

Abstract
Background
Irritable bowel syndrome (IBS) is a chronic and debilitating functional gastrointestinal disorder affecting 9%–23% of the population across the world. The relative efficacy of fecal microbiota transplantation (FMT) on IBS symptoms was demonstrated in a double-blind, randomized study.
Methods
We describe the case of a 73-year-old woman suffering from IBS (abdominal pain, bloating, and abundant and disabling diarrhea, with 10–15 stools a day) and repetitive urinary tract infection (UTI; 5 episodes in 6 months, including 3 the last 2 months) for several years, generating an impaired quality of life. She received an FMT with 400 mL of fecal infusion from a healthy donor via a nasogastric tube after bowel lavage. Her digestive microbiota was analyzed using culturomic and metagenomic targeting 16S rRNA sequencing methods.
Results
Eight months after transplantation, we observed a significant reduction in frequency and improvement in stool consistency (3–4 molded stools a day against 10–15 before the transplant) and no recurrence of urinary infection (as previously reported). Using culturomics, we found 12 bacteria present in the fecal infusion and post-transplant stool; these were absent pretransplant. Three of them (Intestinimonas massiliensis, Oscillibacter massiliensis, and Provencibacter massiliensis) were previously discovered and cultivated in our laboratory using culturomics. Using metagenomics, we also observed 12 bacteria, different from those observed during culture, that could have been transferred to the patient by FMT.
Conclusions
In this case report, IBS symptoms and UTI frequency decreased after FMT UTI. Further studies involving more patients would be relevant to confirm this work and develop bacteriotherapy.
Keywords: culturomics, fecal microbiota transplantation, irritable bowel syndrome, metagenomics, recurrent urinary tract infection
Irritable bowel syndrome (IBS) is the most common functional bowel disorder, affecting 9%–23% of the world population, and is characterized by abdominal pain or discomfort associated with changes in bowel habits and stool characteristics. The etiology of IBS is multifactorial, and the pathophysiology is not completely understood. Nevertheless, several studies over the past few years have reported qualitative and quantitative alterations in the intestinal microbiota of patients with IBS [1]. Tap et al. found that the severity of IBS symptoms was negatively associated with microbial richness, exhaled CH4, the presence of methanogens, and enterotypes enriched with Clostridiales or Prevotella species [2]. Pozuelo et al. found a reduction of butyrate-producing bacteria in patients with IBS [3]. A number of therapeutic approaches have been described in an attempt to modulate the microbiota in IBS, such as nonabsorbable antibiotics (neomycin and rifaximin) and other therapies, including dietary modification, probiotics, prebiotics, and fecal microbiota transplantation [4].
Fecal microbiota transplantation (FMT), the change of the gut microbiota of a patient after transplantation of stool from a healthy donor, has been successfully used to treat recurrent [5], refractory [6], and severe cases of Clostridium difficile infections [7, 8]. Multiple other diseases, associated with Western lifestyles, including especially IBS, constipation, inflammatory bowel disease, neurological diseases, cardiovascular diseases, obesity, the metabolic syndrome, autoimmunity, asthma, and allergic diseases, have taken on epidemic proportions in recent years due to the potential association with the gut microbiota [9]. This has resulted in speculation that FMT may eventually be beneficial in other conditions associated with dysbiosis, such as metabolic syndrome, obesity, food allergies, IBD, and especially IBS [10]. Indeed, Johnsen et al. showed its relative efficacy with a double-blind randomized, placebo-controlled study of patients with moderate to severe irritable bowel syndrome (decreased symptoms for 65% of patients [36/55] with FMT vs 43% with placebo [12/28] at 3 months) [11]. Moreover, in a single-center study in the United States, the authors observed that 70% of patients experienced resolution or improvement of symptoms after FMT, especially those with abdominal pain, dyspepsia, bloating, and flatus [12]. Moreover, FMT resulted in the resolution of recurrent Clostridium difficile, a significant decrease in the frequency of recurrent urinary tract infections (UTIs), and an improvement in the antibiotic susceptibility profile of organisms causing UTIs [13]. Over the past 10 years, most gut microbiota analysis after FMT have been carried out using metagenomic approaches, which allow a relatively rapid assessment of the microbial composition by high-throughput sequencing. More recently, culturomics has emerged as a new approach for the study of complex microbial ecosystems, such as the human intestinal tract, with the potential to detect minority populations and provide information on the viability of detected microorganisms [14, 15]. Indeed, we describe here the case of a patient with irritable bowel syndrome and repetitive urinary tract infections who received an FMT, and we analyzed her digestive microbiota by culturomic and metagenomic targeting 16S rRNA sequencing methods.
METHODS
Case Presentation
We report the case of a 73-year-old woman who presented with the following medical history: 2 vaginal deliveries, hypothyroidism, Meniere's disease, Helicobacter pylori gastritis, depressive syndrome, hemorrhoid sclerosis, and cystopexy. She reported having irritable bowel syndrome, with abdominal pain, bloating, and transit disorders with a capricious evolution, alternating nonbloody liquid diarrhea and uncontrolled molded stools for several years, with an average of 10–15 stools a day associated with anal incontinence. In addition, she reported very poor quality of life, with a daily antidiarrheal intake and a drastic diet without fiber, gluten, or lactose; she indicated global exhaustion due to her disabling symptoms.
In terms of diagnosis, no infectious etiology including the presence of Clostridium difficile was found. Abdominal imaging showed no specific anomalies, and a colonoscopy was performed in 2014, during which polypectomy and mucosectomy were performed, with no significant macroscopic or anatomopathological lesions.
Therapeutically, she had tried various diets, antidiarrheals, probiotics, and antidepressants. Faced with anal incontinence associated with diarrhea, a sacral neurostimulator was implanted to see if it could at least partially improve the symptoms, to no avail. In addition, she had been suffering from repetitive urinary tract infections for several years, with 5 episodes in the past 6 months, including 3 in the 2 months before FMT and treatment with fosfomycin. The last was a month before the FMT. It should be noted that the patient did not take any other antibiotics between the last urinary tract infection and the FMT. A cytobacteriological examination of the urine (CBEU) performed a few days before the transplant showed an Escherichia coli colonization with low-level penicillinase (resistant to ampicillin, trimethoprim-sulfamethoxazole, and doxycycline). It was the same Escherichia coli with the same resistance profile as that found in the last 3 episodes of urinary infection.
For these conditions of refractory transit disorders associated with repetitive urinary tract infections, an FMT was performed.
Fecal Microbiota Transplantation Procedure
The anonymous donor was selected by a questionnaire and microbiological analyses (blood and feces) according to the 2015 French recommendations. Fecal microbiota transplantation consisted of a bowel lavage with 2 glasses of Fast Prep the day before the transplant. The positioning of the nasogastric tube was performed and verified by chest radiography, then 200 mL of 1.4% bicarbonate was instilled 15 minutes before transplantation. The donor delivered fresh stool samples of at least 30 g, which was frozen at –80°C. All manipulations were performed at the bacteriology laboratory using a laminar-flow biosafety hood. Feces were diluted in 400 mL of 0.9% NaCl and mixed using a blender for at least 10 minutes to ensure homogenization, and the solution was then filtered to eliminate debris. The solution was poured into 8 syringes of 50 mL and kept at room temperature until infusion through the nasogastric tube. No antibiotic was prescribed.
Fecal Microbiota Analysis
In parallel, an analysis of the intestinal microbiota was performed using culturomic (a culture-dependent technique with identification of species using matrix-assisted laser desorption ionization time of flight [MALDI-TOF]) and metagenomic (a culture-independent technique targeting 16S rRNA sequencing) approaches. We collected the patient's stool before (pretransplant) and after the transplant (post-transplant), as well as the infusion of the donor's stool as it was administered to the patient (fecal infusion). During follow-up, every 2 months, only clinical monitoring was performed. All manipulations were carried out within the laboratory of the Institute Hospital-University Méditerranée Infection of Marseille. A prior authorization was obtained to carry out this study (IHU Méditerranée Infection Ethics Committee, No. 2016-011).
Culturomics
A sample of the patient's stool was tested the day before the infusion and 10 days after, and a sample of the infusion was collected as well. Four milliliters of each sample was inoculated into blood culture (Biomérieux, Marcy-l'Etoile, France) with the addition of 10 of the 18 standardized conditions of culturomics [16] and 7 others after pretreatment with alcohol to promote bacterial sporulation (Supplementary Data 1) [17]. At regular intervals (D1, D3, D7, D10, D14, D21, and D30) after 10 successive dilutions in Dulbecco's phosphate-buffered saline (DPBS), we seeded the contents of each of the 17 culture bottles on Columbia agar, which incubated for 24 hours in aerobic conditions and 48–72 hours in anaerobic conditions. Then, each bacterial colony was subcultured in COS agar isolation before being identified by MALDI-TOF on a Microflex LT (Bruker, Billerica, MA) [18]. A small amount of bacteria was deposited in at least 2 positions (spot) on an analysis plate and covered on each spot with 2 μL of a lytic matrix composed of α-cyano-4-hydroxycinnamic acid, 500 μL of acetonitrile HPLC, 25 μL of trifluoroacetic acid, and 475 μL of HPLC water. The protein spectra corresponding to each bacterium were acquired using Flexcontrol software (Bruker). A maximum of 100 peaks were used for each spectrum, and the spectra were compared with the Bruker (Biotyper) database and the IHU laboratory database. The laboratory database had an archive of the spectra of new bacterial species discovered in previous studies. An isolate was considered correctly identified at the species level when at least 1 spectrum had a score ≥1.9 and at the genus level for a score ≥1.7 [18]. If the bacterium was identified, it was stored at –80°C. If, despite several tests with good-quality protein spectra, the bacterium could not be identified, a pure culture dish of the bacterium was transmitted to molecular biology lab for identification by sequencing of the 16S gene [18]. Strains of each of the different bacterial species identified in each stool sample were stored in Protect Microorganism Preservation System tubes (Technical Service Consultant Ltd., Lancashire, UK) at –80°C. Potential contamination was limited by the use of negative and positive controls. In addition, in previous work of culturomics on more than 1000 samples, the vast majority of species were detected in many samples [19].
Metagenomic Targeting 16S rRNA Sequencing
Samples were extracted by a mechanical treatment performed with powder glass beads that had been acid washed (G4649-500g Sigma) and 0.5-mm glass beads using cell disruption media (Scientific Industries, Inc.) with a FastPrep BIO 101 instrument (Qbiogene, Strasbourg, France) at maximum speed (6.5 m/sec) for 90 seconds. Then, stools were treated with 2 kinds of lysis methods: method 1, with classical lysis and a protease step following purification using the NucleoSpin Tissu kit (Macherey Nagel, Hoerdt, France), and method 5, using a deglycosylation step and purification on the EZ1 Advanced XL device (Qiagen, Courtaboeuf, France) [20]. Samples were first amplified on these 2 extractions, pooled and barcoded, then sequenced for 16S rRNA sequencing using MiSeq technology (Illumina, Inc, San Diego, CA) and a paired-end strategy, constructed according to the 16S Metagenomic Sequencing Library Preparation (Illumina). For each protocol extraction, metagenomic DNA was amplified for the 16S “V3-V4” regions by polymerase chain reaction (PCR) for 40 cycles, using the Kapa HiFi Hotstart ReadyMix 2x (Kapa Biosystems Inc,Wilmington, MA), and the surrounding conserved region V3_V4 primers with overhang adapters (FwOvAd_341F TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; RevOvAd_785R GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). After purification on AMPure beads (Beckman Coulter Inc, Fullerton, CA), concentration was measured using high-sensitivity Qubit technology (Beckman Coulter Inc, Fullerton, CA), and dilution to 1 ng/µL was performed. During this step, the library of protocol 1 was pooled volume-to-volume to the library for protocol 5, so that 15 ng was involved in a subsequent limited-cycle PCR, where Illumina sequencing adapters and dual-index barcodes were added to the amplicon. After purification on AMPure beads (Beckman Coulter Inc, Fullerton, CA), this library was pooled with 95 other multiplexed samples. The global concentration was quantified by a Qubit assay using the high-sensitivity kit (Life Technologies, Carlsbad, CA). Before loading for sequencing on MiSeq (Illumina Inc, San Diego, CA), the pool was diluted at 8 pM. Automated cluster generation and paired-end sequencing with dual index reads were performed in a single 39-hour run at 2×250 bp. The paired reads were filtered according to the read qualities. The raw data were configured in fastaq files for R1 and R2 reads. The data were uploaded to the institution's server and transferred to the Metagx software.
MetaGX Software
Data Processing
The paired-end reads of the corresponding raw fastq files were assembled into contigs using Pandaseq [21]. The high-quality sequences were then picked for the next steps of analysis by considering only the sequences that hold both primers (forward and reverse). In the following filtering steps, the sequences containing N were removed. Sequences longer than 500 nt were trimmed, sequences with a length shorter than 100 nt were removed, and forward and reverse primers were removed from each of the sequences. The filtering steps were achieved using the QIIME pipeline [22]. Strict dereplication (clustering of duplicate sequences) was performed on the filtered sequences, which were then classified by decreasing order of abundance [23–25]. For each metagenome, the clustering of operational taxonomic units (OTUs) was performed with 97% identity.
Building Reference Databases
We used the Silva SSU and LSU database and released 1.32 from the Silva website, and from this, a local database of predicted amplicon sequences was created by extracting the sequences holding both primers. To this local database, we added 16S sequences of 556 species isolated in our laboratory from diagnostic and research samples. We collected a reference database of 14 459 sequences. We also included all the putative species of our previous analyzes. This yielded a database containing 76 368 sequences to perform our analysis.
Taxonomic Assignments
We applied at least 20 reads per OTU, which were then searched against each database using BLASTN [26]. The best match of ≥97% identity and 100% coverage for each of the OTUs was extracted from the reference database, and taxonomy was attributed down to the species level. Finally, we counted the number of OTUs allocated to unique species.
Statistical Analysis
These data were processed using Excel software and analyzed with “Microbiome Analyst” [27] following the database SILVA's guidelines [28] using a principal coordinate analysis, reflection of NMDS beta diversity, a core microbiome, and heatmap analysis.
RESULTS
Clinical Outcomes
No adverse effects occurred during the FMT procedure. Eight months after transplantation, the evolution was favorable, with a clear reduction in frequency and an improvement in stool consistency (3–4 molded stools a day compared with 10–15 before the transplant), as well as the absence of recurrence of urinary infection and absence of microbial growth on urinalysis performed after transplantation. This was accompanied by an improvement in the patient's quality of life, with a decrease in intestinal incontinence and antidiarrheal drug consumption (1 per day compared with 3–4 before transplantation), return to a normal diet, and an improvement in the patient's state of mind.
Analysis of Fecal Microbiota
Culturomics
Overall, we cultivated 177 different bacteria, 114 in the pretransplant group, 77 in the post-transplant group, and 102 in the fecal infusion group (Supplementary Data 2). The distribution of bacterial species by group is summarized in Figure 1, with, in particular, 42 bacteria initially present that were gone post-FMT and 12 bacteria present in the fecal infusion, present in the post-transplant stool, and absent in the pretransplant sample, potentially transferred from the infusion into post-transplant stool and eventually responsible for the patient's healing (Anaerococcus vaginalis, Bifidobacterium longum, Campylobacter ureolyticus, Intestinimonas massiliensis, Lactobacillus pentosus, Oscillibacter massiliensis, Phascolarctobacterium faecium, Prevotella denticola, Provencibacter massiliensis, Streptococcus agalactiae, Streptococcus constellatus, and Streptococcus parasanguinis). Intestinimonas massiliensis, Oscillibacter massiliensis, and Provencibacter massiliensis were previously discovered and cultivated in our laboratory using culturomics [29].
Figure 1.
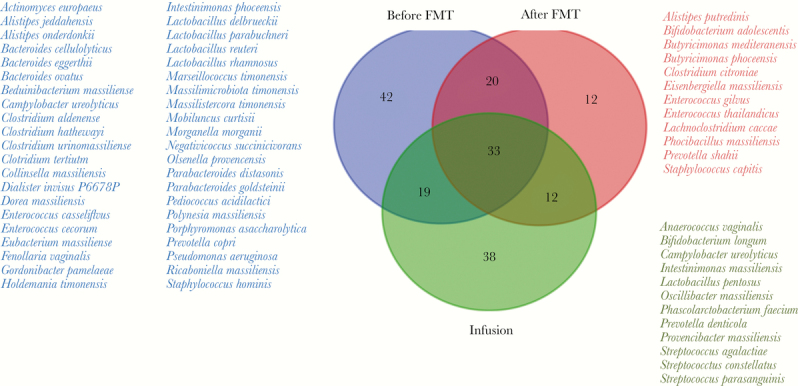
Venn diagram. Bacterial species found by culturomics in pretransplant, post-transplant, and fecal infusion samples.
We observed, respectively, 64.9%, 70.1%, and 57.7% of anaerobic bacteria and 34.2%, 40.3%, and 23.1% of gram-negative bacteria in the pretransplantation, post-transplant, and fecal infusion groups (no significant differences) (Table 1).
Table 1.
Phylum Level, Condition, and Gram Status in Each Stool Sample Group Using Culturomics
| Before FMT,a No. | After FMT,b No. | Infusion,c No. | |
|---|---|---|---|
| Phylum | |||
| Actinobacteria | 13 | 6 | 12 |
| Bacteroidetes | 21 | 16 | 11 |
| Firmicutes | 67 | 49 | 73 |
| Proteobacteria | 6 | 2 | 7 |
| Synergistetes | 1 | 1 | 0 |
| Not specified | 6 | 3 | 1 |
| Condition | |||
| Anaerobic bacteria | 74 | 54 | 60 |
| Aerobic bacteria | 40 | 23 | 44 |
| Gram status | |||
| Gram-postitive bacteria | 67 | 41 | 78 |
| Gram-negative bacteria | 39 | 31 | 24 |
| Gram–not specified bacteria | 8 | 5 | 2 |
Abbreviation: FMT, fecal microbiota transplantation.
aThe day before the FMT.
bTen days after the FMT.
cThe infusion the day of the FMT.
Metagenomic Targeting 16S rRNA Sequencing
Pyrosequencing generated a total of 85 438 reads. In the pretransplant stool, the post-transplant stool, and the fecal infusion, there were, respectively, a total of 11 223, 43 334, and 30 881 reads corresponding to 71, 124, and 113 OTUs. The distribution of OTUs found using 16S rRNA sequencing by group is summarized in Figure 2 with, in particular, 3 bacteria initially present that were lost post-FMT, and were therefore potentially pathogenic in the patient, and 12 bacteria present in the fecal infusion, present in the post-transplant stool, and absent in the pretransplant stool, potentially transferred from the infusion into the post-transplant stool and eventually responsible for the patient's recovery (Akkermansia muciniphila, Bacteroides fragilis, Collinsella aerofaciens, Coprococcus comes, Desulfovibrio piger, Holdemania timonensis, IHU PS 94 Oscillospiraceae 125390, IHU PS Unassigned 109709, IHU PS Unassigned 58217, Ihuella massiliensis, Kineothrix alysoides, Marseillibacter massiliensis, and Prevotella lascolaii). These are different from those found with culturomics. The global heatmap is described in Figure 3. This represents the distribution of the different genera according to the sample and to their correlation index. Note that we find a great dissimilarity between post-transplant stool and the infusion, which is different from published studies on Clostridium difficile [30]. This is difficult to interpret in a single case report, but perhaps it is because of the considerable lack of diversity in the microbiota of the patient with Clostridium difficile colitis before transplant that we do not find in patients with IBS. The analysis of the microbiota at several months after the FMT would make it possible to evaluate if there are more similarities. Supplementary Data 3 summarizes the number of pyrosequencing-trimmed reads obtained for each group, with their respective relative abundance. The 3 most abundant OTUs obtained in the pretransplant stool, the post-transplant stool, and the fecal infusion were, respectively, Escherichia coli, IHU PS Unassigned 1204239, and Bacteroides vulgatus; IHU PS Unassigned 142193, Escherichia coli, and Bacteroides eggerthii; Akkermansia municiphila, Prevotella copri, and IHU PS Unassigned 109709. The family and order diversity in each sample is detailed in Supplementary Data 4. The biodiversity in each group is utterly different, as shown in Supplementary Data 5.
Figure 2.

Venn diagram. Operational taxonomic units found by metagenomic targeting 16S rRNA sequencing in pretransplant, post-transplant, and fecal infusion samples.
Figure 3.
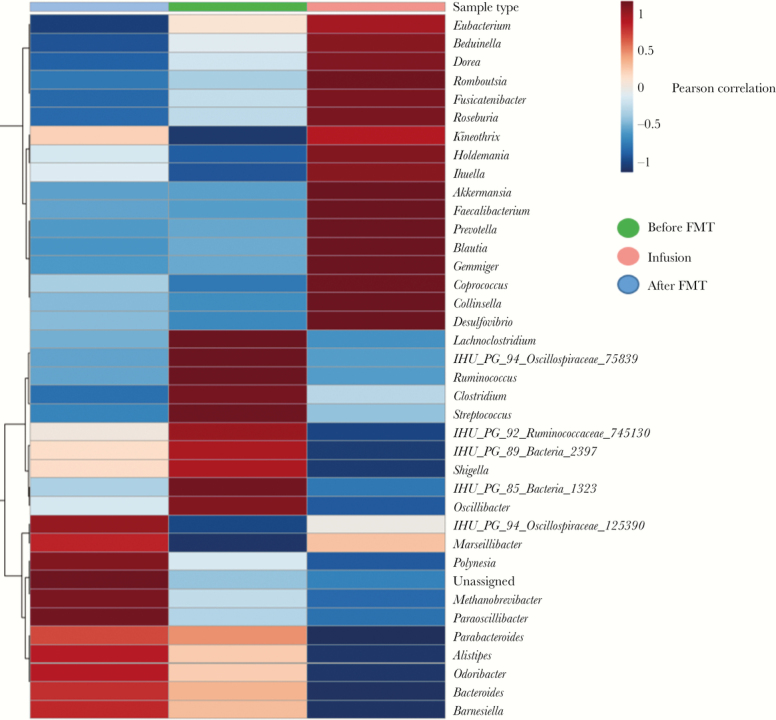
Heatmap comparison of the abundance of all bacterial genera found using 16S rRNA sequencing across pretransplant, post-transplant, and fecal infusion samples.
Culturomics and Metagenomics
We found, respectively, 14, 11, and 13 common species with both techniques, and most of the other species were only found when using 1 of the 2 techniques, which shows their complementarity, as previously described (Supplementary Data 6) [15].
Discussion
We report here the first case of FMT for both IBS and UTI indications with stools' microbiota analyzed by culturomics and 16S rRNA sequencing. Indeed, in the literature, we found articles that report cases of IBS successfully evolving after FMT for IBS with constipation mainly [31] or alternating diarrhea and constipation [32], especially a recent double-blind, randomized, placebo-controlled, parallel-group, single-center trial [11]. Concerning recurrent UTIs, Whiteside et al. suggested that the development of a synthetic urinary microbiota for transplantation might lead to effective treatment for patients with recurrent UTI, as the cause of this condition is probably similar to a recurrent infection caused by C. difficile, in which the infection is likely to be associated with an inability to reconstitute the normal microbiota [33]. Subsequently, Tariq et al. fortuitously demonstrated that FMT may decrease the frequency of UTIs associated with multidrug-resistant organisms, possibly by gut decolonization through the reestablishment of colonization resistance. This effect may lead to a decrease in antibiotic use, morbidity, and cost [13].
It should be noted that we found a discordance of the species by culturomics and metagenomics, as previously described [34]. This is due to biases associated with 16S sequencing such as extraction bias, depth bias (detects only the majority species), and viability bias (it is not known whether species are alive). All this explains the low recovery rate between the 2 techniques. To overcome this, we propose for future studies the use of several methods of extractions concomitantly, an increase in the depth of sequencing, use of the latest technologies of metagenomics, or coupling this with metatranscriptomics [34].
Finally, there are articles in which the authors have tried to characterize the microbiota of patients with IBS, but the preliminary results are not quite superimposable. Jeffery et al. found changes characterized by an increase in Firmicutes-associated taxa and a depletion of Bacteroidetes-related taxa in IBS stool samples [35]. Rajilic-Stojanovic et al. also found an increase in the ratio of Firmicutes and Bacteroidetes, with an increase in the number of Dorea, Ruminococcus, and Clostridium spp. in some samples and a decrease in the number of Bacteroidetes, Bifidobacterium, and Faecalibacterium spp. in others, and, when present, a lower average number of methanogens in IBS stool samples [36].
In analyzing fecal and mucosal microbiota from patients with IBS and healthy individuals, Tap et al. found IBS symptom severity to be negatively associated with microbial richness, exhaled CH4, presence of methanogens, and enterotypes enriched with Clostridiales or Prevotella species [2]. Finally, Pozuelo has shown a reduction of butyrate- and methane-producing microorganisms in patients with irritable bowel syndrome [3].
Conclusions
In this case report, IBS symptoms and UTI frequency decreased after FMT. To date, the exact mechanism of this effect remains unclear and could be the subject of further in-depth studies. We observed 12 bacteria obtained by culturomics and 12 others by metagenomics targeting 16S rRNA sequencing that were present in the fecal infusion and the stool after transplant, which could represent bacteria of interest. A study with more patients, urine microbiota analysis, and dendrogram comparisons and genome sequencing on these common bacteria would be interesting to confirm clinical efficiency and to find bacteria of interest to participate in the healing of IBS and UTI and thus develop the right mix of bacteria to consider targeted bacteriotherapy.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Acknowledgments
We thank Sophie Amrane, Camille Valles, and Pamela Afouda for their help with culture and statistical analysis.
Financial support. This work was supported by the French State, managed by the Agence Nationale pour la Recherche including the Programme d'Investissement d'Avenir under the reference Méditerranée Infection 10-IAHU-03.
Potential conflicts of interest. The authors of this manuscript have no conflicts of interest related to the material presented. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Author contributions. M.H.: proceed to the culture, the statistical analysis, and the writing of the manuscript. E.K. and T.P.: performed the culture. E.T.: proceed to the metagenomics sequencing protocol. J.L. and D.R.: directed and supervised lab manipulations and article writing.
References
- 1. Maharshak N, Ringel Y, Katibian D, et al. . Fecal and mucosa-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Dig Dis Sci 2018; 63:1890–9. [DOI] [PubMed] [Google Scholar]
- 2. Tap J, Derrien M, Törnblom H, et al. . Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology 2017; 152:111–123.e8. [DOI] [PubMed] [Google Scholar]
- 3. Pozuelo M, Panda S, Santiago A, et al. . Reduction of butyrate- and methane-producing microorganisms in patients with irritable bowel syndrome. Sci Rep 2015; 5:12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahmad OF, Akbar A. Microbiome, antibiotics and irritable bowel syndrome. Br Med Bull 2016; 120:91–9. [DOI] [PubMed] [Google Scholar]
- 5. van Nood E, Vrieze A, Nieuwdorp M, et al. . Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013; 368:407–15. [DOI] [PubMed] [Google Scholar]
- 6. Yoon SS, Brandt LJ. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J Clin Gastroenterol 2010; 44:562–6. [DOI] [PubMed] [Google Scholar]
- 7. Hocquart M, Lagier JC, Cassir N, et al. . Early fecal microbiota transplantation improves survival in severe Clostridium difficile infections. Clin Infect Dis 2018; 66:645–50. [DOI] [PubMed] [Google Scholar]
- 8. Lagier JC, Delord M, Million M, et al. . Dramatic reduction in Clostridium difficile ribotype 027-associated mortality with early fecal transplantation by the nasogastric route: a preliminary report. Eur J Clin Microbiol Infect Dis 2015; 34:1597–601. [DOI] [PubMed] [Google Scholar]
- 9. Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol 2011; 9:88–96. [DOI] [PubMed] [Google Scholar]
- 10. Kelly CR, Kahn S, Kashyap P, et al. . Update on fecal microbiota transplantation 2015: indications, methodologies, mechanisms, and outlook. Gastroenterology 2015; 149:223–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnsen PH, Hilpüsch F, Cavanagh JP, et al. . Faecal microbiota transplantation versus placebo for moderate-to-severe irritable bowel syndrome: a double-blind, randomised, placebo-controlled, parallel-group, single-centre trial. Lancet Gastroenterol Hepatol 2018; 3:17–24. [DOI] [PubMed] [Google Scholar]
- 12. Pinn DM, Aroniadis OC, Brandt LJ. Is fecal microbiota transplantation the answer for irritable bowel syndrome? A single-center experience. Am J Gastroenterol 2014; 109:1831–2. [DOI] [PubMed] [Google Scholar]
- 13. Tariq R, Pardi DS, Tosh PK, et al. . Fecal microbiota transplantation for recurrent Clostridium difficile infection reduces recurrent urinary tract infection frequency. Clin Infect Dis 2017; 65:1745–7. [DOI] [PubMed] [Google Scholar]
- 14. Greub G. Culturomics: a new approach to study the human microbiome. Clin Microbiol Infect 2012; 18:1157–9. [DOI] [PubMed] [Google Scholar]
- 15. Lagier JC, Armougom F, Million M, et al. . Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect 2012; 18:1185–93. [DOI] [PubMed] [Google Scholar]
- 16. Lagier JC, Hugon P, Khelaifia S, et al. . The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev 2015; 28:237–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khanna S, Pardi DS, Kelly CR, et al. . A novel microbiome therapeutic increases gut microbial diversity and prevents recurrent Clostridium difficile infection. J Infect Dis 2016; 214:173–81. [DOI] [PubMed] [Google Scholar]
- 18. Seng P, Drancourt M, Gouriet F, et al. . Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Infect Dis 2009; 49:543–51. [DOI] [PubMed] [Google Scholar]
- 19. Lagier JC, Khelaifia S, Alou MT, et al. . Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 2016; 1:16203. [DOI] [PubMed] [Google Scholar]
- 20. Angelakis E, Bachar D, Henrissat B, et al. . Glycans affect DNA extraction and induce substantial differences in gut metagenomic studies. Sci Rep 2016; 6:26276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masella AP, Bartram AK, Truszkowski JM, et al. . PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 2012; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caporaso JG, Kuczynski J, Stombaugh J, et al. . QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boissière A, Tchioffo MT, Bachar D, et al. . Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog 2012; 8:e1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mondani L, Piette L, Christen R, et al. . Microbacterium lemovicicum sp. nov., a bacterium isolated from a natural uranium-rich soil. Int J Syst Evol Microbiol 2013; 63:2600–6. [DOI] [PubMed] [Google Scholar]
- 25. Stoeck T, Behnke A, Christen R, et al. . Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biol 2009; 7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altschul SF, Gish W, Miller W, et al. . Basic local alignment search tool. J Mol Biol 1990; 215:403–10. [DOI] [PubMed] [Google Scholar]
- 27. Dhariwal A, Chong J, Habib S, et al. . Microbiome analyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res 2017; 45:W180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quast C, Pruesse E, Yilmaz P, et al. . The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013; 41:D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durand G, Afouda P, Raoult D, Dubourg G. “Intestinimonas massiliensis” sp. nov, a new bacterium isolated from human gut. New Microbes New Infect 2017; 15:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seekatz AM, Aas J, Gessert CE, et al. . Recovery of the gut microbiome following fecal microbiota transplantation. MBio 2014; 5:e00893–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borody TJ, George L, Andrews P, et al. . Bowel-flora alteration: a potential cure for inflammatory bowel disease and irritable bowel syndrome? Med J Aust 1989; 150:604. [DOI] [PubMed] [Google Scholar]
- 32. Pinn DM, Aroniadis OC, Brandt LJ. Is fecal microbiota transplantation the answer for irritable bowel syndrome? A single-center experience. Am J Gastroenterol 2014; 109:1831–2. [DOI] [PubMed] [Google Scholar]
- 33. Whiteside SA, Razvi H, Dave S, et al. . The microbiome of the urinary tract—a role beyond infection. Nat Rev Urol 2015; 12:81–90. [DOI] [PubMed] [Google Scholar]
- 34. Lagier JC, Dubourg G, Million M, et al. . Culturing the human microbiota and culturomics. Nat Rev Microbiol 2018; 1: 540–50. [DOI] [PubMed] [Google Scholar]
- 35. Jeffery IB, O'Toole PW, Öhman L, et al. . An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut 2012; 61:997–1006. [DOI] [PubMed] [Google Scholar]
- 36. Rajilić-Stojanović M, Biagi E, Heilig HG, et al. . Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011; 141:1792–801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.