

Abstract
Over the past decade, electrochemical carbon dioxide reduction has become a thriving area of research with the aim of converting electricity to renewable chemicals and fuels. Recent advances through catalyst development have significantly improved selectivity and activity. However, drawing potential dependent structure–activity relationships has been complicated, not only due to the ill-defined and intricate morphological and mesoscopic structure of electrocatalysts, but also by immense concentration gradients existing between the electrode surface and bulk solution. In this work, by using in situ surface enhanced infrared absorption spectroscopy (SEIRAS) and computational modeling, we explicitly show that commonly used strong phosphate buffers cannot sustain the interfacial pH during CO2 electroreduction on copper electrodes at relatively low current densities, <10 mA/cm2. The pH near the electrode surface was observed to be as much as 5 pH units higher compared to bulk solution in 0.2 M phosphate buffer at potentials relevant to the formation of hydrocarbons (−1 V vs RHE), even on smooth polycrystalline copper electrodes. Drastically increasing the buffer capacity did not stand out as a viable solution for the problem as the concurrent production of hydrogen increased dramatically, which resulted in a breakdown of the buffer in a narrow potential range. These unforeseen results imply that most of the studies, if not all, on electrochemical CO2 reduction to hydrocarbons in CO2 saturated aqueous solutions were evaluated under mass transport limitations on copper electrodes. We underscore that the large concentration gradients on electrodes with high local current density (e.g., nanostructured) have important implications on the selectivity, activity, and kinetic analysis, and any attempt to draw structure–activity relationships must rule out mass transport effects.
Introduction
Rising atmospheric CO2 levels and associated climate change will put local and global economies under enormous pressure if immediate actions are not taken to escalate significant cuts in carbon emissions.1 Due to the immense scale of the problem, the deployment of multiple technologies will be necessary to combat this problem depending on location, industry, and other societal, economical, and political constraints. One of the most promising technologies to address these needs is the electrochemical reduction of CO2 to CO, hydrocarbons, and multicarbon oxygenates.2 This approach not only bypasses hydrogen production by supplying protons from solution, but also reduces energy transformation and separation costs when compared to thermocatalytic processes.
Nanostructured metal electrodes have provided encouraging results in their ability to decrease the required overpotentials to form products such as formic acid, CO, and ethylene.3−6 Regardless of the catalyst material and morphology, nanostructured and porous catalysts typically shift the onset potential of CO2 reduction toward less negative potentials and/or provide better selectivity toward carbon-based products over hydrogen evolution.4,7 Although the nanoscale effects and surface structure to the electrocatalytic activity cannot be overlooked on these electrodes,8−11 the selectivity toward CO and C2 products over hydrogen at high currents can be very susceptible to mass transport effects.12−15 One of the key reasons for this delicate interplay is the participation or absence of protons in the rate-determining step of the formed product. The proton decoupled electron transfer during the formation of C2 products on copper surfaces has been well established independently by different research groups.16−18 Although less established, there is a noteworthy amount of evidence that the formation of CO on gold surfaces may also take place via a rate-determining step where electron and proton transfer is decoupled.19 Subsequently, there is a particular trend for the products that are formed via a pH independent (vs NHE) pathway compared to the ones that are pH dependent, e.g., methane and hydrogen,20,21 as the (local) pH of the solution is increased.22−24 The evaluation of different catalysts by minimizing the concentration difference between the bulk and surface is of key importance to not only compare the intrinsic activity and selectivity of a catalyst, but to also accurately test and provide meaningful input to computational calculations.25
During the electrochemical reduction of CO2, the concentration of protons and CO2 near the electrode surface is counterbalanced between consumption from the electrode, diffusion from the bulk solution, and homogeneous buffer reactions.26 When the reactions are carried out at a high current density or in electrolytes that have poor buffer action and/or mass transport, the pH near the cathode surface is well-known to increase compared to the bulk value.27,28 Although the impact of local conditions on the selectivity of metal electrodes is well recognized, simulated, and reproduced,18,26,27,29,30 it is still a common practice to test high surface area electrodes in very low buffer capacity solutions. While the surface structure of a catalyst is very important to determine its intrinsic selectivity and activity, the distinction between the effect of surface structure and mass transport effects on the electrocatalytic activity is not explicitly clear and needs to be urgently clarified in order to improve fundamental understanding of reaction mechanisms.31 Recently, Dunwell et al. studied the concentration gradients near the electrode surface by using SEIRAS in bicarbonate solutions with special emphasis on the implications of the kinetic analysis.29 However, the activity and selectivity toward CO2 reduction products has not been extensively described. Moreover, a comparison of simulation results with physical measurements is currently missing in the literature.7,23
We studied the near-surface concentrations of reactants, e.g., protons and CO2, during CO2 electrolysis by using in situ surface enhanced infrared absorption spectroscopy (SEIRAS) under densely buffered neutral pH concentrations in phosphate electrolytes. These electrolytes are commonly used as high buffer capacity solutions to evaluate and exclude local pH effects on the catalytic selectivity, and thus it is vital to understand their buffer capacity and mass transport to cathode during CO2 electrolysis.7,32 In our work, we would not only like to test the buffer strengths of these solutions, but also attempt to find a suitable electrolyte that can minimize the difference between local and bulk environment for kinetic studies. The experimental results were supported by a computational model that takes into account bubble induced mass-transport and the concentration dependent solubility of CO2 in order to mimic the conditions created during in situ SEIRAS measurements. In addition, the activity and selectivity of a sputtered copper electrode and a very high surface area electrode composed of densely packed copper nanowires were tested with variable buffer concentrations, and the implications of these findings are thoroughly discussed. The experimental and computational data show that all concentrations of phosphate buffer breakdown quickly in a narrow potential window, with the lowest concentrations having the steepest pH gradient. These results suggested that most CO2 electroreduction experiments on the formation of hydrocarbons and oxygenates have been done in mass transport limited configurations in CO2 saturated aqueous solutions, which severely complicates the mechanistic insights that have been derived from them.
Results
An atomic force microscopy (AFM) image of a typical 25 nm thick Cu film sputtered on Ge is given in Figure 1(a). The film was composed of partially merged metal islands with an average diameter of 30 nm that were initially nucleated onto the Ge surface. Surface features ranging from 1 to 5 nm in height were present on top of a thin continuous Cu underlayer (Figure S1). Calibration and experiments were conducted in a custom designed spectroelectrochemical cell in which the copper coated Ge internal reflection element (IRE) was mounted to the bottom of cell serving as the working electrode (Figure 1(b)). The SEIRA spectra of phosphate species on a 25 nm thick Cu film for different pH values were measured, and the results are shown in Figure 1(c). The SEIRA spectrum taken at pH 4.34 exhibited three major peaks at 1152, 1076, and 941 cm–1, which were assigned to asymmetrical νa(PO), symmetrical νs(PO), and asymmetrical νa(POH) vibrations of H2PO4–, respectively.33 After complete deprotonation of H2PO4– around pH 9, the spectra exhibited the same vibrational modes that slightly shifted to lower frequencies belonging to HPO42–.33 As the solution was changed to become more alkaline, the spectra became dominated by the asymmetrical stretching of the PO43– molecule around 1005 cm–1. To monitor the pH changes during electrolysis, calibration spectra were recorded between pH 5–12 by the addition of KOH to the parent H2PO4– solution and recording a spectrum every 0.2 ± 0.1 pH units (Figure S2). The overlapping bands were deconvoluted to resolve the contributions from individual bands (Figure S2), and peak heights were derived from νa(PO) of H2PO4– and νs(PO) of HPO42– between pH 5–9 and νa(PO) of HPO42– and PO43– between pH 9–12. The deconvoluted peak contributions are plotted as a function of pH in Figure 1(d). It is important to note that it is difficult to obtain SEIRAS active films with the same enhancement factors, and therefore the ratios of peaks are taken into account to determine the pH during electrochemical experiments. The changes in the spectra of phosphate species were recorded upon sweeping the potential in the cathodic direction with a scan rate of 1 mV/s for CO2 purged phosphate solutions containing equimolar amounts of H2PO4– and of HPO42– (Figure 1(e)). The initial concentrations of H2PO4– and HPO42– were slightly changed upon purging CO2 due to the shift of the equilibrium toward H2PO4–. As the potential was scanned toward more negative potentials, the shift of the equilibrium toward HPO42– was apparent from the increase in the concentration of HPO42– (νs(PO), 990 cm–1) at the expense of H2PO4– (νa(PO), 1152 cm–1). Potentials more negative than −1.0 V vs RHE gave rise to the νa(PO) mode of PO4– as a shoulder around 1010 cm–1 indicating that the pH near the surface is above 9. The pH near the electrode surface was calculated for various potentials and current densities by correlating the ratio of phosphate peaks in the sample spectra to the calibration spectra after assuming the resultant spectra are the linear combination of the phosphate species.34
Figure 1.

(a) AFM image of sputtered Cu film on to Ge. (b) Schematic representation of the spectroelectrochemical cell used for in situ SEIRAS measurements. Reference electrode (RE) is Agl/AgCl and counter electrode (CE) is a graphite rod. (c) pH dependent SEIRA spectra of phosphate solutions indicating the dominating species. (d) Equilibrium of phosphate species constructed by absorption in intensity in SEIRA spectrum. (e) Potential dependent changes in the concentrations of phosphate species near the electrode surface.
Near surface concentrations of molecules during CO2 electroreduction can be approximated computationally by the Nernst–Planck equation.35,36 However, recent in situ SEIRAS measurements in bicarbonate solutions implied that physical measurements might be necessary to accurately quantify the local concentrations of molecules.34 For SEIRA active smooth surfaces, the concentration of molecules within the boundary layer were calculated as a function of current density by including diffusion, bubble induced convective mixing, and bulk reactions of buffer molecules in this study.35,37−39 The details of the simulation methodology are provided in the electronic Supporting Information.
The electric field leading to the surface enhancement effect on SEIRA active thin metal electrodes decays in very short distances (5–10 nm) from the surface when compared to the thickness of the diffuse layer (30–300 μm), which allows the ability to monitor local concentrations of species as schematically depicted in Figure 2(a).34,40 Considering the high concentrations of the electrolytes, infrared absorption from the bulk electrolyte can be significant in addition to the SEIRA effect. However, the metal films further dampen infrared light, and for copper films the penetration depth of the evanescent wave was approximated to not extend longer than 50 nm.41 The experimentally measured (Figure 2(b)) and simulated (Figure 2(c)) pH near the electrode surface is shown as a function of current density for different concentrations of phosphate buffers, 0.2, 0.5, and 1 M, respectively. In spite of the discrepancies between the experimental data and the simulation results, the overall agreement is reasonable, considering the assumptions and simplifications made during the simulation which will be discussed in the next section. Three distinct regions for the local pH as a function of current density were observed, which was more apparent for high electrolyte concentrations, including two buffering regions, indicated by plateaus in local pH, and a breakdown of the parent buffer solution (H2PO4–/HPO42–). In the first region of low current density, the buffer solutions sustained the local pH with a reasonable difference (<1.5) with respect to the bulk pH depending on the buffer capacity. Considering the fact that buffers are only effective in a certain range near the pKa, the steep increase in local pH between 8 and 10 can be seen as a result of the breakdown of the buffer. The current density where this was observed is particularly important because the steep increase in the pH in a very narrow window of current density will cause a huge overestimation of the kinetic overpotential. Strikingly, the current density where this happens was very low (<10 mA/cm2) for the commonly used 0.2 M phosphate buffer solution. In fact, the 0.2 M phosphate buffer barely showed a noticeable buffering region at all, and as the current density was increased the local pH also increased considerably fast.
Figure 2.

(a) Schematic representation of buffer reactions, pH gradient, and probed area SEIRA. (b) Experimentally measured and (c) simulated cathode surface pH as a function of phosphate buffer concentration and current density. (d) Current vs potential curve for different electrolyte concentrations (e) Experimentally measured cathode surface pH as a function of potential. All solutions are composed of equimolar H2PO4– and HPO42– mixtures. Shaded areas in b, c, e represent the buffering region that are relevant to local pH measurements.
Increasing the electrolyte concentration had a substantial effect on the buffering ability of the solution as anticipated. The buffer breakdown current density for 0.5 M and1M phosphate was around −30 and 75 mA/cm2, respectively, which is reasonably high for an electrode tested in CO2 saturated aqueous solution. However, the overall impact of the wider buffering region on the evaluation of the performance of electrocatalysts was remarkably dependent on the total current density at a specific potential. In Figure 2(d), the recorded current–potential curves during SEIRAS measurements are given for different electrolyte concentrations. All three electrolyte concentrations exhibited a similar current–potential profile until the current density was sufficient for breakdown of the particular buffer. The breakdown of each individual buffer leads to a plateau in the current–potential curves due to a sudden increase in the concentration overpotential. Please note that this region overlaps with the broad shoulder observed in phosphate buffers that was attributed to CO2 reduction to formate and/or CO on copper electrodes.42 More importantly, the rise in the current density at high potentials with increasing concentration of the buffer solution had a large impact on the buffering ability at a specific potential. This is more clearly reflected inFigure 2(e), where the local pH near the electrode surface is plotted as a function of the potential for different electrolyte concentrations. Remarkably, despite the large changes in the phosphate concentrations, the buffering ability at high potentials is limited due to the increase in the overall current density. The expansion of the buffer potential window with increasing electrolyte concentration was more evident between 0.2 and 0.5 M, while doubling the concentration from 0.5 to 1 M enlarged the potential window of the buffers only ∼50 mV. Nevertheless, high phosphate concentrations (>0.5M) exhibited an acceptable pH gradient at potentials between −0.7 to −1.0 V vs RHE, which partially overlap with the formation of hydrocarbons. In addition, even though all the electrodes attained an alkaline pH near the electrode surface at potentials more negative than −1 V vs RHE, the high buffer capacity solutions still had a slightly lower local pH.
CO2 concentrations near the electrode surface were quantified by monitoring the change in the CO2(aq) band intensity with respect to its initial intensity while sweeping the potential negatively (Figure S3). The experimentally measured (Figure 3(a)) and simulated (Figure 3(b)) CO2(aq) concentrations near the electrode surface were plotted as a function of current density for different concentrations of phosphate buffers, respectively. The initial decrease in the concentration of CO2 with increasing current density was the result of electrochemical consumption on the electrode surface following similar rates at lower current density (<5 mA/cm2), which was more apparent in the simulation results.26 More importantly, the consumption rate of CO2 changed dramatically at particular current densities for different electrolyte concentrations. At this specific current density, nearly all H2PO4– was converted to HPO42– (local pH ∼9) for all electrolyte concentrations, and thus the increase in the consumption rate of CO2 was result of the homogeneous reaction of CO2 with the cathodically produced hydroxide ions. This observation is also supported by the steady increase in (bi)carbonate concentrations near the electrode surface determined by the SEIRA spectra (Figure S4). The change in the consumption rate was more evident in the simulation results, most likely due to the constant faradaic efficiency assumed at different current densities. Nevertheless, experimental and theoretical results for different electrolyte concentrations exhibited very similar patterns as a function of current density. However, the apparent beneficial role of high buffer capacity solutions is again not reflected in the potential curves. In Figure 3(c), a change in the CO2(aq) concentration near the electrode surface is plotted against potential. The corresponding potential where CO2 begins to be consumed appreciably and the formation of adsorbed CO were in good agreement with the onset potentials of hydrocarbons (−0.75 V vs RHE) and CO (−0.45 V vs RHE) on polycrystalline copper (Figure S5). More importantly, similar to the local pH, the CO2 concentration near the electrode surface exhibited only a slight dependence on the electrolyte concentration against the applied potential when compared to plots against current density. This observation challenges the common presumption that increasing the buffer capacity leads to a higher CO2(aq) concentration near the electrode surface at the same applied potential.
Figure 3.

(a) Experimentally measured and (b) simulated cathode surface concentrations of CO2(aq) as a function of phosphate buffer concentration and current density. (c) Experimentally measured changes in CO2 concentration near the electrode surface as a function of potential. All solutions are composed of equimolar H2PO4– and HPO42– mixtures. CO2 concentrations are normalized to initial equilibrium concentrations.
In order to link the aforementioned results to the performance of copper electrodes, the CO2 reduction activity and selectivity of a sputtered flat copper electrode was evaluated. Additionally, a high surface area electrode composed of copper nanowires was tested to attempt to correlate the results obtained on flat copper surfaces to high surface electrodes. Scanning electron microscope (SEM) images of the sputtered and nanowire copper electrodes are shown in Figure 4(a) and 4(b), respectively. The roughness factors of the electrodes were found to be 2.4 and 55, respectively, by comparing to an electropolished copper surface (Figure S6). Due to the extensive bubbling of hydrogen causing a disruption of electrochemical experiments at high electrolyte concentrations, and a relatively lower impact of doubling the concentration from 0.5 to 1 M (see Figure 2(e)), lower electrolyte concentrations were used (0.1–0.5 M) in our experiments, which cover the commonly used ranges in the literature. The partial current density of CO2 electroreduction and H2 evolution for smooth and nanowire copper electrodes are shown in Figure 4(c,d). The incremental effect of the electrolyte concentration on the partial current density of HER was evident in a broad range of applied potentials for both smooth and nanowire electrodes, which is predominantly the result of a decrease in the concentration overpotential. However, the partial current density for CO2 reduction products exhibited a very small dependence on the electrolyte concentration and peaked between 10 and 12 mA/cm2 and 50 and 60 mA/cm2 on smooth and nanowire copper electrodes, respectively. This is most likely a result of mass transport limitations (Figure S7),23,43 in which the lowest concentration of the electrolyte (0.1 M) showed a slightly lower mass transfer limited current density. Although the CO2 solubility is lower at higher electrolyte concentrations, a slightly lower pH near the electrode surface and more extensive bubbling are considered to result in a slightly higher mass transfer limited current density for CO2 reduction. It is important to note that the mass transport limited current density, normalized to the number electrons transferred for the formation of each product, is highly dependent on the cell design, distribution of the CO2 gas and stirring efficiency.44 Although the electrolysis experiments were conducted in the same cell for both electrodes, the nanowire electrodes showed a 4–5-fold increase in mass transport limited current density. This is a result of enhanced bubble induced mass transport found on the nanowire electrodes. For instance, Burdyny et al. recorded a 4-fold increase in mass transport limited current density for CO2 electroreduction to CO when the morphology of gold electrodes was changed from nanoparticles to nanoneedles.37 Therefore, we underscore that maintaining the consistency in hydrodynamics of the electrochemical cells may not necessarily lead to the same mass transfer limited current density depending on the morphology of the electrodes.
Figure 4.
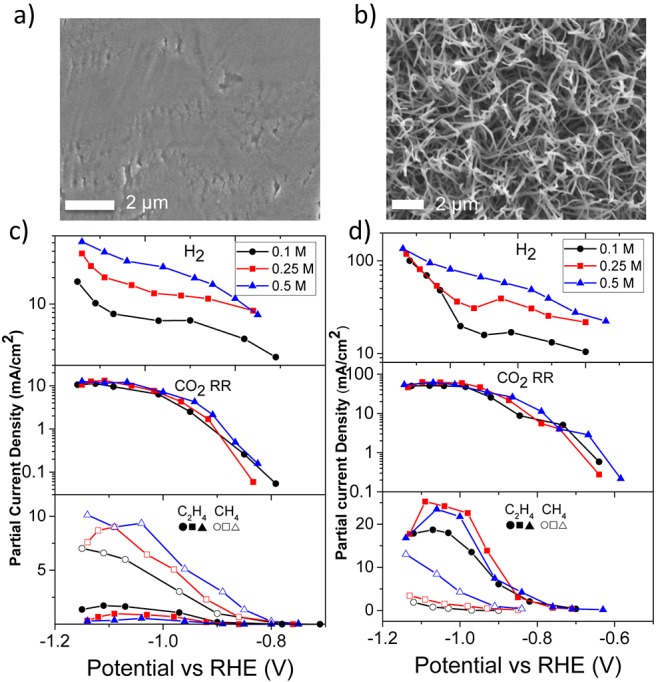
SEM image of the (a) sputtered and (b) nanowire copper electrodes. Partial current density of H2 and CO2 ethylene and methane as a function of potential and phosphate buffer concentration for (c) sputtered and (d) nanowire copper electrode.
The potential dependent partial current density and FE of the all gas and liquid products for both electrodes were measured, and are presented and discussed in the Supporting Information. The partial current density versus potential plots are considered to be a better representation of the activity because a change in the selectivity of products can be influenced dramatically by the changes in the rates of HER. For instance, a higher selectivity toward CO2 reduction products (∼72%) was obtained on the nanowire electrodes at low electrolyte concentrations when compared to the selectivity at high electrolyte concentrations (∼30%) at a potential of −0.95 V vs RHE in (Figure S8). Although this was found to be less prominent for sputtered copper electrodes, a change in selectivity toward CO2 reduction products from 40% to 20% was also recorded by increasing the electrolyte concentration at a potential of −1.05 V vs RHE (Figure S9). However, the increased selectivity at low electrolyte concentrations was largely a result of lower hydrogen production rather than an increase in the production rate of CO2 reduction, as manifested from the partial current density plots.
Moreover, we would like to highlight the suppression of methane on the nanowire electrodes compared to the sputtered electrodes, which has led to a longstanding debate in the literature.7,23 The onset potential of methane on sputtered copper surfaces was around −0.85 V vs RHE, while the onset potential was observed around −1.1 V vs RHE at about at the same electrolyte concentration (0.1 M) on nanowire electrodes, as shown in Figure 4c and 4d. A high concentration of phosphate buffer and very negative potentials (−1.1 V) were necessary to produce appreciable amounts of methane with respect to ethylene on rough copper surfaces. Therefore, ethylene formation, along with other C2 and C3 products (Figure S10), surpassed methane formation on rough copper surfaces at potentials between −0.75 and −1.0 V vs RHE in contrast to smooth copper electrodes where methane formation was dominant.
Discussion
On the basis of the above experimental and computational results, in this section we discuss the correlation between these results and the insights that they provide in conjunction with previous studies for understanding, interpreting, and implementing electrochemical CO2 reduction. First, bicarbonate is by far the most commonly used electrolyte in aqueous electrochemical and is known to have lower buffer capacity than the phosphate buffers used in this study.26,32,45 For instance, the local pH in 0.5 M NaHCO3 solution on a gold electrode was measured to be around 9 at ∼5 mA/cm2,29 while the same local pH was recorded around 40 mA/cm2 for the 0.5 M phosphate buffer in this study. In spite of this fact, we explicitly showed that the pH near the electrode surface can be dramatically different from the bulk solution (>5 pH units) in “well buffered” phosphate solutions (0.2 M) at potentials that are relevant to the formation of hydrocarbons (−0.9 V) and at current densities <20 mA/cm2, which can be considered as very low for high surface area copper electrodes.46 This clearly demonstrates that simply switching the buffer employed from bicarbonate to phosphate solutions to examine the effect of local pH at high potentials and/or high currents is not a straightforward method and most of the previous studies in aqueous electrochemical CO2 reduction on copper electrodes in H-cells are taking place under mass transport limitations of protons. Increasing the buffer capacity of the electrolyte up to 1 M provided a great improvement on the buffer breakdown current density; however, the potential window that can be tested with a minimal pH gradient only slightly improved due to the increase in the rate of HER at the same applied potential. In other words, the increase in the buffer capacity of the solution promotes the development of a relatively lower local pH that correspondingly increases the rate of HER. Subsequently, higher current densities achieved in the same applied potential leads to only a minor improvement in the potential ranges that can be tested with a minimal concentration gradient.
Simulation results indicated that the higher local pH is predominantly governed by slow transport of buffer ions and a fast release of hydroxide ions from HER and CO2 electroreduction. An exact match between the experimental and simulation was not obtained due to the potential dependent and broad distribution of CO2 reduction products, which complicates the bubble induced mass transport model. Any rigorous evaluation of the accuracy of electrochemical models should be performed in CO2 free phosphate (or bicarbonate) buffer solutions by assuming near unity FE for hydrogen and avoiding bicarbonate formation due to CO2 purging and breakdown of the buffer. In our work, we focus on the product formation trends and their implications in CO2 electroreduction. Nevertheless, the simulations performed remarkably well on predicting the trends and relative magnitudes of the local pH in different electrolyte concentrations, though there are small discrepancies between the experimental and simulation results especially after the breakdown of the parent phosphate buffer (H2PO4–/HPO42–). At concentrations of 0.2 and 0.5 M of the phosphate buffer, the simulation slightly deviated from the measured local pH with an average of 0.57 and 0.43 pH units, respectively. One of the reasons might be the formation and accumulation of bicarbonate during the slow voltammetry scan, which contributed to the buffer capacity of the solution at lower electrolyte concentrations, while during simulations galvanostatic steps were applied. Interestingly, when all the H2PO4– was converted to HPO42–, OH– ions reacted simultaneously with CO2, bicarbonate and HPO42–. Therefore, any buffer performing better than HPO42–/PO43– might reduce the concentration overpotential due to diminishing CO2(aq) concentrations near the electrode surface. Using a 1 M concentration of phosphate buffer, the simulation results mostly underestimated the local pH with an average of 0.47 pH units in contrast to lower concentrations. This is most likely a result of the assumptions made during the bubble induced mass transport model, i.e., constant bubble coverage and bubble departure diameter. It is important to note that this model drastically overestimates the local pH at high current densities, i.e., underestimates the buffer capacity, in the absence of bubble induced convection even after the addition of convective mass transport via stirring (Figure S11). Therefore, it is highly important to take into account the bubble induced mass transport while performing simulations at high current densities. However, the mass transport models for bubble induced convection are highly dependent on the departure diameter and coverage of the bubbles.38 Small changes in bubble departure diameter and coverages make a noticeable change in the simulated values, especially after the breakdown of the buffer (Figure S12).
The production of SEIRA active films that are more analogous to the nanowire electrodes is restricted by the percolation limit of the metal films which allows only very thin films to be studied, typically below 100 nm.47 While a direct quantitative comparison of the near surface concentrations of molecules between the sputtered Cu films and copper nanowire electrodes is not possible, a qualitative comparison of the two different electrodes revealed striking correlations. The alkaline pH near the electrode surface at potentials relevant to the formation of hydrocarbons was clearly observed by SEIRAS experiments on sputtered copper electrodes. Since the local current density on the nanowire electrodes is much higher due to their large electrochemically active surface area (ECSA), it is anticipated that the local pH is higher on the surface of these electrodes even in the presence of highly improved mass transport from the bubbles. This is indirectly evident from the dramatic increase in the partial current density of hydrogen with increasing electrolyte concentration (Figure 4(d)). Moreover, due to the 3-D structure of the nanowire electrodes, a gradient of CO2 and pH likely exists along the catalyst layer.39 Subsequently, the production rate of CO2 products likely to vary along the catalyst layer and normalizing the measured activity by ECSA is an average of catalytic activity containing partially exploited and nonexploited areas. Moreover, depending on the porosity and thickness of the catalyst layer, the activation controlled region might be very narrow as charge transfer limited and mass transport limited regions are convoluted and hard to differentiate (Figures S7 and S13). These types of comparisons might be potentially misleading especially when the catalyst layer is too thick and/or CO2 electroreduction takes place via pH dependent pathways. Similarly, using Tafel analysis to verify reaction mechanisms is only appropriate under activation controlled potentials where the impact of mass transfer limitations on the measured rates is minimized. Considering the concentration gradient along the catalyst layer and a relatively low current density for the breakdown of commonly used buffer solutions, this type of analysis must be done with great care. The implication of the poor mass transport on the kinetic analysis of CO2 electroreduction catalysts has been thoroughly discussed recently by Dunwell et al.25
The improved selectivity toward C2 and/or C3 products has been shown numerous times on roughened copper electrodes, regardless of the preparation method and morphology.23 Therefore, suppression of methane on these surfaces has led to some debate over whether there are undercoordinated active sites48,49 or as a result of a change in local conditions, e.g., local pH, CO coverage and readsorption.21,50,51 Although higher local pH itself is a result of poor mass transfer and/or buffer actions, for some products the electron and proton transfer is decoupled during the rate-determining step. For instance, the pH dependent and independent formation of methane and ethylene were recognized in the early studies of Hori.17,26 Therefore, the rate of formation of ethylene is essentially blind to the pH changes near the electrode surface, unless the alkalinity leads to a concentration overpotential by the reaction between OH– and CO2(aq). Since this nonelectrochemical reaction is typically slow, it can be minimized by having favorable mass transport to the electrode surface.25 In addition, although the convective forces are less prominent inside porous structures, this reaction most likely induces less concentration overpotentials inside the pores as well, when compared to pH induced concentration overpotentials, due to higher diffusion coefficient of CO2(aq) than buffer anions. Moreover, CO coverage, and hence competition between *CO and *H, has been identified recently as a potential parameter to sustain ethylene formation over a broad range of potentials, which still requires more independent studies.50,52 However, it is still not clear which factor plays the primary role in the suppression of methane and the formation of C2 compounds in a broad range of potentials. Nevertheless, we have shown that a high buffer capacity solution of 0.5 M phosphate was necessary to initiate an appreciable amount of methane formation on nanowire electrodes, while solutions with much lower buffer capacity were usually employed in the literature to test high surface area electrodes.23 The onset potential of methane is sometimes shifted to more negative potentials and often not even observed (depending also on the tested potential region and hydrodynamics of the cell) when low buffer capacity solutions are used to test electrodes with high local current density.
Conclusions
Local concentration of molecules during CO2 electroreduction was physically measured by SEIRAS and compared with the widely used mass transport simulations. The slightly extended mass transport model used in this study was quantitative enough to account for the relative magnitudes of what is recorded by SEIRAS, and prescriptive enough to predict new experiments when bubble induced mass transport effects were included. SEIRAS measurements provided crucial additional information as a function applied potential in contrast to simulations results, which usually give information only as a function current density. Both experimental and calculation results strongly suggested that immense concentration gradients exist between the electrode surface and bulk in neutral solutions during CO2 electroreduction to hydrocarbons on copper electrodes, which is not able to contend with high buffer capacity solutions (1 M phosphate buffer). The resulting local pH on the surface of flat sputtered copper electrodes in densely buffered electrolytes were much higher than initially anticipated. Remarkably, even though increasing electrolyte concentration has a substantial effect on the buffering capacity of the solutions as a function of current density, a dramatic increase in HER resulted in only a slight improvement on the potential window that can be tested with minimal pH and CO2(aq) gradients. It is important to reemphasize that although the morphology of the catalyst can provide significant changes in the activity and selectivity of the electrocatalysts, the distinct contributions of intrinsic activity and the local environment are currently far from being resolved. Considering that electrolysis in CO2 saturated aqueous solutions cannot attain commercially viable rates of production at ambient pressure, evaluation of intrinsic activity and extraction of kinetic parameters in the absence of mass transport limitations should be the primary goal, which can be potentially transferred to other type of electrochemical cell designs and practical applications.
Methods
Preparation of SEIRA Active Films
Twenty-five nm thick copper thin films were deposited onto a 60° bevelled germanium internal reflection element (IRE) by magnetron sputtering deposition. Although silicon is a better choice in terms of electrochemical stability, the transparency of silicon IRE does not allow measurements below 1000 cm–1, which includes the crucial phosphate bands. Nevertheless, Ge IREs were suitable for this study due to the range of applied cathodic potentials that were used.2 Before sputtering, Ge IRE was mirror polished by using 1.0, 0.3, and 0.05 μm grain sized alumina paste, respectively, which was followed by thoroughly rinsing with isopropyl alcohol and water. The IRE was transferred into the sputtering chamber in which a pressure of 1.8 × 10–7 mbar was maintained. Then it was placed to a suitable working distance and rotated at a velocity of 15°/s to enable a uniform deposition of copper. Following this, argon was allowed to flow through the sputtering system maintaining a pressure of 3 μbar, and copper was sputtered using a plasma power of 20 W, which resulted in a growth rate approximately 0.04 nm s–1. The sputtering process was performed for 10 min to produce 25 nm thick copper films that resulted in a mirror finish similar to that of the germanium IRE. Side to side resistance of the films along the 2 cm wide IRE was measured after the sputtering and was found to be typically between 25 and 35 Ω.
Spectroelectrochemical Measurements
All spectroelectrochemical measurements were performed in a custom-made modular cell schematically depicted in Figure 1b. Copper thin films sputtered on a Ge internal reflection element served as the cathode/working electrode. A Ag/AgCl (BASI) reference electrode in 3 M NaCl solution was used as the reference electrode, while the counter electrode was a graphite rod. Upon assembly, the cell was placed in a Bruker Vertex 70 spectrometer equipped with a liquid nitrogen cooled LN-MCT detector and a Veemax III ATR accessory. The spectrometer and sample compartment were purged with nitrogen (5.0 grade) for 30 min to remove atmospheric water vapor and CO2. All electrochemical measurements were performed using a BioLogic SP-200 potentiostat. In order to obtain an appreciable signal, for all Cu thin films, an activation routine was applied consisting of three voltammetry cycles between 0.5 and −0.4 V vs RHE in 0.1 M NaCl. After activation of the surface, the spectroelectrochemical cell was rinsed thoroughly with ultrapure water and then a background spectrum was collected in electrolyte free water solutions. Voltammetry measurements were performed between 0 and −1.25 V vs RHE with a scan rate of 1 mV/s in various concentrations of CO2 saturated phosphate buffers prepared from K2HPO4 and KH2PO4 salts (Sigma, ACS reagent grade). Simultaneously, IR spectra were collected in Kretschmann configuration at a resolution of 4 cm–1 and 32 coadded scans were recorded for each 10 mV. Solution resistance was measured by electrochemical impedance spectroscopy and extracted from a high frequency intercept of the Nyquist plot in the frequency range from 100 kHz to 10 Hz (Figure S14). All the potentials were corrected for IR losses after the experiments.
Preparation of Copper Electrodes
Cu nanowires were synthesized on a Cu wire (99.9%, 1 mm diameter, Sigma-Aldrich) and used as cathodes/working electrodes. The Cu wires were cleaned with acetone and ethanol in an ultrasonicator, each for 5 min separately, then rinsed with deionized water several times and dried with nitrogen. Cu(OH)2 nanowires were first fabricated on the copper wire substrates using a wet chemical method. The cleaned copper substrates were immersed into a solution mixture containing 0.133 M (NH4)2S2O8 (98%, Sigma-Aldrich) and 2.667 M NaOH (99.99%, Alfa Aesar) for 10 min. The color of wires changed from reddish brown to blue, indicating the formation of Cu(OH)2 on the surface of Cu wire substrate. After 10 min, the Cu wires were taken out of the solution, thoroughly cleaned with deionized water and dried with nitrogen. Finally, Cu wires were placed into a tube oven at 180 °C for 2 h in air flow to form CuO nanowires. Smooth Cu electrodes were fabricated by magnetron sputtering deposition (AJA International Inc.)100 nm thick Cu on Ti substrate (99.5%, Alfa Aesar) with the rate of 0.14 nm/s using 50 W DC power supply. The morphologies of catalysts before and after electrochemical CO2 reduction were characterized using scanning electron microscope (Figure S15) (SEM, JEOL, JSM-6010LA).
CO2 Electrolysis and Analysis
Electrochemical CO2 reduction measurements were conducted in a custom-made two-compartment H type of cell (Figure S16). The Cu electrodes were placed in the cathodic compartment (12 mL of electrolyte, ∼3 mL headspace) close to the Ag/AgCl reference electrode and served as the working electrode. A Pt mesh was used as the counter electrode and positioned in the anodic compartment, which was separated from the cathodic part by a Nafion-115 proton exchange membrane. Different concentrations of phosphate buffer (0.1, 0.25, and 0.5 M, KH2PO4/K2HPO4 = 1:1) were used as the electrolytes. Electrolytes were saturated with CO2 by purging CO2 gas into the solutions for at least 4 h. During electrochemical CO2 reduction tests, CO2 was continuously purged into the catholyte and anolyte, both at a rate of 10–20 sccm through mass flow controllers (Bronkhorst High-Tech). A PARSTAT 4000A potentiostat was used for all electrolysis experiments. Before electrochemical measurements, a potential of −0.8 V vs Ag/AgCl was applied to reduce the CuO nanowires to Cu nanowires (Figure S17). All potentials reported in this work were IR corrected and converted to the reversible hydrogen electrode (RHE) for convenience. Each electrochemical measurement was conducted over a time span of 2200s by applying a constant current under 1000 rpm stirring with a magnetic bar. During the experiments, gas products were collected and quantified by using online gas chromatography (GC) at 3 min intervals (Compact GC 4.0, GAS) (Figure S18). An average of six measurements were used unless any deactivation was observed, which was the case typically at high potentials (Figure S19). In such cases, the faradaic efficiency of H2 increased gradually while the FE of C2H4 decreased as potential became less negative. After experiments, 5 mL liquid products were collected for nuclear magnetic resonance (NMR 400 MHz, Agilent) and high performance liquid chromatography (HPLC, Agilent 1260 Infinity) analysis. In a typical NMR measurement, 540 μm catholyte was mixed with 60 μm D2O (99.9%, Sigma-Aldrich) containing 5 mM DMSO (99.7%, Sigma-Aldrich) as internal standard. 1H NMR spectrum was measured with 2 s presaturation delay, 2 s relaxation delay, 2 s acquisition time and water suppression. 50 μL liquid sample was injected to HPLC apparatus equipped with Hi-Plex H column with 0.02 M H2SO4 mobile phase. The surface roughness factor of the electrodes was determined by measuring the double layer capacitance of the Cu nanowires. Cyclic voltammograms with different scan rates between 0.05 to 0.2 V (vs RHE) were performed in a nonfaradaic region in 0.1 M NaClO4 solution (Figure S6). The capacitance was obtained by plotting the geometric current density against the scan rate of the CVs. The surface roughness factor was calculated by comparing this capacitance value to that of a reported smooth polycrystalline Cu electrode.53
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 759743 – WUTANG). We thank Sanjana Chandrashekar and Dr. Nathan Nesbitt for AFM measurements. We would like to thank Dr. Tom Burdyny for fruitful discussions and Herman Schreuders for technical support during the sputtering process.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07000.
Complete simulation and experimental details, electrochemical and nonelectrochemical characterization of the electrodes, and additional discussion on selectivity (PDF)
Author Contributions
† KY and RK contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Robinson M.; Shine T. Achieving a climate justice pathway to 1.5 C. Nat. Clim. Change 2018, 8 (7), 564. 10.1038/s41558-018-0189-7. [DOI] [Google Scholar]
- Kondratenko E. V.; Mul G.; Baltrusaitis J.; Larrazábal G. O.; Pérez-Ramírez J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6 (11), 3112–3135. 10.1039/c3ee41272e. [DOI] [Google Scholar]
- Zhu D. D.; Liu J. L.; Qiao S. Z. Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 2016, 28 (18), 3423–3452. 10.1002/adma.201504766. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhao Z. J.; Gong J. Nanostructured materials for heterogeneous electrocatalytic CO2 reduction and their related reaction mechanisms. Angew. Chem., Int. Ed. 2017, 56 (38), 11326–11353. 10.1002/anie.201612214. [DOI] [PubMed] [Google Scholar]
- Higgins D.; Hahn C.; Xiang C.; Jaramillo T. F.; Weber A. Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Letters 2019, 4 (1), 317–324. 10.1021/acsenergylett.8b02035. [DOI] [Google Scholar]
- Burdyny T.; Smith W. A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12 (5), 1442–1453. 10.1039/C8EE03134G. [DOI] [Google Scholar]
- Pander J. E. III; Ren D.; Huang Y.; Loo N. W. X.; Hong S. H. L.; Yeo B. S. Understanding the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide on Oxide-Derived Catalysts. ChemElectroChem 2018, 5 (2), 219–237. 10.1002/celc.201701100. [DOI] [Google Scholar]
- Li Y.; Cui F.; Ross M. B.; Kim D.; Sun Y.; Yang P. Structure-sensitive CO2 electroreduction to hydrocarbons on ultrathin 5-fold twinned copper nanowires. Nano Lett. 2017, 17 (2), 1312–1317. 10.1021/acs.nanolett.6b05287. [DOI] [PubMed] [Google Scholar]
- Lum Y.; Ager J. W. Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction. Nat. Catal. 2019, 2, 86. 10.1038/s41929-018-0201-7. [DOI] [Google Scholar]
- De Luna P.; Quintero-Bermudez R.; Dinh C.-T.; Ross M. B.; Bushuyev O. S.; Todorović P.; Regier T.; Kelley S. O.; Yang P.; Sargent E. H. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nature Catalysis 2018, 1 (2), 103. 10.1038/s41929-017-0018-9. [DOI] [Google Scholar]
- Hoang T. T.; Ma S.; Gold J. I.; Kenis P. J.; Gewirth A. A. Nanoporous copper films by additive-controlled electrodeposition: CO2 reduction catalysis. ACS Catal. 2017, 7 (5), 3313–3321. 10.1021/acscatal.6b03613. [DOI] [Google Scholar]
- Pang Y.; Burdyny T.; Dinh C.-T.; Kibria M. G.; Fan J. Z.; Liu M.; Sargent E. H.; Sinton D. Joint tuning of nanostructured Cu-oxide morphology and local electrolyte programs high-rate CO2 reduction to C 2 H 4. Green Chem. 2017, 19 (17), 4023–4030. 10.1039/C7GC01677H. [DOI] [Google Scholar]
- Ma M.; Djanashvili K.; Smith W. A. Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem. 2016, 128 (23), 6792–6796. 10.1002/ange.201601282. [DOI] [PubMed] [Google Scholar]
- Liu X.; Schlexer P.; Xiao J.; Ji Y.; Wang L.; Sandberg R. B.; Tang M.; Brown K. S.; Peng H.; Ringe S.; et al. pH effects on the electrochemical reduction of CO (2) towards C 2 products on stepped copper. Nat. Commun. 2019, 10 (1), 32. 10.1038/s41467-018-07970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kas R.; Kortlever R.; Milbrat A.; Koper M. T.; Mul G.; Baltrusaitis J. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16 (24), 12194–12201. 10.1039/C4CP01520G. [DOI] [PubMed] [Google Scholar]
- Schouten K. J. P.; Qin Z.; Pérez Gallent E.; Koper M. T. Two pathways for the formation of ethylene in CO reduction on single-crystal copper electrodes. J. Am. Chem. Soc. 2012, 134 (24), 9864–9867. 10.1021/ja302668n. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Takahashi R.; Yoshinami Y.; Murata A. Electrochemical reduction of CO at a copper electrode. J. Phys. Chem. B 1997, 101 (36), 7075–7081. 10.1021/jp970284i. [DOI] [Google Scholar]
- Varela A. S.; Kroschel M.; Reier T.; Strasser P. Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH. Catal. Today 2016, 260, 8–13. 10.1016/j.cattod.2015.06.009. [DOI] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (32), E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten K. J. P.; Perez Gallent E.; Koper M. T.M. The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes. J. Electroanal. Chem. 2014, 716, 53–57. 10.1016/j.jelechem.2013.08.033. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Yılmaz H.; Koper M. T.; Mul G. Manipulating the hydrocarbon selectivity of copper nanoparticles in CO2 electroreduction by process conditions. ChemElectroChem 2015, 2 (3), 354–358. 10.1002/celc.201402373. [DOI] [Google Scholar]
- Dinh C.-T.; Burdyny T.; Kibria M. G.; Seifitokaldani A.; Gabardo C. M.; Garcia de Arquer F. P.; Kiani A.; Edwards J. P.; De Luna P.; Bushuyev O. S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360 (6390), 783–787. 10.1126/science.aas9100. [DOI] [PubMed] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E.; Chan K.; Hahn C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119 (12), 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Lee S.; Ocon J. D.; Son Y.-i.; Lee J. Alkaline CO2 electrolysis toward selective and continuous HCOO–production over SnO2 nanocatalysts. J. Phys. Chem. C 2015, 119 (9), 4884–4890. 10.1021/jp512436w. [DOI] [Google Scholar]
- Dunwell M.; Luc W.; Yan Y.; Jiao F.; Xu B. Understanding surface-mediated electrochemical reactions: CO2 reduction and beyond. ACS Catal. 2018, 8 (9), 8121–8129. 10.1021/acscatal.8b02181. [DOI] [Google Scholar]
- Hori Y. I., Electrochemical CO2 reduction on metal electrodes. In Modern aspects of electrochemistry; Springer: 2008; pp 89–189. [Google Scholar]
- Gattrell M.; Gupta N.; Co A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594 (1), 1–19. 10.1016/j.jelechem.2006.05.013. [DOI] [Google Scholar]
- Ryu J.; Wuttig A.; Surendranath Y. Quantification of Interfacial pH Variation at Molecular Length Scales Using a Concurrent Non-Faradaic Reaction. Angew. Chem., Int. Ed. 2018, 57 (30), 9300–9304. 10.1002/anie.201802756. [DOI] [PubMed] [Google Scholar]
- Dunwell M.; Yang X.; Setzler B. P.; Anibal J.; Yan Y.; Xu B. Examination of Near-Electrode Concentration Gradients and Kinetic Impacts on the Electrochemical Reduction of CO2 using Surface-Enhanced Infrared Spectroscopy. ACS Catal. 2018, 8 (5), 3999–4008. 10.1021/acscatal.8b01032. [DOI] [Google Scholar]
- Yang K. D.; Ko W. R.; Lee J. H.; Kim S. J.; Lee H.; Lee M. H.; Nam K. T. Morphology-directed selective production of ethylene or ethane from CO2 on a Cu mesopore electrode. Angew. Chem., Int. Ed. 2017, 56 (3), 796–800. 10.1002/anie.201610432. [DOI] [PubMed] [Google Scholar]
- Handoko A. D.; Wei F.; Yeo B. S.; Seh Z. W. Understanding heterogeneous electrocatalytic carbon dioxide reduction through operando techniques. Nat. Catal. 2018, 1 (12), 922. 10.1038/s41929-018-0182-6. [DOI] [Google Scholar]
- Ren D.; Deng Y.; Handoko A. D.; Chen C. S.; Malkhandi S.; Yeo B. S. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper (I) oxide catalysts. ACS Catal. 2015, 5 (5), 2814–2821. 10.1021/cs502128q. [DOI] [Google Scholar]
- Klähn M.; Mathias G.; Kötting C.; Nonella M.; Schlitter J.; Gerwert K.; Tavan P. IR spectra of phosphate ions in aqueous solution: predictions of a DFT/MM approach compared with observations. J. Phys. Chem. A 2004, 108 (29), 6186–6194. 10.1021/jp048617g. [DOI] [Google Scholar]
- Ayemoba O.; Cuesta A. Spectroscopic Evidence of Size-Dependent Buffering of Interfacial pH by Cation Hydrolysis during CO2 Electroreduction. ACS Appl. Mater. Interfaces 2017, 9 (33), 27377–27382. 10.1021/acsami.7b07351. [DOI] [PubMed] [Google Scholar]
- Gupta N.; Gattrell M.; MacDougall B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2006, 36 (2), 161–172. 10.1007/s10800-005-9058-y. [DOI] [Google Scholar]
- Hashiba H.; Weng L.-C.; Chen Y.; Sato H. K.; Yotsuhashi S.; Xiang C.; Weber A. Z. Effects of Electrolyte Buffer Capacity on Surface Reactant Species and the Reaction Rate of CO2 in Electrochemical CO2 Reduction. J. Phys. Chem. C 2018, 122 (7), 3719–3726. 10.1021/acs.jpcc.7b11316. [DOI] [Google Scholar]
- Burdyny T.; Graham P. J.; Pang Y.; Dinh C.-T.; Liu M.; Sargent E. H.; Sinton D. Nanomorphology-enhanced gas-evolution intensifies CO2 reduction electrochemistry. ACS Sustainable Chem. Eng. 2017, 5 (5), 4031–4040. 10.1021/acssuschemeng.7b00023. [DOI] [Google Scholar]
- Vogt H.; Stephan K. Local microprocesses at gas-evolving electrodes and their influence on mass transfer. Electrochim. Acta 2015, 155, 348–356. 10.1016/j.electacta.2015.01.008. [DOI] [Google Scholar]
- Raciti D.; Mao M.; Wang C. Mass transport modelling for the electroreduction of CO2 on Cu nanowires. Nanotechnology 2018, 29 (4), 044001 10.1088/1361-6528/aa9bd7. [DOI] [PubMed] [Google Scholar]
- Osawa M. Dynamic processes in electrochemical reactions studied by surface-enhanced infrared absorption spectroscopy (SEIRAS). Bull. Chem. Soc. Jpn. 1997, 70 (12), 2861–2880. 10.1246/bcsj.70.2861. [DOI] [Google Scholar]
- Christensen P.; Hamnett A.. In-situ Infrared Studies of the Electrode–Electrolyte Interface. In Comprehensive Chemical Kinetics; Elsevier, 1989; Vol. 29, pp 1–77. [Google Scholar]
- Kortlever R.; Tan K.; Kwon Y.; Koper M. Electrochemical carbon dioxide and bicarbonate reduction on copper in weakly alkaline media. J. Solid State Electrochem. 2013, 17 (7), 1843–1849. 10.1007/s10008-013-2100-9. [DOI] [Google Scholar]
- Resasco J.; Chen L. D.; Clark E.; Tsai C.; Hahn C.; Jaramillo T. F.; Chan K.; Bell A. T. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 2017, 139 (32), 11277–11287. 10.1021/jacs.7b06765. [DOI] [PubMed] [Google Scholar]
- Lobaccaro P.; Singh M. R.; Clark E. L.; Kwon Y.; Bell A. T.; Ager J. W. Effects of temperature and gas–liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalysts. Phys. Chem. Chem. Phys. 2016, 18 (38), 26777–26785. 10.1039/C6CP05287H. [DOI] [PubMed] [Google Scholar]
- Resasco J.; Lum Y.; Clark E.; Zeledon J. Z.; Bell A. T. Effects of Anion Identity and Concentration on Electrochemical Reduction of CO2. ChemElectroChem 2018, 5 (7), 1064–1072. 10.1002/celc.201701316. [DOI] [Google Scholar]
- Ren D.; Fong J.; Yeo B. S. The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction. Nat. Commun. 2018, 9 (1), 925. 10.1038/s41467-018-03286-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-T.; Zhou Z.-Y.; Broadwell I.; Sun S.-G. In-situ infrared spectroscopic studies of electrochemical energy conversion and storage. Acc. Chem. Res. 2012, 45 (4), 485–494. 10.1021/ar200215t. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jiang K.; Fan S.; Kanan M. W. A direct grain-boundary-activity correlation for CO electroreduction on Cu nanoparticles. ACS Cent. Sci. 2016, 2 (3), 169–174. 10.1021/acscentsci.6b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaguer-Casadevall A.; Li C. W.; Johansson T. P.; Scott S. B.; McKeown J. T.; Kumar M.; Stephens I. E.; Kanan M. W.; Chorkendorff I. Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts. J. Am. Chem. Soc. 2015, 137 (31), 9808–9811. 10.1021/jacs.5b06227. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Handoko A. D.; Hirunsit P.; Yeo B. S. Electrochemical reduction of CO2 using copper single-crystal surfaces: effects of CO* coverage on the selective formation of ethylene. ACS Catal. 2017, 7 (3), 1749–1756. 10.1021/acscatal.6b03147. [DOI] [Google Scholar]
- Wang X.; Varela A. S.; Bergmann A.; Kühl S.; Strasser P. Catalyst particle density controls hydrocarbon product selectivity in CO2 Electroreduction on CuOx. ChemSusChem 2017, 10 (22), 4642–4649. 10.1002/cssc.201701179. [DOI] [PubMed] [Google Scholar]
- Schreier M.; Yoon Y.; Jackson M. N.; Surendranath Y. Competition between H and CO for Active Sites Governs Copper-Mediated Electrosynthesis of Hydrocarbon Fuels. Angew. Chem., Int. Ed. 2018, 57 (32), 10221–10225. 10.1002/anie.201806051. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Kanan M. W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134 (17), 7231–7234. 10.1021/ja3010978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.