ketamine’s mechanism of action [17]. In a separate study, low-dose ketamine in combination with low-dose lithium induced antidepressant effects and increased the number and function of synapses in the medial prefrontal cortex (mPFC), indicating that the interaction of lithium with ketamine may involve the activation of the mTOR signal pathway and inhibition of GSK-3 activity [18]. Overall, this evidence supports the mechanistic rationale to study lithium as a pharmacotherapeutic strategy to continue the rapid antidepressant response to ketamine.
In the current study we conducted a randomized, double-blind, placebo-controlled trial investigating lithium up to 1200 mg daily compared to placebo as a continuation strategy following an acute course of ketamine administered intravenously. The primary outcome of the study was depression severity as measured by the Montgomery–Åsberg Depression Rating Scale (MADRS) at Day 28, occurring approximately 2 weeks following the final ketamine infusion, compared between the lithium and placebo treatment arms.
Materials and Methods
Study Participants and Design
Study participants were recruited from Internet and newspaper advertising as well as physician referrals between June 2013 and September 2016. Participants were between the ages of 21 and 65 and had a primary diagnosis of MDD without psychotic features, as assessed by a trained rater using the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders—Fourth Edition Text Revision (DSM-IV-TR) Axis I Disorders—Patient Edition (SCID-I/P). To be eligible, participants had to have an ongoing major depressive episode (MDE) of at least 4 weeks duration and of at least moderate severity as defined by a Quick Inventory of Depressive Symptomatology - Self Report (QIDS-SR) score ≥14 and a Clinical Global Impression Severity (CGI-S) score of ≥4. In addition, participants had to report a history of at least one previous episode of depression prior to the current episode (recurrent MDD) or chronic MDD of at least 2 years duration. Finally, participants must have failed to respond to two or more adequate trials of an FDA-approved antidepressant lifetime, as determined using the antidepressant treatment history form (ATHF). A lack of response was defined by a failure to improve by at least 50% based on the available data and patient report. Exclusionary diagnoses included substance use disorder in the past 2 years, lifetime history of schizophrenia or other psychotic disorder, bipolar disorder, pervasive developmental disorder or mental retardation, current diagnosis of obsessive compulsive disorder or eating disorder, or any clinically significant personality disorder, including, but not limited to, schizotypal or antisocial personality disorder. Lifetime history of recreational use of ketamine or phencyclidine (PCP) was also exclusionary. Other exclusion criteria included pregnancy, urine toxicology positive for illicit drugs, history of at least one seizure without clear and resolved etiology, uncontrolled hypertension, renal or thyroid impairment or abnormal electrocardiogram (EKG) reading at the time of the screening. Treatment with antidepressants within 1 week [2 weeks for monoamine oxidase inhibitors (MAOI) and 4 weeks for fluoxetine] of randomization was exclusionary. Thus, study participants were not receiving any antidepressant medications at the time of ketamine infusion and for the duration of the study. Table S1 “Concomitant Psychotropic and other Medications at Randomization in Study Sample” of the supplementary materials includes a summary of the concomitant medications.
Following screening (Day −28 to Day −2), all eligible participants received a single open-label infusion of ketamine (Day −1, Test infusion). The ketamine infusion was administered intravenously over 40 min by a study anesthesiologist at the dosage of 0.5 mg/kg. Participants who showed an initial antidepressant response at 24 h after the infusion (Day 0), as defined by an improvement of 25% or greater on the MADRS score from baseline (Day −1, pre-test infusion assessment), were eligible for randomization and entered into a double-blind trial of lithium/placebo prescribed in conjunction with ketamine for the initial week and thereafter in isolation. A daily lithium dose ranging from 600 to 1200 mg was used, with the goal of achieving a target blood level in the range of 0.6–0.9 mEq/L, but not to exceed 1.0 mEq/L. Medication changes were made on the basis of blood levels and clinical side effects. In the event of suspected lithium-related adverse effects, the dose of lithium was reduced and a minimum dose of 600 mg daily was required for continued study participation. Subsequent to randomization, participants received additional ketamine infusions (ketamine 0.5 mg/kg infused IV over 40 min) on study Day 7, 9, and 11. Following the final ketamine infusion on Day 11, study participants entered the post-ketamine continuation phase (Days 12–41) and remained on the assigned treatment (lithium or placebo). Participants were assessed twice weekly (once in person and one by phone) over the 4-week continuation phase. The primary outcome was measured at Day 28, which occurred ~2 weeks following the final ketamine infusion. Participants who continued to meet at least partial response criteria were eligible to enter the extension phase (Day 42–91), during which time participants were assessed weekly in the clinic. During this phase, study participants remained in the trial as long as they continued to meet criteria for partial response; those who failed to meet the threshold were exited from the study. Lithium levels were obtained in the morning on Days 7, 11, 28, and 42 ~12 h following participants’ last lithium dose. At the exit visit, participants underwent a physical exam and psychiatric evaluation. At this visit, EKG, clinical hematological and biochemical blood analyses, urine toxicology and urine pregnancy test for female participants were also repeated. See Fig. 1 for study flow diagram.
Fig. 1.

Study flow diagram. Day 0: Participants are evaluated for response to ketamine (MADRS > 25% above baseline); responders were randomized to receive lithium or placebo beginning that evening; non-responders were exited per protocol. Continuation Phase: 4 weeks, weekly in-person and phone-call follow-ups. Day 28: primary outcome. Day 42: secondary outcome and end of Continuation phase. Extension phase: 8 weeks, weekly in-person follow-ups
At each visit, participants completed self-report questionnaires, underwent clinician-administered rating scales performed by a trained rater, and met with a study psychiatrist who assessed suicidal thinking or behavior, adverse events (AE), and changes in concomitant medications. The MADRS score at Day 28 represented the primary outcome measure. Secondary outcomes measures included global illness severity measured using the Clinical Global Impression—Improvement and Severity Scales (CGI-I and CGI-S), self-reports of depression measured using the QIDS-SR, anxiety measured using the Hamilton Anxiety Scale (HAM-A), and suicidal thinking measured using the Beck Scale for suicide ideation (BSS). AEs were summarized according to the Medical Dictionary for Regulatory Activities (MedDRA) system organ class and preferred terms. Safety and tolerability were assessed by discontinuation rate, frequency of adverse events, and change in score on the Columbia Suicide Severity Rating Scale (C-SSRS), the Brief Psychiatric Rating Scale (BPRS), the Clinician Administered Dissociative States Scale (CADSS), and the first item of the Young Mania Rating Scale (YMRS-1). Secondary outcome measures were represented by change in depression severity as measured by MADRS score from Day −1 (baseline, pre-test infusion assessment) to Day 42 (secondary outcome and end of continuation phase), and by change in additional efficacy measures (QIDS-SR, CGI, BSS, HAM-A) over the ketamine study period ranging from Day −1 (baseline, pre-test infusion assessment) to Day 28 (primary outcome). Change in measurement of psychiatric safety using the C-SSRS, CADSS, BPRS, and YMRS-1 during the ketamine study period were also reported. All study procedures were conducted at the Icahn School of Medicine at Mount Sinai in New York City. The Program for the Protection of Human Subjects at Mount Sinai approved the protocol and study procedures, and written informed consent was obtained from all participants prior to the performance of any study procedures. Participants were compensated for their time and effort. The study was registered at clinicaltrials.gov (NCT01880593). See Supplemental Material for additional details related to screening procedures, randomization and masking.
Statistical methods
The analytic plan was that of a futility (also called non-superiority) study, an early phase approach well suited to early screening of potential therapies.
Attention to the special formulation of the null and alternative hypotheses under this design is worthwhile since the formulation of null and alternative hypotheses is inverted compared to a traditional approach: briefly, in a futility study, the null hypothesis states that the experimental therapy is sufficiently promising to warrant definitive, phase III testing, whereas the alternative hypothesis states that the experimental therapy lacks the pre-specified superiority. For good discussion of the design’s strengths as a programmatic screening device to weed out unpromising new treatments as well as some of its limitations we refer the reader to a recent article by Levin [19]. The primary null hypothesis (Ho) of this futility study was that lithium would reduce the mean MADRS score at Day 28 by at least 5 points compared to placebo—consistent with a clinically meaningful effect. This was tested against the alternative (Ha), that Li reduces the mean MADRS by less than 5 points as compared to placebo (one-sided alpha = 0.15). Rejection of the null hypothesis would indicate that our intervention is not sufficiently promising to warrant further investigation, and, in that sense, is futile.
Following common practices, the test statistic was adjusted for baseline MADRS scores (MADRSbaseline) and the initial response to the ketamine test infusion (MADRS24 h) through a linear model as follows: the test statistic was , the estimated effect of lithium on Day 28 MADRS scores from the regression model:
where ILithium Assignment is the indicator function entered as “1” for participants assigned to the lithium arm and “0” otherwise, and ϵ is a normally distributed error term. Ho would be rejected as preliminary evidence of effect in favor of Ha: futility of proposed therapy if exceeded a cutoff value of −0.5; otherwise, the conclusion would be non-futility. Under the assumptions of the previous analysis plan, including a normally distributed endpoint with standard deviation 10.8, this choice of cutoff value corresponds to the previous operating characteristics for the study. The value of α—the probability of falsely rejecting a promising therapy that reduced mean MADRS score at Day 28 by at least 5 points—accordingly less than or equal to 0.15. The value of β—the probability of incorrectly endorsing an ineffective therapy—accordingly less than or equal to 0.2 at the pre-specified alternative where such a therapy actually increased mean MADRS score at Day 28 by 3 or more points. In our case we anticipated that correction for baseline and test infusion scores might further reduce both error rates beyond these stated values, though we lacked adequate test data to precisely estimate how large an improvement might be expected.
The design specifies power of more than 80% to reject the null hypothesis if the true mean MADRS score in the lithium arm is 3 points higher than in the placebo group and effect sizes have been estimated based on Murrough et al. [12]. With N = 14 patients randomized to each arm, the one sided Mann–Whitney Test at alpha = 0.15 meets the specified power requirement assuming that the MADRS scores in the placebo and lithium arms are both normally distributed with standard deviation 10.8. These assumed variances were conservatively selected as the maximum variance estimate at any point from baseline to day 13 in the data reported by Murrough et al. [12]. See Supplemental Material for additional details.
Results
Sixty-seven participants were assessed for eligibility; of those, 42 individuals with TRD met all the inclusion criteria and none of the exclusion criteria and received a single open-label infusion of ketamine (test infusion). Thirty-five participants who showed an initial partial antidepressant response at 24 h after the test infusion were eligible for randomization and entered into the double-blind trial of lithium/placebo. Of these, n = 34 continued in the study and were exposed to at least one dose of lithium or placebo, and represent the intention to treat analyzed sample (ITT) (see Supplemental Material for Figure S1. CONSORT Diagram). On average, study participants were 45 years old, in a moderately severe current depressive episode (average MADRS score: 32), experiencing recurrent or chronic MDD, and had failed to respond to a median of five lifetime adequate antidepressant trials (range: 2–12). Seven subjects underwent ECT in their lifetime (two randomized to lithium) and received an adequate course of either unilateral or bilateral ECT, with no benefit reported among the participants randomized to lithium or placebo. No significant difference emerged between the two groups. Additional clinical and demographic details are reported in Table 1, and a summary of concomitant medications is reported in Table S1 of the supplementary materials. On the primary outcome visit, the mean lithium blood level was 0.61 mEq/L (SD = 0.19) and the average dosage was 800 mg/day.
Table 1.
Characteristics of study sample
| Total sample | Lithium | Placebo | ||||
|---|---|---|---|---|---|---|
| N | 34 | 18 | 16 | |||
| Age at enrollment, years (mean, SD) | 45.4 | 11.7 | 45 | 12.6 | 45.8 | 10.9 |
| Male (frequency, percent) | 16 | 47.0% | 9 | 50.0% | 7 | 43.7% |
| White (frequency, percent) | 29 | 85.2% | 17 | 94.4% | 12 | 75.0% |
| Hispanic ethnicity (frequency, percent) | 3 | 8.8% | 0 | 0% | 3 | 18.7% |
| College degree, at least 2-year (frequency, percent) | 27 | 79.4% | 15 | 83.3% | 12 | 75.0% |
| Employed, at least part-time (frequency, percent) | 15 | 44.1% | 10 | 55.6% | 5 | 31.2% |
| Married (frequency, percent) | 5 | 14.7% | 1 | 5.6% | 4 | 25.0% |
| Age at first episode (mean, SD) | 16.0 | 9.9 | 19.0 | 12.1 | 12.6 | 5.3 |
| Years since first episode (mean, SD) | 29.2 | 15.1 | 25.8 | 14.7 | 33.0 | 15.2 |
| Number of episodes (median, range) | 3 | 1–53 | 3.5 | 1–53 | 2 | 1–32 |
| Duration of current MDE, months (median, range) | 78 | 1–708 | 30 | 6–576 | 190.5 | 1–708 |
| Chronic MDE (frequency, percent) | 25 | 73.5% | 12 | 66.7% | 13 | 81.2% |
| Recurrent MDD (frequency, percent) | 25 | 73.5% | 14 | 77.8% | 5 | 68.7% |
| Prior hospitalization (frequency, percent) | 9 | 26.4% | 5 | 27.8% | 4 | 25.0% |
| # Psychiatric hospitalizations (mean, SD) | 1.0 | 2.8 | 1.3 | 3.4 | 0.7 | 2.0 |
| Prior suicide attempt (frequency, percent) | 8 | 23.5% | 6 | 33.3% | 2 | 12.5% |
| Number of suicide attempts (median, range) | 0 | 0–7 | 0 | 0–7 | 0 | 0–1 |
| No. lifetime ADT failures (median, range) | 5 | 2–12 | 5 | 2–8 | 4.5 | 2–12 |
| Baseline QIDS-SR Score (mean, SD) | 17.1 | 3.8 | 16.2 | 4.3 | 18.1 | 3.0 |
| Baseline MADRS score (mean, SD) | 32.5 | 4.6 | 32.3 | 3.8 | 32.8 | 5.6 |
ADT antidepressant treatment, MADRS Montgomery–Åsberg Depression Rating Scale, MDD Major depressive disorder, MDE major depressive episode, QIDS-SR Quick Inventory of Depressive Symptomatology - Self Report
Efficacy
Change in MADRS scores over time for the lithium and placebo groups are reported in Fig. 2. The estimated coefficient for the indicator of lithium treatment was 0.051 (SD = 3.89). The p-value for testing the difference between the means of the two treatment groups was highly non-significant, p = 0.99, two sided. With respect to the formulation of the hypotheses in the futility test, the corresponding p-value against the null hypothesis of non-futility was 0.099. This value was less than the 0.15 significance threshold and consistent with the conclusion of futility. The model did not show recognizable departures from model assumptions; however, the predictive power of the included explanatory variables on MADRS score at the primary outcome visit model was low (R-sq = 0.154). The estimated coefficient of the treatment indicator, 0.051, fell above the stated cutoff of −0.5 for declaring futility of the lithium treatment. The addition of lithium does not appear to have led to reduction in MADRS scores compared to treatment with ketamine alone.
Fig. 2.
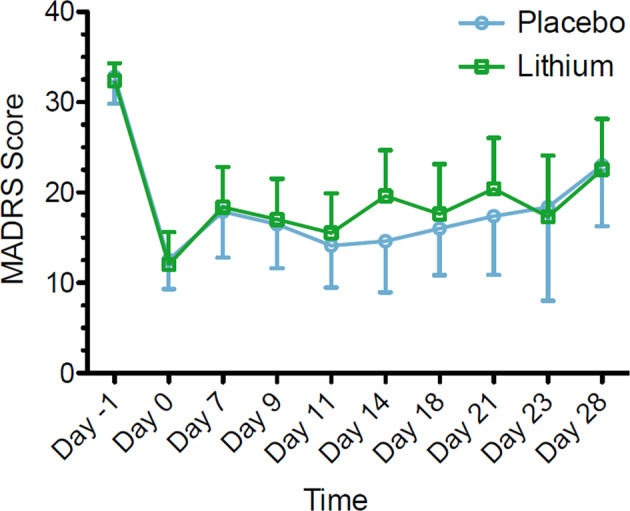
Change in MADRS score during clinical trial of lithium/placebo continuation therapy following ketamine in patients with treatment-resistant depression (n = 34). Values reflect means with associated 95% CI. Only upper or lower bounds are shown for clarity. MADRS, Montgomery–Åsberg Depression Rating Scale
Descriptive analyses on the secondary outcome measures by treatment group were performed and, as with MADRS scores, no statistically significant differences emerged between the lithium and placebo treatment groups (results not shown). Change in these measures, including QIDS-SR, CGI-S/I, BSS, HAM-A, and C-SSRS over the ketamine study period ranging from Day −1 (baseline, pre-test infusion assessment) to Day 28 (primary outcome) are reported in Table 2.
Table 2.
Secondary outcome measuresa
| Mean (estimate; 95% CI) | Standard deviation (estimate; 95% CI) | Variance (estimate; 95% CI) | |
|---|---|---|---|
| MADRS | |||
| Lithium | −7.2 (−12.7; −1.6) | 11.1 (8.3; 16.7) | 124.6 (70.1; 280.1) |
| Placebo | −7.6 (−14.1; −1.2) | 12.0 (8.8; 18.6) | 144.6 (78.9; 346.4) |
| QIDS-SR | |||
| Lithium | −6.3 (−9.0; −3.5) | 5.5 (4.1; 8.3) | 30.7 (17.2; 69.0) |
| Placebo | −8.0 (−11.6; −4.4) | 6.7 (4.9; 10.4) | 45.1 (24.6; 108.1) |
| CGI | |||
| Lithium | −0.8 (−1.2; −0.4) | 0.8 (0.6; 1.2) | 0.7 (0.4; 1.6) |
| Placebo | −1.1 (−1.8; −0.3) | 1.4 (1.0; 2.1) | 1.9 (1.0; 4.7) |
| BSS | |||
| Lithium | −0.9 (−2.6; 0.7) | 3.4 (2.6; 5.2) | 12.1 (6.8; 27.3) |
| Placebo | −4.8 (−8.8; −0.9) | 7.3 (5.4; 11.4) | 54.6 (29.8; 130.9) |
| HAM-A | |||
| Lithium | −5.2 (−8.2; −2.2) | 6.0 (4.5; 9.0) | 36.8 (20.7; 82.7) |
| Placebo | −9.1 (−13.0; −5.2) | 7.3 (5.4; 11.3) | 53.7 (29.3; 128.7) |
| C-SSRS | |||
| Lithium | −1.6 (−2.8; −0.5) | 2.3 (1.7; 3.4) | 5.2 (2.9; 11.9) |
| Placebo | −1.6 (−2.7; −0.5) | 2.0 (1.5; 3.2) | 4.3 (2.3; 10.5) |
| CADSS | |||
| Lithium | −0.1 (−0.5; 0.3) | 0.9 (0.6; 1.3) | 0.8 (0.4; 1.8) |
| Placebo | −0.1 (−0.5; 0.1) | 0.6 (0.4; 1.0) | 0.43 (0.2; 1.0) |
| BPRS | |||
| Lithium | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) |
| Placebo | −0.1 (−0.3; 0.1) | 0.5 (0.3; 0.7) | 0.2 (0.1; 0.6) |
| YMRS-1 | |||
| Lithium | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) |
| Placebo | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) | 0.0 (0.0; 0.0) |
BPRS Brief Psychiatric Rating Scale, BSS Beck Scale for suicide ideation, CADSS Clinician Administered Dissociative States Scale, CGI Clinical Global Impression Scale, C-SSRS Columbia Suicide Severity Rating Scale, HAM-A Hamilton Anxiety Rating Scale, MADRS Montgomery–Åsberg Depression Rating Scale, QIDS-SR Quick Inventory of Depressive Symptomatology - Self Report, YMRS-1 Young Mania Rating Scale—item 1
aThe secondary outcome measures were represented by change in depression severity as measured by MADRS score from Day −1 (baseline, pre-test infusion assessment) to Day 42 (secondary outcome and end of Continuation Phase), and by change in additional efficacy measures (QIDS-SR, CGI, BSS, HAM-A) over the ketamine study period ranging from Day −1 (baseline, pre-test infusion assessment) to Day 28 (primary outcome). Change in measurement of psychiatric safety using the C-SSRS, CADSS, BPRS, and YMRS-1 during the ketamine study period were also reported
Safety and tolerability
Dropout rates were relatively low and did not differ significantly between groups; of the 34 participants who received at least one dose of lithium or placebo, 29 completed the primary outcome visit procedures at Day 28, yielding a retention rate of 85%. Of the 18 participants allocated to lithium, 15 completed the primary outcome visit, while three discontinued early (16.7% dropout). Of the 16 participants allocated to placebo, 14 completed the primary outcome while two discontinued early (12.5% dropout). Five patients discontinued subsequent to randomization (Day 0) but prior to reaching and completing the primary outcome on Day 28. Of the five participants who discontinued from the study, three were discontinued due to lack efficacy, two due to adverse effects, and one for unspecified for reasons.
In the entire sample the most common AE was headache, which occurred in 17 participants; AEs were more frequent in the lithium group compared to placebo. In the lithium group, the most common AEs were nausea and headache, which occurred in eight individuals; dry mouth, constipation, abdominal pain, pollakiuria, and cutaneous rash occurred in three participants. Within the placebo group the most common AEs were headache (reported in nine participants) and nausea (reported in four participants). Altogether, four subjects elected to discontinue the study in association with an AE, two in the lithium group and two in the placebo group. The participant who was withdrawn from the trial due to AEs (skin rash and nystagmus) was receiving lithium. No serious adverse event (SAE) occurred during the course of the trial. A summary of study AEs is reported in Table 3.
Table 3.
Summary of adverse events by treatment group
| Lithium (n = 18) | Placebo (n = 16) | |||
|---|---|---|---|---|
| N events | N participants (%) | N events | N participants (%) | |
| Gastrointestinal disorders | ||||
| Nausea | 19 | 8 (44.4%) | 5 | 4 (25.5%) |
| Dry mouth | 4 | 3 (16.6%) | 0 | 0 (0.0%) |
| Constipation | 3 | 3 (16.6%) | 0 | 0 (0.0%) |
| Vomiting | 4 | 2 (11.1%) | 0 | 0 (0.0%) |
| Abdominal pain | 3 | 3 (16.6%) | 0 | 0 (0.0%) |
| Nervous system disorders | ||||
| Headache | 17 | 8 (44.4%) | 14 | 9 (5.6%) |
| Dizziness | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| Tinnitus | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| Tremor | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| Musculoskeletal and connective tissue disorders | ||||
| Arthralgia | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| General disorders and administration site conditions | ||||
| Fatigue | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| Renal and urinary disorders | ||||
| Pollakiuria | 3 | 3 (16.6%) | 1 | 1 (6.2%) |
| Polyuria | 2 | 2 (11.1%) | 0 | 0 (0.0%) |
| Psychiatric disorders | ||||
| Insomnia | 2 | 2 (11.1%) | 1 | 1 (6.2%) |
| Skin and subcutaneous tissue disorders | ||||
| Rash | 3 | 3 (16.6%) | 0 | 0 (0.0%) |
Table summarizes occurrence of adverse events (AE) in the trial that occurred subsequent to at least one dose of study medication and were associated with a causality rating of at least “possibly.” AEs are reported for N participants (%) >10%
There were no significant differences in BPRS, CADSS, or YMRS-1 scores between the lithium and placebo group during the course of the ketamine infusions (results not shown). In the overall sample there was no emergence of serious suicidal ideation compared to baseline, as defined by an increase in the maximum suicidal ideation score to three or greater on the C-SSRS during the trial. No participants experienced emergence of suicidal behavior during the study.
Discussion
The current study tested the efficacy and feasibility of lithium as a continuation therapy following an acute treatment of four IV infusions of ketamine in patients with TRD. Participants were suffering from moderate to severe depression at the time of enrollment and failed to respond to two or more adequate trials of an FDA-approved antidepressant. The primary research objective was to test if participants randomized to lithium compared to placebo exhibited less severe depression at 2 weeks (primary outcome) following the cessation of the ketamine infusions. Participants randomized to lithium failed to show a prolonged reduction in MADRS score compared to those randomized to placebo.
Key to our hypothesis, both ketamine and lithium are potent inhibitors of GSK-3. As GSK-3 is a key regulator of intracellular neuroplasticity pathways in the central nervous system (CNS) and a functional target of both ketamine and lithium, this combination represented a rational pharmacotherapeutic strategy for patients with TRD who are in urgent need of new and more effective treatments [15, 18]. Moreover, lithium is also used as an augmentation strategy for TRD [13], and treatment guidelines for patients who fail to respond to monotherapy recommend combining antidepressants and lithium as a first-line treatment strategy [20]. Lithium is also recommended in the continuation phase following a successful acute course of ECT and the combination of lithium and nortriptyline [21] has been shown to be superior to nortriptyline alone or placebo in preventing relapse while being comparable to continuation ECT in maintaining remission [22].
Few studies have explored strategies to extend the antidepressant effect of ketamine, and most are limited by open-label design, lack of a control group, and use of concomitant psychotropic medications, which circumscribe the ability to draw any definitive conclusions. One study by Kantrowitz et al. [23], explored the effect of d-cycloserine, a partial agonist of the NMDA receptor and FDA–approved antituberculotic drug, to prevent the relapse of depressive symptomatology after an initial infusion of ketamine. A treatment-resistant bipolar depression sample (seven participants) on a stable regimen of mood stabilizers received open-label ketamine followed by 8 weeks of d-cycloserine and pyridoxine. Four participants remained in remission after treatment with d-cycloserine, and the clinical improvement correlated with the magnitude of improvement 24 h after the ketamine infusion, suggesting a synergistic effect between d-cycloserine and ketamine. Another study investigated the effect of cognitive behavioral therapy (CBT) in extending the antidepressant effect of ketamine [24] in a cohort of subjects suffering from MDD (n = 16). In this study participants initiated a 10-week course of CBT concurrently with four infusions of ketamine over the course of 2 weeks. Among ketamine responders (eight participants), the relapse rate at the end of the CBT course was 25%, suggesting that CBT may enhance the durability of the antidepressant response to ketamine. Two randomized, double-blind studies examining relapse prevention following IV ketamine investigated the effect of riluzole, a glutamate-modulating agent with neuroprotective properties approved for amyotrophic lateral sclerosis (ALS), compared to placebo [25, 26]. In one study, participants who showed durable antidepressant responses following a single ketamine infusion were randomized to double-blind treatment with either riluzole (100 mg/day) or placebo [25]. Riluzole did not prevent relapse in the first month following ketamine, with no significant differences emerging between riluzole and placebo in time-to-relapse. Similarly, in the second study, 42 patients with TRD received a single infusion of ketamine and were randomized to either riluzole (100–200 mg/day) or placebo under double-blind conditions. The two groups did not show significant differences in depressive symptoms 28 days after a single infusion of ketamine, and these results remained stable when the analyses were limited to ketamine responders alone [26]. Very recently, the efficacy and tolerably of long term administration of the S- enantiomer of racemic ketamine, S-ketamine or ‘esketamine,’ has been studied in patients with TRD and showed a persistent antidepressant response for more than two months to the intranasal esketamine [27].
The current study has several limitations. Chiefly, the treatment resistance of the sample limits the generalizability of these findings to a broader population of participants suffering from MDD. Although repeated ketamine infusions may be a feasible strategy for the treatment of TRD, the safety and efficacy of prolonged courses of ketamine should be established in controlled trials. In addition, the risk and burden conferred by IV administration of an anesthetic suggests the need for further controlled trials of alternative interventions that have the potential to extend the initial antidepressant response to ketamine. As a separate limitation, the dosing of lithium in the current study was on the low end of the recommended range for the treatment of TRD [28]. The mean daily dose of lithium on the primary outcome visit in the current study was 800 mg (SD = 244.9), similar to that reported in the STAR*D on augmentation therapies for TRD [29] (lithium mean daily doses = 859.8 ± 373.1 mg), and the mean serum level of lithium in our sample was 0.61 ± 0.19 mEq/L. The serum level therefore was toward the inferior limit of the recommended therapeutic range for its action as mood stabilizer for the treatment of bipolar disorder (0.6–1.2 mEq/L). The lithium blood level required to enhance the effects of antidepressants is not firmly established [30], and it is unclear if a higher level of lithium exposure would have conferred a therapeutic advantage following ketamine compared to placebo in the current trial. Another potential limitation is that the time point of the primary outcome occurring only ~2 weeks following the end of the ketamine treatment period, which may not have enough time to observe a separation between lithium and placebo. Finally, in the current trial the effect of lithium as adjunct therapy to ketamine was not investigated. Rather, lithium was tested in monotherapy to prolong the antidepressant effect of ketamine following an acute course of infusions. Further studies are warranted to investigate the combination of lithium and an FDA-approved antidepressant, in comparison to lithium monotherapy, to extend the antidepressant effect of ketamine.
In the current study, a course of four IV administrations of ketamine was well tolerated in TRD. Lithium was not superior to placebo in continuing the antidepressant response to ketamine at 2 weeks following the cessation of the ketamine infusions. Given the high risk for relapse of this population and the limited durability of the antidepressant effect of ketamine, future larger randomized controlled trials to identify interventions that maintain the initial antidepressant response of ketamine are warranted.
Research dataset and protocol
Study protocol and de-identified participant data collected during the trial will be available for data meta-analysis upon review of the project proposal by the corresponding author, Dr. James Murrough, MD, PhD. Proposals should be directed via email to james.murrough@mssm.edu.
Funding and disclosure
This study was supported by Grant 2013098 from the Doris Duke Charitable Foundation Grant to Dr. James Murrough. In the past 5 years, Dr. Murrough has provided consultation services to FSV7, Boehreinger Ingelheim, Sage Therapeutics, Novartis, Allergan, Fortress Biotech, Janssen Research and Development, Genentech, Medavante-Prophase, and Global Medical Education (GME) and has received research support from Avanir Pharmaceuticals, Inc. Dr. Murrough is named on a patent pending for neuropeptide Y as a treatment for mood and anxiety disorders.The Icahn School of Medicine (employer of Dr. Murrough) is named on a patent and has entered into a licensing agreement and will receive payments related to the use of ketamine if it is approved for the treatment of depression. Drs. Feder and Murrough are not named on this patent and will not receive any payments. Dr. Feder is named on a separate, pending patent for the use of ketamine to treat posttraumatic stress disorder. Dr. Collins is a paid independent rater for Medavante-Prophase. Dr Charney has a patent US 9,592,207—Intranasal Administration of Ketamine to Treat Depression (issued 14 March 2017) licensed to Janssen Pharmaceutical, Inc., a patent US 9,539,220—Methods for Treating Suicidal Ideation (issued 10 January 2017) licensed to Janssen Pharmaceutical, Inc., a patent US 8,785,500—Intranasal Administration of Ketamine to Treat Depression (issued 22 July 2014) licensed to Janssen Pharmaceuticals, Inc., a patent WO 2016/049234—Systems and Methods for Treating a Psychiatric Disorder licensed to Click Therapeutics, a patent U.S. Serial No. 14/783,686 and related foreign patent applications—Ketamine—As a Rapid Treatment for Post-Traumatic Stress Disorder (PTSD) pending, a patent U.S. Serial No. 14/889,746 and related foreign patent applications—Intranasal Neuropeptide Y (NPY)—for the Treatment of Mood and Anxiety Disorders pending, a patent WO 2016/172672—Method of Reducing Risk of Suicidal Ideations with Combined Ketamine/Lithium Therapy pending, and a patent U.S. Serial No. 15/379,013 and U.S. Serial No. 15/417,689—Intranasal Administration of Ketamine to Treat Depression (Continuation patent applications in the same patent family as issued U.S. Patents 8,785,500 and U.S. 9,539,220 and U.S. 9,592,207 pending). In the past 5 years, Dr. Iosifescu has received consulting fees from Alkermes, Axsome, Centers for Psychiatric Excellence (COPE), MyndAnalytics (CNS Response), Jazz, Lundbeck, Otsuka, Sunovion, and has received research support (through his academic institutions) from Alkermes, Astra Zeneca, Brainsway, LiteCure, Neosync, Roche, Shire. All other authors report no conflicts of interest.
Supplementary information
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-019-0365-0).
References
- 1.Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJL, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10:e1001547. doi: 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 3.Cipriani A, Geddes JR, Furukawa TA, Barbui C. Metareview on short-term effectiveness and safety of antidepressants for depression: an evidence-based approach to inform clinical practice. Can J Psychiatry. 2007;52:553–62. doi: 10.1177/070674370705200903. [DOI] [PubMed] [Google Scholar]
- 4.Cuffel BJ, Azocar F, Tomlin M, Greenfield SF, Busch AB, Croghan TW. Remission, residual symptoms, and nonresponse in the usual treatment of major depression in managed clinical practice. J Clin Psychiatry. 2003;64:397–402. doi: 10.4088/JCP.v64n0406. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg PE, Fournier AA, Sisitsky T, Pike CT, Kessler RC. The economic burden of adults with major depressive disorder in the United States (2005 and 2010) J Clin Psychiatry. 2015;76:155–62. doi: 10.4088/JCP.14m09298. [DOI] [PubMed] [Google Scholar]
- 6.Insel TR, Scolnick EM. Cure therapeutics and strategic prevention: raising the bar for mental health research. Mol Psychiatry. 2006;11:11–17. doi: 10.1038/sj.mp.4001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 8.Mathew SJ, Shah A, Lapidus K, Clark C, Jarun N, Ostermeyer B, et al. Ketamine for treatment-resistant unipolar depression: current evidence. CNS Drugs. 2012;26:189–204. doi: 10.2165/11599770-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caddy C, Amit BH, McCloud TL, Rendell JM, Furukawa TA, McShane R, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst. Rev. 2015;9:CD011612. [DOI] [PubMed]
- 10.Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 11.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74:250–6. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bschor T. Lithium in the treatment of major depressive disorder. Drugs. 2014;74:855–62. doi: 10.1007/s40265-014-0220-x. [DOI] [PubMed] [Google Scholar]
- 14.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–9. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31. doi: 10.1016/j.pharmthera.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–70. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–77. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin B. The futility study—progress over the last decade. Contemp Clin Trials. 2015;45:69–75. doi: 10.1016/j.cct.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam RW, Kennedy SH, Grigoriadis S, McIntyre RS, Milev R, Ramasubbu R, et al. Canadian network for mood and anxiety treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults. III. Pharmacotherapy. J Affect Disord. 2009;117(Suppl 1):S26–43. doi: 10.1016/j.jad.2009.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Kellner CH, Knapp RG, Petrides G, Rummans TA, Husain MM, Rasmussen K, et al. Continuation electroconvulsive therapy vs pharmacotherapy for relapse prevention in major depression: a multisite study from the Consortium for Research in Electroconvulsive Therapy (CORE) Arch Gen Psychiatry. 2006;63:1337–44. doi: 10.1001/archpsyc.63.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sackeim HA, Haskett RF, Mulsant BH, Thase ME, Mann JJ, Pettinati HM, et al. Continuation pharmacotherapy in the prevention of relapse following electroconvulsive therapy: a randomized controlled trial. J Am Med Assoc. 2001;285:1299–307. doi: 10.1001/jama.285.10.1299. [DOI] [PubMed] [Google Scholar]
- 23.Kantrowitz JT, Halberstam B, Gangwisch J. Single-dose ketamine followed by daily D-Cycloserine in treatment-resistant bipolar depression. J Clin Psychiatry. 2015;76:737–8. doi: 10.4088/JCP.14l09527. [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson ST, Wright D, Fasula MK, Fenton L, Griepp M, Ostroff RB, et al. Cognitive behavior therapy may sustain antidepressant effects of intravenous ketamine in treatment-resistant depression. Psychother Psychosom. 2017;86:162–7. doi: 10.1159/000457960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2010;13:71–82. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37:1526–33. doi: 10.1038/npp.2011.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75:139–48. doi: 10.1001/jamapsychiatry.2017.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauer M, Dopfmer S. Lithium augmentation in treatment-resistant depression: meta-analysis of placebo-controlled studies. J Clin Psychopharmacol. 1999;19:427–34. doi: 10.1097/00004714-199910000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Nierenberg AA, Fava M, Trivedi MH, Wisniewski SR, Thase ME, McGrath PJ, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163:1519–30. doi: 10.1176/ajp.2006.163.9.1519. [DOI] [PubMed] [Google Scholar]
- 30.Gelenberg A, Freeman C, Markowitz J, Rosenbaum J, Thase M, Trivedi M, et al. Treatment of Patients With Major Depressive Disorder. Practice Guideline. 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.