
Figure 1.
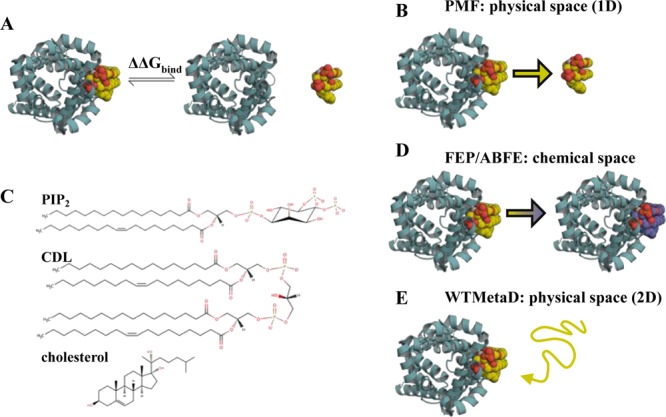
Introduction to free energy calculations. (A) Overview of free energy calculations. A membrane protein, as viewed from above the membrane, is shown in cyan cartoon, and a lipid in yellow, orange and red spheres. Two states are modelled: the left state is the protein bound to the target lipid, the right is the protein bound to a generic lipid (not shown), with the target lipid unbound. Free energy calculations aim to compute the difference in free energy between these states (ΔΔGbind). (B) PMF calculations create a reaction coordinate in physical space by pulling the lipid away from or towards the binding site. This coordinate can then be sampled, for example with umbrella sampling, to provide a 1D energetic landscape, allowing calculation of ΔΔGbind between the target and a generic lipid. (C) Chemical structures of PIP2, CDL and cholesterol. (D) FEP and ABFE calculations build alchemical pathways which either change the bound lipid into a different species, in this study to that of the bulk membrane, or fully remove the lipid from the simulation box. This provides the binding free energy difference between the target lipid and a generic lipid, ΔΔGbind. (E) WTMetaD biases the diffusion of a target lipid around the protein through addition of a time-dependent Gaussian of energy to the CV. These Gaussians can then be reconstructed into a full 2D energy landscape, with comparison of binding regions and the bulk membrane giving ΔΔGbind.