

Abstract
RNA editing critically regulates neurodevelopment and normal neuronal function. We surveyed RNA editing across 364 schizophrenia cases and 383 control postmortem brain samples from the CommonMind Consortium, comprising two regions: dorsolateral prefrontal cortex (DLPFC) and anterior cingulate cortex. In schizophrenia, RNA editing sites in genes encoding AMPA-type glutamate receptors and post-synaptic density proteins were less edited, while those encoding translation initiation machinery were more edited. These sites replicate between brain regions, map to 3’UTR and intronic regions, share common sequence motifs, and map to binding sites for RNA binding proteins crucial for neurodevelopment. These findings cross-validate in hundreds of non-overlapping DLPFC samples. Furthermore, ~30% of RNA editing sites associate with cis-regulatory variants (edQTLs). Fine-mapping edQTLs with schizophrenia GWAS loci revealed co-localization of 11 edQTLs with 6 GWAS loci. Our findings demonstrate widespread altered RNA editing in schizophrenia and its genetic regulation, and suggest RNA editing mechanisms of schizophrenia neuropathology.
Keywords: RNA editing quantitative trait loci, disease risk, molecular mechanisms, neurodevelopment
INTRODUCTION
Schizophrenia (SCZ) is a severe psychiatric disorder affecting ~0.7% of adults and is characterized by abnormalities in thought and cognition1. While the onset of SCZ typically does not occur until late adolescence or early adulthood, strong support from clinical and epidemiological studies suggests that SCZ reflects a disturbance of neurodevelopment2. There is clear and consistent evidence that SCZ is largely a genetic disorder. Large-scale mapping of genetic risk variants has identified multiple rare copy number variants3, several rare single nucleotide variants4,5, and >100 common genetic loci6, the latter exerting small polygenetic effects on disease risk. This observation of a highly polygenic architecture has been widely replicated7,8. However, the role of sequence variation arising as a result of post-transcriptional events, such as RNA editing, remains largely unexplored.
RNA editing is a modification of double-stranded pre-mRNA that introduces codon changes in mRNA through insertions, deletions or substitutions of nucleotides and hence can lead to alterations in protein function. Adenosine to inosine (A-to-I) editing is the most common form of RNA editing, affecting the majority of human genes and is highly prevalent in the brain9,10. These base-specific changes to RNA result from site-specific deamination of nucleotides catalyzed by adenosine deaminases acting on RNA (ADAR) enzymes, whereby a genetically encoded adenosine is edited into an inosine, which is read by the cellular machinery as a guanosine. Editing sites in coding regions can be conserved across species and are commonly located in genes involved in neuronal function11,12. RNA editing has been reported to modulate excitatory responses, permeability of ion channels and other neuronal signaling functions13,14. These sites have been shown to be tightly and dynamically regulated throughout pre- and post-natal human cortical development15. Aberrant RNA editing has also been reported in several neurological disorders, including major depression16, Alzheimer’s disease17, and amyotrophic lateral sclerosis18.
In SCZ, the role of RNA editing in serotonin and glutamate receptors has drawn significant attention largely due to the serotonergic and glutamatergic hypotheses of mood disorders. To this end, RNA editing research in SCZ has focused on targeted approaches of serotonin 2C receptor (5-HT2CR)19–22 and two classes of ionotropic glutamate receptors, 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)-propanoic acid (AMPA) and kainate receptors23–25. Consequently, there is no consensus on the type of editing nor how pervasive altered RNA editing is in the brain of SCZ patients. Moreover, the underlying cis-acting genetic variants, which are associated with RNA editing levels (edQTLs) in the brain and whether these variants are also implicated in disease risk also remain poorly understood.
The primary goal of the current investigation was to clarify the relevance of RNA editing in SCZ pathophysiology using an unbiased, genome-wide approach applied to a large cohort of SCZ cases and control samples generated from the CommonMind Consortium (CMC), which is orders of magnitude larger than prior RNA editing studies. Two brain regions implicated in neurodevelopment and SCZ neuropathology were examined, including the dorsolateral prefrontal cortex (DLPFC; Brodmann areas 9 and 46) and anterior cingulate cortex (ACC). Results from this cohort were then reproduced in a separate, non-overlapping DLPFC cohort generated through the National Institute of Mental Health (NIMH) Human Brain Collection Core (HBCC). By applying a multi-step analytic framework and including genome-wide characterization of common genetic variation (Figure 1), we generated a resource of the genetics of RNA editing in the brain. We use this resource to identify: (1) genes and RNA editing sites with significant differences in RNA editing levels between subjects with SCZ and control subjects; (2) coordinated editing (co-editing) of RNA editing sites implicated in SCZ; and (3) specific effects on RNA editing of genetic variants previously implicated in disease risk. In doing so, these findings substantially refine our understanding of the RNA editing mediated mechanism involved in the neurobiology of SCZ.
Figure 1. Overview of the study design and analytic pipeline.
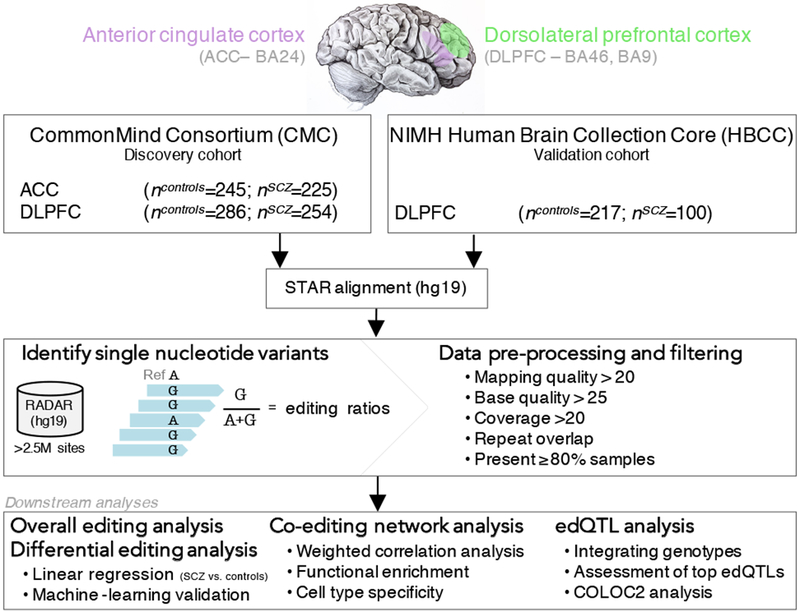
The samples used in this study were from participants in two large studies of schizophrenia in the United States who donated their brains upon death. A total of 364 unique schizophrenia cases and 383 unique controls were sampled in at least one brain region. Genome-wide RNA-seq data from the CommonMind Consortium (CMC) covered two brain regions, the ACC and the DLPFC, and these samples served as the discovery cohort. RNA-seq data of post-mortem DLPFC tissue was generated on behalf of NIMH Human Brain Collection Core (HBCC) and this second resource served as a validation cohort. Fastq files were aligned to the human reference genome and transcriptome (hg19) using STAR and bam files were sorted using samtools. RNA editing events were called from sorted bam files using the mpileup function in samtools together with customized perl scripts, which integrated all known RNA editing sites from the RADAR database. A series of internal filtering. quality control and imputation metrics were computed before moving downstream to overall RNA editing, differential RNA editing, co-editing and edQTL analyses.
RESULTS
Discovery and validation samples
In order to quantify RNA editing events, we leveraged RNA-sequencing data from post-mortem brain tissue collected and generated on behalf of the CMC. Two brain regions, including the ACC (SCZ=225, Controls=245) and the DLPFC (SCZ=254, Controls=286) were investigated, and together these samples served as the discovery cohort (Figure S1). These samples were also genotyped on the Illumina Infinium HumanOmniExpressExome array. In parallel, we also leveraged a completely separate, non-overlapping cohort consisting of post-mortem DLPFC tissue (SCZ=100, Controls=217) collected and generated on behalf of NIMH HBCC. This second resource served as a validation cohort so as to cross-validate the discovery of SCZ-related editing events.
Overall RNA editing levels in SCZ
Overall RNA editing levels were computed for each sample and was defined as the percentage of edited nucleotides at all known editing sites (Materials and Methods). Higher levels of overall RNA editing in SCZ cases were observed compared to controls in the ACC and DLFPC (p=0.0001, p=7.2×10−6, respectively) (Figure 2A). Approximately 10% of the variation in overall RNA editing levels was explained by ADAR1 (p=<2.2×10−16) and ADAR2 expression explained ~3% of variation in overall RNA editing (p=1.2×10−08) (Figure 2B,C). ADAR3 expression had no significant effect on overall editing levels (p=0.10) (Figure 2D), although recently demonstrated a negative association with overall editing when measured across several brain regions10. Marked increases in overall editing levels were observed within definite genic regions, specifically 3’UTR and intergenic regions in SCZ, which replicated across the ACC and DLPFC (Figure S2A). Moreover, as previous research has quantified RNA editing levels explicitly in serotonergic and glutmatergic receptors, we computed overall editing levels in serotonergic and glutmatergic receptor activity genes using a priori defined gene-sets (GO:009589 and GO:0008066, respectively). Higher levels of overall editing were found in glutamatergic receptors in SCZ cases relative to controls in the ACC (p=0.001) and DLPFC (p=2.2×10−5), while no significant differences were found in the levels of overall editing in serotonergic receptors (Figure S2B). Expression of ADAR1 and ADAR2 were also significantly higher in SCZ compared to control samples (Figure S2C–E). Collectively, these observations, apart from the expression of ADAR2, were reproduced in our independent DLPFC validation cohort, and collectively highlight higher overall RNA editing levels in SCZ, primarily within 3’UTR and intergenic regions, as well as within genes encoding glutamatergic receptors.
Figure 2. Overall RNA editing profiles.
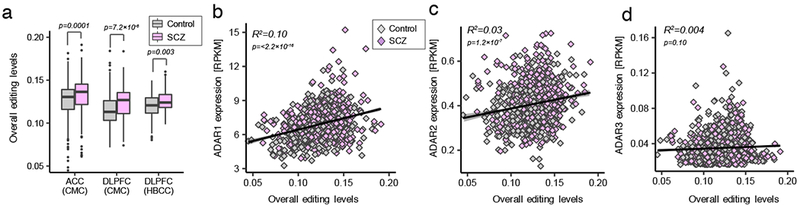
(a) Overall RNA editing levels across for the CMC ACC (ncontrol=245, nSCZ=225) and DLPFC (ncontrol=286, nSCZ=254) and HBCC DLPFC (ncontrol=217, nSCZ=100). A two-sided Mann-Whitney U test with continuity correction was used to test significance between diagnostic groups. Whisker box plots show median, lower and upper quartiles, and whiskers represent minimum and maximum of the data. Associations between expression levels of (b) ADAR1, (c) ADAR2 and (d) ADAR3 (quantified as the number of RNA-seq reads per kilobase of transcript per million mapped reads (RPKM)) and overall editing levels across all available ACC and DLPFC samples (including CMC and HBCC data). These concordance analyses were made across all samples (ncontrol=735, nSCZ=579) as the ACC and DLPFC showed highly collinear relationships. R2 values were calculated by robust linear regressions on overall editing levels and logarithmic transformed RPKM values.
To rule out the possibility that these reproducible differences in overall RNA editing levels may be driven by medication effects, we examined overall editing levels in postmortem DLPFC tissue derived from an RNA-sequencing study of 34 Rhesus macaque monkeys treated with high doses of haloperidol (10 mg/kg/d), low doses of haloperidol (4mg/kg/d), clozapine (5.2 mg/kg/d), and vehicle. We found no associations between overall RNA editing levels with medication or dosage (Figure S3), indicating that antipsychotic treatments likely do not have a strong effect on the amount of overall RNA editing observed in SCZ cases.
Discovery of altered RNA editing sites in SCZ
To identify RNA editing sites associated with SCZ, a compendium of high quality and high confident RNA editing sites was assembled by imposing a series of detection-based thresholds (see Materials and Methods). After thorough quality control, we identified a high confidence set of 11,242 RNA editing sites in the ACC and 7,594 sites in the DLPFC with no systematic differences in the mapping, base quality, and read coverage between SCZ and control samples. A significant fraction of these RNA editing events replicated across brain regions (∩sites=6,999, OR=21.05, p=<2.0×10−50). A large fraction of the sites were located in Alu repeat elements, mapped to 3’UTR regions and were enriched for A-to-I conversions (Figure S4).
Following this curation of editing events, differential RNA editing analysis was carried out. It is likely that genome-wide RNA editing events, similar to gene expression, may be influenced by differences in biological and technical factors. To this end, a linear mixed effect model was applied to quantify the total amount of RNA editing variance explained by various biological and technical factors, which collectively displayed little influence on RNA editing profiles, with individual age having the largest genome-wide effect and explained a median 0.79% of the observed variability (Figure S5A). These factors, however, explained a much higher amount of median variability in matching gene expression profiles than observed RNA editing profiles (Figure S5B). Subsequently, differential editing analysis covarying for individual age, RIN, PMI, sample site and sex identified 182 sites in the ACC and 194 sites in the DLPFC significantly associated with SCZ (Adj. P < 0.05) (Figure 3A; Table S1A–C). Among the top-ranked sites, were those encoding for genes ATRLN1, AKAP5 and RPS20 in the ACC and KCNIP4, VPS41 and ZNF140 in the DLPFC. A high degree of concordance was observed between the altered RNA editing sites in the ACC and DLPFC (R2=0.59) and a significant overlap of differentially edited sites replicated between brain regions (∩sites=29, OR=12.6 p=9.6×10−20) (Figure 3B). Differentially edited sites were also enriched in genes found to be highly expressed in the ACC (∩sites=42, OR=5.4 p=2.0×10−13) and DLPFC (∩sites=54, OR=8.6 p=2.7×10−22) and that the majority of genes with differential RNA editing sites did not display differential gene expression (Figure S6A–F). Moderate, yet significant, correlations were also observed between RNA editing levels and gene expression, implying RNA editing as a possible posttranscriptional mechanism for the regulation of gene expression (Figure S6G–I).
Figure 3. Identification of differentially edited sites in SCZ.
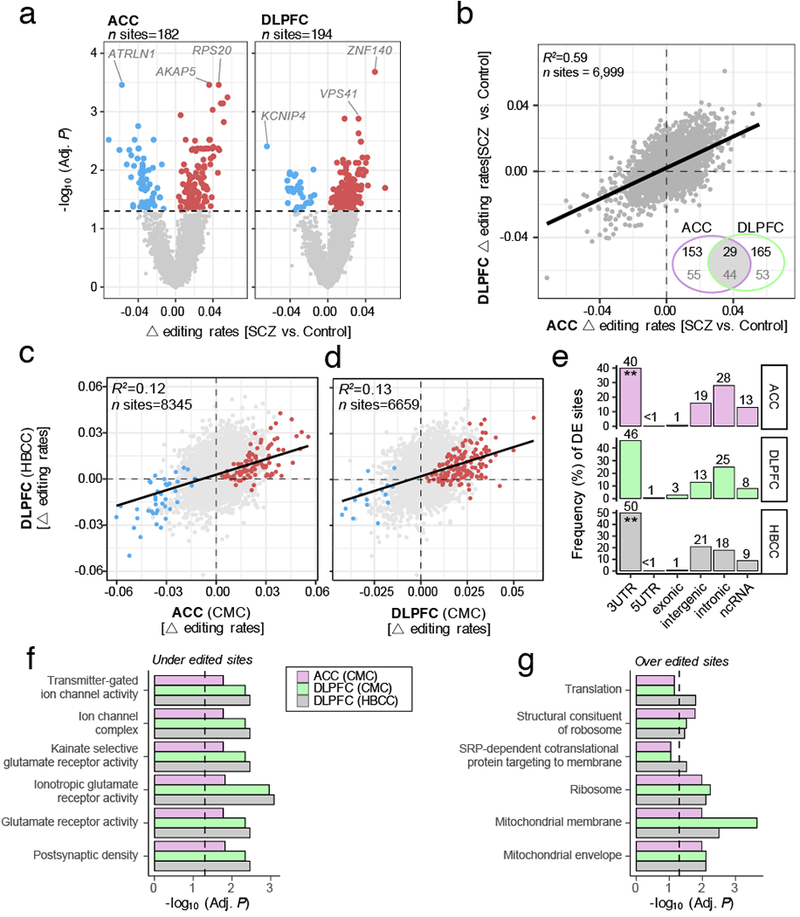
Differential editing sites in the (a) ACC (ncontrol=245, nSCZ=225) and DLPFC (ncontrol=286, nSCZ=254). Dotted line marks a multiple test corrected level of significance (Adj. P < 0.05, limma, linear regression with Benjamini-Hochberg (BH) correction). Red points indicate over-edited sites and blue points indicate under-edited sites. For the top three sites, we outline their respective gene body. (b) Scatterplot of change (△) in editing rates for RNA editing sites in the ACC compared to the DLPFC. Inset Venn Diagram indicates the total number of significant overlapping sites (top value) and respective gene symbols (bottom value). Results were cross-validated for the (c) ACC (x-axis) and (d) DLPFC (x-axis), respective to △ editing rates within independent HBCC DLPFC samples (y-axis) (ncontrol=217, nSCZ=100). R2 values were calculated by robust linear regressions on △editing rates. Red and blue points indicate sites passing BH correction in the discovery sample. (e) Significantly differentially edited sites by genic region indicates a significant depletion of sites mapping to 3’UTR regions (Fisher’s Exact Test, P < 0.05,* Alternative hypothesis=less; ACC p=0.02; DLPFC p=0.01; NIMH HBCC DLPFC p=0.04). Functional annotation of the top five enrichment terms for (f) under-edited sites and (g) over-edited sites in SCZ were computed by a one-sided hypergeometric test and adjusted for multiple comparisons using Bonferroni correction.
Validation of altered RNA editing sites in SCZ
Next, we asked whether these differential editing patterns in SCZ replicate within our independent DLPFC validation cohort. These samples underwent matching quality-control metrics to identify a collection of high confidence RNA editing events, as noted above. A total of 15,000 RNA editing events were detected across these validation samples and a significant fraction of sites were also detected in the ACC (74%, ∩sites=8354, OR=4.01 p=5.6×10−251) and DLPFC (87%, ∩sites=6659, OR=7.73 p=4.67×10−248) discovery samples (Table S2). Differential RNA editing analysis was carried out on these independent samples as previously described and 137 sites (75%) were detected in the ACC and 165 sites (85%) in the DLPFC. In order to assess replication, we first measured the concordance between directionality of change in editing rates for all RNA editing sites identified in the ACC and the DLPFC discovery samples relative to these independent DLPFC validation samples. High levels of concordance were observed across all RNA editing sites in both the ACC and DLPFC (R2=0.12, R2=0.13, respectively) (Figure 3C–D). Subsequently, two prediction models were built based on differentially edited sites from the (1) DLPFC and (2) ACC discovery samples using regularized regression models and evaluated their performance to predict class labels (i.e. distinguish between SCZ and control samples) on withheld DLPFC validation samples. Classification accuracies were reported as area under the receiver operative curve on withheld DLPFC samples. When distinguishing between SCZ and control samples, classification accuracies reach 78% and 72% on withheld, independent DLPFC samples when using differentially edited sites derived from DLPFC and ACC discovery samples, respectively (ridge regression outperformed other methods: Figure S7). Overall, these results suggest a moderate level of cross-validation of SCZ-related editing events across brain regions and independent cohorts.
Characterization of differentially edited sites
Differentially edited sites derived from discovery and validation samples were comprehensively annotated. While the majority of differentially edited sites map to 3’UTR regions across brain regions and cohorts, a moderate depletion was observed when adjusting for the total number of non-differentially edited sites in 3UTRs for each brain region and cohort (Figure 3E). Functional enrichment analysis revealed that under-edited sites consistently mapped to postsynaptic density genes as well as genes encoding kainate and glutamate receptor activity and over-edited sites mapped to genes implicated in protein translation and mitochondrial-related terms (Figure 3F–G). We also examined whether these differentially edited sites map to genes with specific developmental expression profiles using gene expression data from the BrainSpan Project and found that differentially edited sites in SCZ consistently mapped to genes that are predominately postnatally biased in expression (Figure S8). These genes were found to peak in brain expression during young and middle adulthood, developmental windows when SCZ often becomes clinically recognizable. Moreover, a substantial fraction of our editing sites (n=612) were also previously found to have increasing rates of editing throughout brain development, 11 of which are significantly over-edited and 3 significantly under-edited in SCZ (Table S1D).
As these sites share several sequence and functional features, we explored whether differential editing sites may share a common sequence motif potentially important for editome recognition (20±nt centered on target A). Consistent enrichment was found for a 10-nt motif (CGGGATTACA) in region adjacent to most differential and non-differential editing sites located in 3’UTR regions (Figure S9, Table S3). Notably, this short sequence has been reported to occur frequently within non-coding regions and is also found to overlap fragments of Alu repeat elements. Subsequently, we examined whether differentially edited sites found to share this sequence motif also mapped to any known human RNA binding protein (RBP) binding sites (30±nt centered on target A). A large fraction of these sites (>61% in each brain region) significantly coincided with binding sites specific for RBP serine/arginine (SR)-rich splicing factor 5 (SRSF5) (Table S4). Interestingly, this protein is associated with pyruvate carboxylase deficiency, a disorder that is associated with developmental delay and recurrent seizures. Other significant RBP binding sites included additional members of the SR-rich family of pre-mRNA splicing factors, such as SRSF2 and SRSF3 as well as CUGBP, an RBP found to mediate neuronal toxicity.
Genes enriched with differential RNA editing in SCZ
We examined whether any genes contained an enrichment of differentially edited sites beyond what could be expected by chance. As expected, gene length functions as a correlate of the total number of RNA editing sites per gene (Figure S10). Therefore, we computed over-representation of differential RNA editing sites within each gene by setting a rotating background specific to the total number of known RNA editing events for a particular gene in order to systematically correct for gene length (Table S5). Genes harboring a significant fraction of under edited sites in SCZ primarily mapped to intronic regions (Figure 4A,B), while genes harboring 3’UTR sites were over-edited in SCZ (Figure 4B–E). Three genes, including KCNIP4, HOOK3 and MRPS16 displayed enrichment for altered editing sites across the ACC, DLPFC and our independent validation DLPFC cohort (Figure 4C–E). KCNIP4 harbored 13 unique differentially edited sites spread over its first and second introns, which were predominately under-edited in SCZ compared to control samples. KCNIP4 is a member of the voltage-gated potassium channel-interacting proteins and has been shown to interact with presenilins and modulate pacemaker neurons in the reward circuitry of the brain26,27. Genome-wide association studies (GWAS) have also found KCNIP4 to be associated with SCZ, suicidal ideation and attention-deficit/hyperactivity disorder28–30. HOOK3 harbored 22 unique sites and MRPS16 harbored 19 unique sites both within their respective 3’UTR regions, which were predominately over edited in SCZ compared to control samples. HOOK3 is a microtubule tethering protein essential for centrosomal assembly during neurogenesis and brain development31 and MRPS16 is a mitochondrial ribosomal protein involved in mitochondrial protein translation32.
Figure 4. Genes enriched with differential editing sites that replicate across two brain regions or across two cohorts.
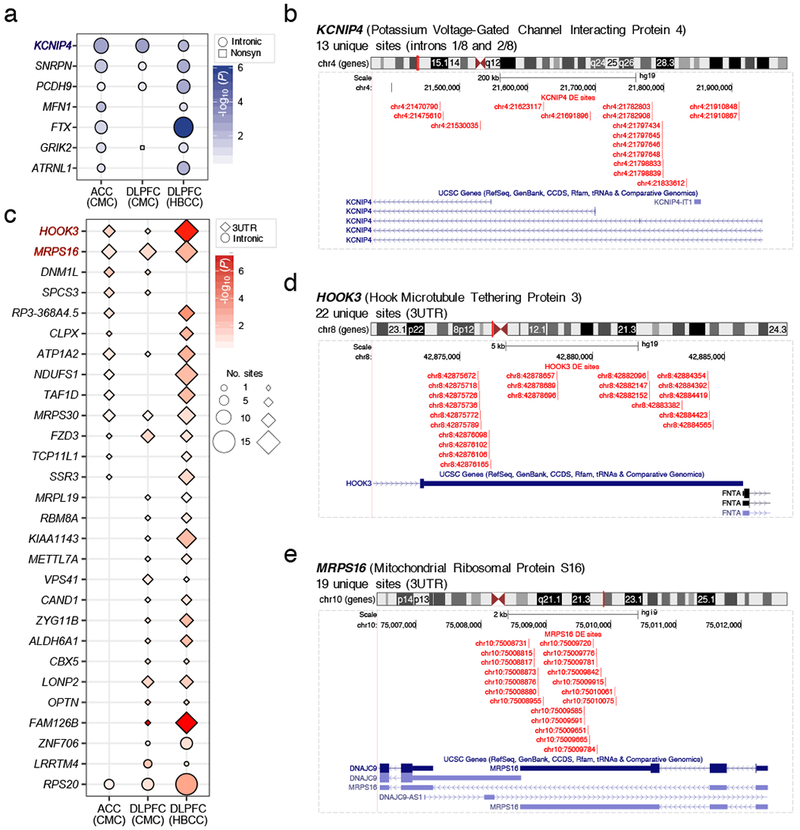
Genes containing enrichment of differentially edited sites from the ACC (ncontrol=245, nSCZ=225) and DLPFC (ncontrol=286, nSCZ=254) as well the NIMH HBCC DLPFC sample (ncontrol=217, nSCZ=100) were examined. (a) Genes enriched for under-edited sites primarily map to intronic regions. (b) KCNIP4 contains 13 unique differential RNA editing sites, which are under-edited and span its first and second intron. These sites replicate across brain regions and withheld validation samples. Enrichment was calculated using the phyper hypergeometic function (lower.tail=FALSE). (c) Genes enriched for over-edited sites in SCZ. Over-edited sites primarily map to 3’UTR regions. (d) HOOK3 contains 22 unique differential RNA editing sites and (e) MRPS16 contains 19 unique differential RNA editing sites which are over-edited within their respective 3’UTR region. Note that genes FTX and NDUFS1 contain sites in more than one genic region, see Table S5 for full details. UCSC Genome Browser customized track options display the precise locations of editing sites within each gene.
Co-editing networks associate with SCZ
Discrete groups of coordinately edited (co-edited) sites were identified and tested for association to SCZ using an unbiased network approach. A total of five co-editing modules were detected in each brain region and displayed a near one-to-one mapping between the ACC and DLPFC (Figure 5A), indicating highly similar co-editing network topology. Modules were assessed for over-representation of differential RNA editing sites and two modules were identified in the ACC (M1a and M4a) and two modules in the DLPFC (M1d and M4d) (Figure 5B). Module eigengene (ME) values for these modules elucidated higher levels of editing in modules M1a and M1d and lower levels of editing in modules M4a and M4d in SCZ compared to control subjects (Figure 5C). Functional annotation of over-edited modules M1a and M1d revealed strong enrichment for regulation of translation and translation initiation, while under edited modules M4a and M4d were enriched for AMPA glutamate and ionotropic receptors (Figure 5D). Cell type enrichment analysis revealed modules M1a and M1d were enriched for pyramidal neurons while modules M4a and M4d were enriched for interneurons (Figure 5E). Notably, these findings were also reproduced in our independent DLPFC validation cohort (Table S6, Figure S11, see Supplemental File). Moreover, M1a and M1d were positively associated with ADAR1 and ADAR2 expression and modules M4a and M4d were negatively associated with ADAR2 expression (Figure S12). Upon closer inspection, several sites located within modules M4a and M4d mapped to nonsynonymous sites in genes NOVA1, UNC80, GRIA2, GRIA3, GRIA4, GRIK2 and ANKD36, and these sites were predominately under-edited in SCZ compared to control samples (Figure 5F). Several of these sites, particularly the Q/R and R/G sites in GRIA2, are well documented as fully edited sites under normal conditions whereby loss of editing in these sites leads to enhanced Ca2+ permeability and cellular dysfunction, and this has been suggested to play a role in SCZ23,24. NOVA1 is essential for normal postnatal motor function and regulates alternative splicing of multiple inhibitory synaptic targets32. NOVA1 has been reported to be dysregulated at the gene level in independent SCZ postmortem brain samples32 and RNA editing in NOVA1 has been shown to influence protein stability33, but has yet to be associated with SCZ.
Figure 5. Unsupervised co-editing network analysis.
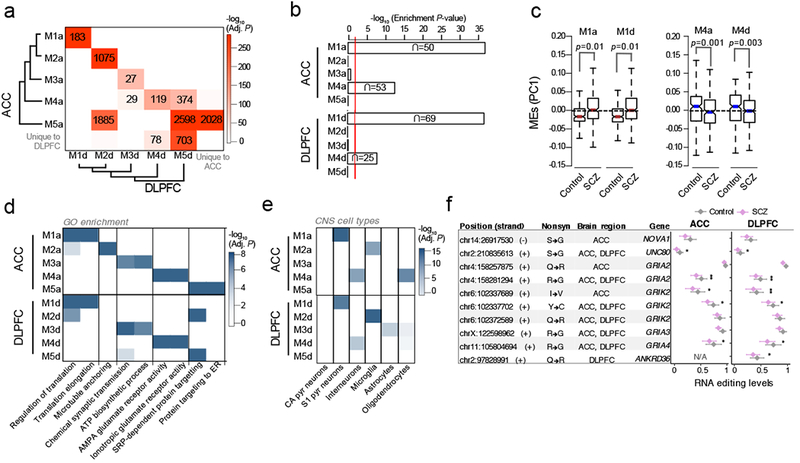
(a) Overlap analysis of co-editing modules identified within the ACC and DLPFC. Unsupervised clustering was used to group modules by module eigengene (ME) values using Pearson’s correlation coefficient and Ward’s distance method. Significance of overlap was computed (one-sided Fisher exact test, Bonferroni correction) and p-values were colored on a continuous scale (bright red, strongly significant; white, no significance). The number of overlapping sites are displayed in each cell with a significant overlap. (b) Enrichment analysis of differentially edited sites within co-editing networks (one-sided hypergeometric test). (c) Assessment of ME values for modules M1a and M1d (over-edited) and M4a and M4d (under-edited). Differential ME analysis was conducted using a linear model and covarying for age, RIN, PMI, sample site and gender. (d) The top functional enrichment terms and (e) brain cell-type enrichment results for all identified modules, verifying similar functional and cell-type properties of co-editing networks in the ACC and DLPFC. Enrichment was computed using one-sided hypergeometric test and adjusted for multiple comparisons using Bonferroni correction. (f) A collection of nonsynonymous sites within SCZ-related AMPA glutamate receptor modules M4a and M4d (** indicates Adj. P <0.05, * indicates P < 0.05 derived from differential RNA editing analysis, see Table S1 for details). Whisker dot plots show mean and whiskers represent minimum and maximum standard error of the ACC (ncontrol=245, nSCZ=225) and DLPFC (ncontrol=286, nSCZ=254).
Identification and characterization of brain cis-edQTLs
Whole-genome genotype data were available for ACC and DLPFC samples used in our discovery cohort and were imputed using standard techniques, as previously described7. Genotype data were used to detect SNPs that have an effect on RNA editing levels (edQTL, editing quantitative trait loci). RNA editing levels from European-ancestry samples (ACC N=360; DLPFC N=421) were adjusted to fit a standard normal distribution and to reduce systematic sources of variation. Adjusted editing levels were then fit to impute SNP genotypes, covarying for individual age, sample site and gender, PMI, RIN and diagnosis, using an additive linear model implemented in MatixEQTL. To identify genetic variants that could explain the variability of RNA editing, we first ran association tests between editing levels and genotypes by restricting the variant search space to only those within the same gene as each editing site and found an abundance of low P-values (Figure S13). Subsequently, we relaxed this assumption to define a broader window and identified 188,778 cis-edQTL (i.e. SNP-editing pairs ± 100kb of a site) in the ACC and 156,865 cis-edQTLs in the DLPFC at a genome-wide FDR < 5% (Figure 6A). A total of 3224 editing sites in the ACC and 2500 editing sites in the DLPFC have edQTLs. Many of the edQTLs for the same site were highly correlated, due to linkage disequilibrium, and 70.9% of edQTL SNPs (edSNPs) in the ACC and 68.9% of edSNPs in the DLPFC predicted editing of more than one site. A high level of concordance was observed for the effect sizes (beta values) of edQTLs between the ACC and DLPFC (Figure S14). Notably, edQTLs tend to be present for editing sites with greater variance in editing levels (Figure S15). Each max-edQTL (defined as the most significant edSNP per site, if any) meeting a genome-wide significance threshold was located close to their associated editing site and acting in cis (5kb±nt) (Figure 6B, Figure S15). We reasoned that due to the propensity of edQTLs to be located close to their associated editing site, they should also influence additional editing sites nearby. This reasoning was strengthened by the observation that editing levels of editing sites within the same gene are more closely correlated than editing levels of editing sites in different genes (Figure S16).
Figure 6. Brain cis-edQTL analysis.
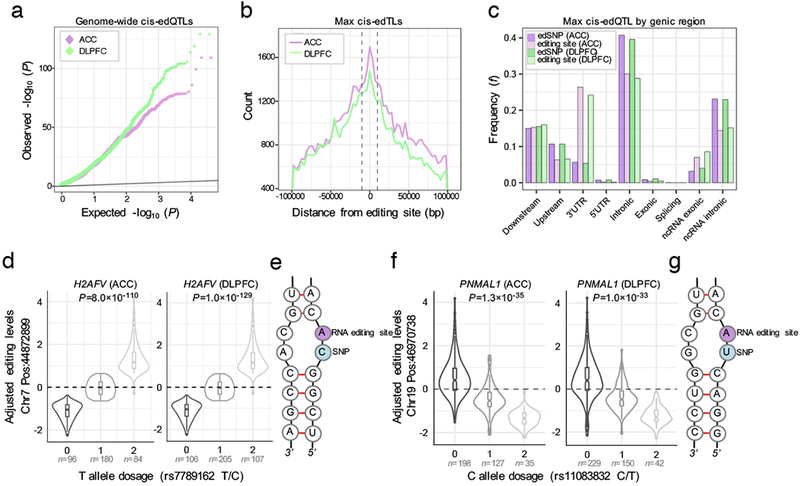
(a) Quantile-Quantile plot for association testing genome-wide P-values between imputed genotype dosages and 11,242 RNA editing sites in the ACC (ncontrol=180, nSCZ=180) and 7,594 RNA editing sites in the DLPFC (ncontrol=210, nSCZ=211) (linear regression and FDR correction via matrixEQTL). (b) Distribution of the association tests in relation to the distance between the editing site and variant for max cis-edQTLs (that is, the most significant edSNP per site, if any). Vertical dotted lines indicate ± 5KB relative to the editing site. (c) Genic locations of edSNPs and corresponding editing sites. (d-g) Two examples of top cis-edQTLs with nearby editing sites replicating between brain regions with (e,g) predicted local RNA secondary base-pairing structures (dosage sample sizes are listed below each violin plot). Whisker violin plots show median, lower and upper quartiles, and whiskers represent minimum and maximum of adjusted RNA editing levels (y-axis) according to imputed genotype dosages (x-axis; linear regression and FDR correction via matrixEQTL).
Max-edQTLs in the ACC and DLPFC were enriched within genic elements and noncoding RNAs, particularly within intronic regions, while the corresponding editing sites were also enriched in intronic regions and depleted from 3’UTR regions (Figure 6C). Max-edQTLs edSNPs were also examined for tissue-specific enhancer specify using data from the FANTOM project across 40 different human tissues. edSNPs in the ACC and DLPFC were strongly enriched for brain-specific enhancer sequences more so than any other tissue (Figure S17). A significant fraction of max-edQTLs edSNPs replicated between the ACC (62%) and DLPFC (70%) (∩=34,367, Z-score=17,443, p=0.0009). Among the most significant associations identified in both brain regions were those in genes H2AFV and PNMAL1, where the edSNP is located immediately upstream of the RNA editing site (Figure 6D–G). In both cases, the alternative allele is unable to pair with the opposite base within the double-stranded RNA hairpin, introducing two consecutive mismatches in the local RNA secondary structure.
In addition, edSNPs were examined for association with gene expression levels by calculating the overlap between max-edQTLs and previously computed max expression QTL (max-eQTL) summary statistics derived from the ACC and DLPFC. A total of 29,335 edSNPs (54.4%) in the ACC were also associated with variation in gene expression, for which 31.3% were associated with a gene and one or more editing sites within the same gene (e.g. SNPx is associated with Geney and one or more editing sites located within Geney). Similarly, a total of 27,133 edSNPs in the DLPFC (55.6%) were also associated with gene expression variation, for which 30.1% were associated with a gene and one or more editing sites within the same gene.
edQTL signatures co-localize with SCZ GWAS associations
It has previously been shown that a substantial proportion of SCZ GWAS associations (~20%) may be mediated by differential gene expression regulation7. RNA editing may represent an additional biological mechanism through which associated variants exert their effects on disease risk. Here, we leverage our edQTL resource to identify RNA editing sites that potentially alter SZC risk. Of the 108 SCZ GWAS loci reported previously, 14 harbor edQTL eSNPs for one or more RNA editing sites identified in either ACC or DLPFC. However, the presence of an edQTL within a GWAS locus does not imply disease causality. We therefore implemented coloc2, a Bayesian approach that integrates over statistics for all variants within a specified locus and estimates posterior probabilities of co-localization between two sets of association signatures, in order to identify RNA editing sites likely to contribute to SCZ etiology. We applied coloc2 to our ACC and DLPFC edQTL data in conjunction with summary statistics for the 108 genome-wide significant schizophrenia GWAS loci. We found evidence for co-localization (posterior probability > 0.5) of ACC edQTL and GWAS signatures at four loci comprising four unique edQTL and of DLPFC edQTL and GWAS signatures at four loci comprising seven unique edQTL (Table S7). Two of these loci are co-localized in both ACC and DLPFC; therefore, a total of six GWAS associations, representing approximately five percent of all genome-wide significant loci, are potentially mediated by aberrant RNA editing (Figure S18). Of the six GWAS loci harboring SCZ-associated cis-edQTLs, these findings include genes NGEF and ARL6IP4, which replicate between brain regions, as well as PCCB and RP11-890B15.3, which are unique to the ACC, and genes ENSA and DGKI, which are unique to the DLPFC; co-localization of the DGKI locus is highlighted in Figure 7.
Figure 7. Coloc2 fine-mapping analysis.
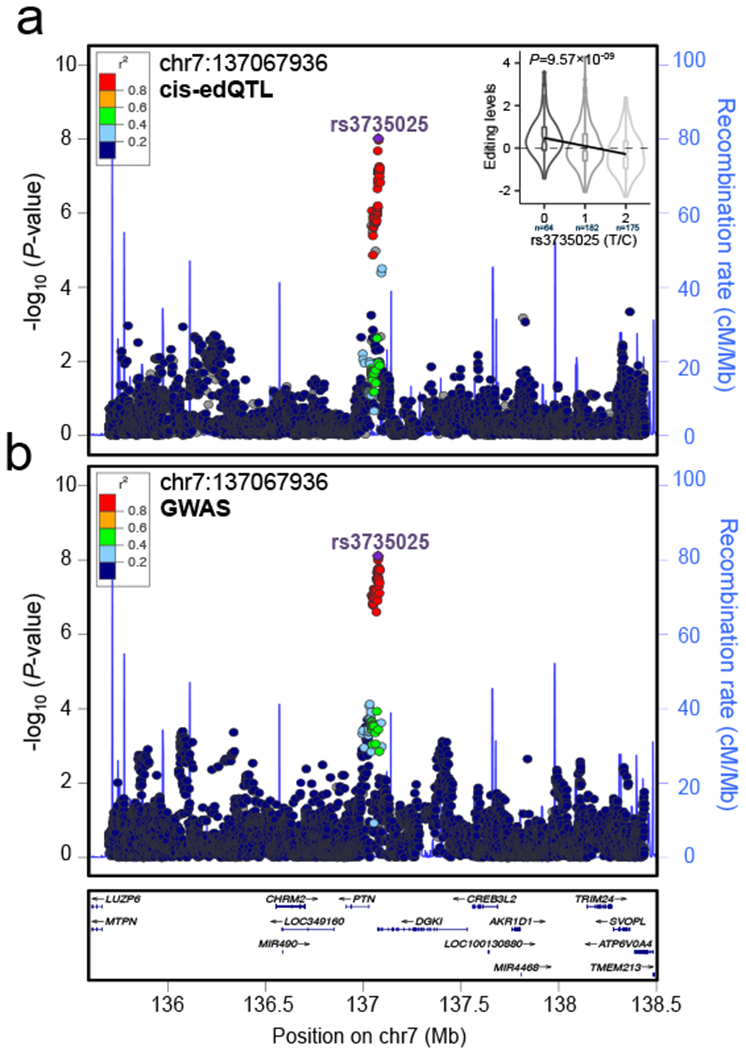
GWAS and edQTL summary statistics (beta, standard error) for SNPs within each GWAS locus were used as input for coloc2. Loci with posterior probability for hypothesis H4 (PPH4) greater than 0.5 were considered to have co-localized GWAS and edQTL signals. One example of co-localization between (a) cis-edQTL and (b) GWAS signal on chromosome 7 DGKI locus (PPH4=0.99). This specific co-localization event is specific to the DLPFC. LD estimates are colored with respect to the GWAS lead SNP (rs3735025) and coded as a heatmap from dark blue (0≥r2>0.2) to red (0.8≥r2>1.0). Recombination hotspots are indicated by the blue lines (recombination rate in cM Mb−1). (a) Inset violin plots reflects the association of editing between RNA editing site chr7:127067936 with SCZ risk allele at the GWAS index SNP in the respective loci (rs3735025; DLPFC, n0=64, n1=182, n2=175; P = 9.5×10−09, linear regression and FDR correction via matrixEQTL). Whisker violin plots show median, lower and upper quartiles, and whiskers represent minimum and maximum of adjusted RNA editing levels (y-axis) according to imputed genotype dosages (x-axis).
DISCUSSION
The recent expansion of RNA sequencing data sets has led to the identification of a huge number of RNA editing events, which affect the majority of human genes and are highly prevalent in the brain. Many such sites are commonly located in genes involved in neuronal maintenance and aberrant editing events have been associated with various neurological disorders. However, it has yet to be understood how pervasive RNA editing events are in the brain of SCZ patients and what are genetic forces guiding the regulation of these events. The ACC and DLPFC have been shown to play an important role in neurodevelopment and have been implicated in the pathophysiology of SCZ through abnormal regulation of executive function, social cognition, emotion, and self-reference. Here we have used genome-wide RNA-sequencing data derived from these tissues to advance our understanding of RNA editing mediated mechanisms involved in the molecular etiology of SCZ.
Lower levels of RNA editing were associated with postsynaptic density and glutamatergic genes as well as kainate and glutamate receptor activity genes (Figure 3). The majority of these sites are A-to-I conversions, are located in Alu elements and map to 3’UTR regions and hence the stability of the resulting RNA structure is likely to be reduced34. These genes comprise some of the most prominent and well published genes in SCZ biology, including GRIA2, GRIA3, GRIK1, GRIK2, for which aberrant RNA editing levels have been documented23–25,35, as well as NRXN1 and KALRN, which have been less studied for mechanisms related to RNA editing. NRXN1 generates multiple splice variants of the longer α-neurexin and shorter β-neurexin proteins, all of which function in synaptic adhesion, differentiation, and maturation36,37. KALRN is known to regulate neurite initiation, axonal growth, dendritic morphogenesis, and spine morphogenesis and is a key factor responsible for reduced densities of dendritic spines on pyramidal neurons in the DLPFC38, as previously reported in SCZ39. We also found enrichment for additional postsynaptic density and ion channel complex genes, including KCNIP4, which contained several altered RNA editing sites spanning its first intron (Figure 4). A major function attributed to KCNIP4 is the regulation of the potassium channel Kv4, which are significant contributors to action potential activity in neurons. The first intron of KCNIP4 is involved in alternative splicing events leading to Var IV of KCNIP4, which has been found to disrupt this current through failure to properly interact with presenilins, a component of the γ-secretase complex40,41. It is plausible that RNA editing may influence splice-site choice in KCNIP4, lending to aberrant neuronal functioning through modulation of Kv4 channel functions.
Higher levels of RNA editing were observed in genes that are essential for mitochondrial protein translation. One of these genes harboring over edited sites in SCZ was RNA Binding Motif Protein 8A (RBM8A), which has been shown to control mRNA stability and splicing, translation and is located in the 1q21.1 copy-number variation associated autism spectrum disorder, SCZ and microcephaly42,43. Moreover, several independent reports indicate mitochondrial dysfunction in schizophrenia44, which can severely affect neuronal activity, including synaptic connection, axon formation, and neuronal plasticity45. A future concerted approach of sites encoding mitochondrial genes will provide a more complete understanding of how editing in these genes impact SCZ neurobiology.
We detected that edQTLs are widespread in brain tissue and a substantial portion replicate between two brain regions. Approximately 30% of all RNA editing sites were associated with one or more nearby cis-regulatory variants. It is expected that the genomics of cis-edQTLs and their RNA editing sites align with context-specific regulation of editing, as indicated through overlap of edSNPs with regulatory elements, such as tissue-specific enhancers (Figure S17) and mapping of RNA editing sites on genes, which are predominately postnatally biased in neocortical gene expression (Figure S8). Moreover, six GWAS loci demonstrate co-localization with edQTLs and show moderate effect sizes (ß, 0.82 ± 0.26). Notably, genes NGEF and ARL6IP4 replicated between brain regions. The edSNPs and editing sites for NGEF are located within 3’UTR regions and enhancer elements. NGEF is predominantly brain expressed, particularly during early development, and shows substantial homology with the Dbl family, which are implicated in human cognitive function46. The editing site in ARL6IP4 (also known as, splicing factor SRp25) causes a non-synonymous amino acid substitution (K/R) and affects a basic region in the protein that has not been ascribed a specific function. We also identified GWAS-edQTL co-localization for ENSA, a gene which belongs to a highly conserved cAMP-regulated phosphoprotein family and is considered an endogenous regulator of ATP-sensitive potassium (KATP) channels, which rest at the intersection of cell metabolism and membrane excitability47,48. The diversity of KATP channel properties allows for exploitation by differential pharmacology, creating in-roads towards new targeted pharmacological interventions.
In conclusion, our study reveals dynamic aspects of RNA editing in human brain tissue covering hundreds of SCZ cases and control samples, including two brain regions and two large primary cohorts used for discovery and validation. Strong reproducible evidence was identified for widespread dysregulation of RNA editing in SCZ, including under-editing of glutamate receptor activity and post-synaptic density genes, which show pyramidal neuronal cell type specificity as well as over-editing in genes involved in regulation of translation and translation initiation which are specific to interneuronal cell types. Moreover, we characterize a large portion of RNA editing sites to be involved in cis-edQTLs in human brain tissue and further perform GWAS-edQTL co-localization analysis, which identified co-localization of 11 edQTLs with 6 GWAS loci. This result is supportive of a causal role of RNA editing in risk for SCZ. While these results shed new light into the mechanisms underlying the neuropathophysiology of SCZ, additional molecular studies of aberrant RNA editing sites identified in the current study and their molecular mechanisms are required to fully appreciate their functional importance for SCZ neurobiology.
MATERIALS AND METHODS
Identification of RNA editing sites from human RNA-sequencing data
RNA-sequencing data generated from the human post-mortem ACC (ncontrol=245, nSCZ=225) and DLPFC (ncontrol=286, nSCZ=254) were obtained through the CommonMind Consortium (CMC; number of uniquely mapped reads, 33,988,367 ± 12,959,625). Additional RNA-sequencing data from human post-mortem DLPFC (ncontrol=217, nSCZ=100) were obtained through the NIH Human Brain Collection Core (HBCC; number of uniquely mapped reads, 105,426,854 ± 10,0005,209). All fastq files were mapped to human reference genome hg19 using STAR version 2.4.049 and the following parameters were optimized: chimSegmentMin=15; chimJunctionOverhangMin=15; outSAMstrandField=intronMotif. For each sample, this produced a coordinate-sorted BAM file of mapped paired end reads including those spanning splice junctions. Known RNA-edited sites were curated using the publicly available database, Rigorously Annotated Database of A-to-I RNA editing (RADAR)50. Nucleotide coordinates for these well documented editing sites were then used to extract reads from each sample using a customized perl script and the samtools mpileup function51. This approach quantifies the total amount of edited reads and the total amount of un-edited reads, which map to each RNA editing site in the RADAR database for each individual sample, thereby producing a rich source of editing information both within and across all samples.
In order to identify a collection of high quality and high confidence sites, a series of detection-based thresholds were placed for each brain region and cohort, separately: 1) The minimum base quality of 25; 2) minimum mapping quality of 20 (that is, probability that a read is aligned to multiple locations); 3) probability of misalignment = 0.01 (i.e., 99% probability that a read is correctly aligned in the genome); 4) minimum read coverage per edited site to be 20. The identification of RNA editing sites has previously been reported to be prone to these biases, therefore, it is likely that changing these parameters to be more lenient would increase the number of falsely predicted editing events; 5) We also removed all known single nucleotide polymorphisms (SNPs) present in the SNP database (dbSNP; except SNPs of molecular type ‘cDNA’) and those within the 1000 Genomes Project; 6) Finally, we required that an editing site must be present in at least 80% of all samples and subsequently, must have no more than 20% missing values per sample. The resulting RNA editing data frames for the CMC ACC and DLPFC samples contained 8.3% and 9.8% missing data respectively, and the data frame for HBCC DLPFC samples contained 7.6% missing data. All missing values were imputed using predictive mean matching method in the mice R package52, using five multiple imputations and 30 iterations. The resulting sets of sites identified from these RNA-sequencing data were subsequently referred to as known RNA editing sites and were used for downstream analysis.
No statistical methods were used to pre-determine sample sizes, however our sample sizes are the largest to be reported. All samples used in this study were from participants in two large studies of schizophrenia in the United States who donated their brains upon death. Data collection and analysis were not performed blind to the conditions of the experiments. No animals or data points were excluded from the analyses for any reason.
Identification of RNA editing sites from macaque RNA-sequencing data
To examine whether drug treatment effects were responsible for overall RNA editing levels observed in SCZ, we computed overall editing derived from an RNA-sequencing study of DLPFC tissue from Rhesus macaque monkeys. Antipsychotic administration, tissue dissection and RNA-sequencing data generation was previously described elsewhere53. In brief, subjects were randomly selected for four treatment groups: (1) high doses of haloperidol (4mg/kg/d), (2) low doses of haloperidol (0.14mg/kg/d), (3) clozapine (5.2mg/kg/d), (4) vehicle. Treatments were administered orally for six months. Following a six-month treatment regime, monkeys were sacrificed using an overdose of barbiturate and transcardinally perfused with ice cold saline. DLPFC tissue was dissected from the dorsal and ventral banks of the principal sulcus (Area 46) and pulverized. Finally, gene expression data was generated using an identical RNA-sequencing protocol. Raw RNA-sequencing data was aligned to the macaque reference genome and transcriptome (mmul1) using STAR. Next, all well documented RNA editing sites in the RADAR database, which were annotated to the human reference hg19, were lifted over to the macaque reference mmul1 using the R library package rtracklayer54. These nucleotide coordinates were used to extract reads from each sample using the same customized perl script and the samtools mpileup function. We also carried out a series of matching thresholds in order to identify a collection of high confidence sites across all samples, as noted above. Notably, few differentially edited sites in SCZ are conserved in rhesus macaque and the vast majority of these conserved sites reside within Alu regions (see Table S1), which undergo significant sequence divergence and Alu retrotransposition activity among primates.
Quantifying RNA editing levels
RNA editing levels were calculated for each sample, as previously described55. In brief, we define editing levels as the total number of edited reads at a specific RNA editing site (i.e., reads with G nucleotides) over the total number reads covering the site (i.e, reads with A and G nucleotides). The resulting metric is a continuous measure, ranging from 0 (i.e., a totally un-edited site) to 1 (i.e., a completely edited site). When computing overall RNA editing levels per sample, we did not impose any sequencing coverage criteria, but instead took all known sites from the RADAR database into account that were identified in each sample in our study to obtain the total amount of editing in each sample. In this way, overall RNA editing is defined as the total number of edited reads at all known RNA editing sites over the total number reads covering all sites for each sample. These measures were used to identify relationships between editing levels and SCZ and between editing levels and expression of editing enzymes. In this way, this approach also takes into account hyper editing events, which are often RNA editing events detected at very low coverage in standard RNA-seq studies.
Differential RNA editing analysis
It is possible that RNA editing levels, similar to that observed in gene expression studies, are influenced by a number of biological and technical factors. By properly attributing multiple sources of RNA editing variation, it is possible to partially correct for some variables. Therefore, prior to differential RNA editing analysis, the editing variance for each site was partitioned into the variance attributable to each variable using a linear mixed model implemented in the R package variancePartition56. Under this framework, categorical variables (i.e., sample site, biological sex) are modeled as random effects and continuous variables (i.e., individual age, PMI) are modeled as fixed effects. Each site was considered separately and the results for all sites were aggregated afterwards. This approach enabled us to rationally include leading covariates into our downstream analysis, which may ultimately have an influence on differential RNA editing analysis. Subsequently, to identify sites with differential RNA editing levels between SCZ and control samples, we implemented linear model though the limma R package57 covarying for the possible influence of individual age, RNA integrity number (RIN), postmortem interval (PMI), sample site and sex. Significance values were adjusted for multiple testing using the Benjamini and Hochberg (BH) method to control the false discovery rate (FDR). Sites passing a multiple test corrected P-value < 0.05 were labeled significant.
To further rule out the possible confounding effects of antipsychotic medications on editing levels, we examined SCZ-related sites that were also conserved in the rhesus macaque samples, as noted above. Of these sites, we identified 24 unique sites with sufficient coverage (> 10 reads/site) across all rhesus macaque DLPFC samples that were also significantly differentially edited in at least one SCZ brain region. Using these sites, a series of pairwise comparisons were made using a linear model implemented through limma57 in order to compute the change in editing rates associated with 5.2 mg clozapine, 0.14 mg haloperidol and 4 mg haloperidol relative to vehicle treatment (Table S1E). Subsequently, we evaluated the concordance between antipsychotic induced changes in editing rates in macaques relative to SCZ-related changes in humans using a robust linear regression and found no significant associations between candidate SCZ sites and antipsychotic medications for this subset of conserved RNA editing events (Table S1E).
We also took additional measures to ensure that the landscape of RNA editing in SCZ was not confounded by differences in cellular composition. RNA-seq fastq files of adult human brain single cells were downloaded from the Gene Expression Omnibus database using the accession number GSE67835. Raw RNA-sequencing files were aligned to the human reference (hg19) using STAR alignment with default paired-end parameters. We identified 35 unique sites with sufficient coverage (> 5 reads/site) across at least 70% or more of all adult human cells that were also detected in at least one SCZ brain region. Note that a lower coverage threshold was implemented due to RNA-seq coverage being orders of magnitude lower than our bulk postmortem tissue RNA-seq samples. Next, no more than 80% missing values were allowed for each individual cell type thereby yielding a total of 181 cells dissociated from adult brain cortex which were in our analysis, including oligodendrocytes (n=16), oligodendrocyte precursors (OPCs; n=7), astrocytes (n=39), and neurons (n=119). To identify changes in editing rates associated with cell type differences, levels of RNA editing were compared between neuronal and non-neuronal cell types using a linear model, as described above. Similarly, we evaluated the concordance between neuronal-related differences in editing rates relative to SCZ-related changes in humans using a robust linear regression and found no significant associations for this subset of RNA editing events (Table S1F).
Supervised class prediction methods
In order to assess cross-validation of the SCZ-related sites, two prediction models were built using the differentially edited sites in the (1) DLPFC and (2) ACC derived from the CMC (here referred to as, training set) to predict case/control status (i.e. SCZ cases from control samples) from withheld DLPFC data derived from the HBCC (here referred to as, test set). Regularized regression models, including ElasticNet, Lasso and Ridge Regression were fit using the glmnet R package58. The penalty parameter lambda (λ) was estimated using 10-fold cross validation on each training set using the caret package in R, and ultimately set to lambda.min, the value of λ that yields minimum mean cross-validated error of the regression model. Once the models were fit, they were applied to RNA editing levels from the test set using the predict() function, which calculates the predicted log-odds of diagnostic status. Subsequently, area under the receiver operative curve (ROC) analysis was performed using the pROC package in R59. Classification accuracies were reported as area under the curve (AUC) on test samples to assess the precision of the models.
Identification of enriched sequence motifs and RNA binding protein sites
Previous studies suggest that RNA editing events are mediated by RNA-binding proteins that recognize specific sequence motifs around the RNA editing sites. Therefore, we extracted ±20 bp long sequences relative to each differentially edited and non-differentially edited site in the ACC and DLPFC, both the discovery and validation cohorts, to discover potential motifs that may determine its interaction with the RNA editing enzyme complexes. These sequences were subjected to the Multiple Em Motif Elicitation (MEME) algorithm60 (http://meme-suite.org/). This method aims to detect motifs that are significantly enriched within user defined list of sequences, regardless of their relative location to the editing sites. MEME was run using classic mode limiting the search to only the top 5 motifs whereby enrichment is measured relative to a (higher order) random model based on frequencies of the letters in the submitted sequences. As a control, we also compared these results to a motif enrichment analysis using a random selection of sequences, of equal number compared to differential and non-differential edited sites per brain region and cohort with matching GC content, in order to determine whether the enriched motifs are specific to RNA editing sites.
Sites enriched that shared a common sequence motif were then used to map binding sites of human RNA binding proteins (RBPs) using the RBPmap database61 (http://rbpmap.technion.ac.il/index.html). This produced a list of 37 motifs in the ACC and 36 motifs in the DLPFC discovery samples and 201 motifs in the DLPFC validation samples, which were independently submitted to RBPmap to identify motifs enriched in RBP targets from a database of 114 experimentally defined human motifs. The algorithm for mapping motifs on the RNA sequences is based on the Weighted-Rank approach, previously exploited in the SFmap web-server for mapping splicing factor binding, and was run in default mode.
Genes enriched with differentially edited sites
In order to identify genes enriched with differentially edited sites, we corrected each gene for gene length. As gene length is strongly correlated with the number of detectable sites in each gene, we used a hyper-geometric test to examine over-representation of differentially edited sites within a particular gene while setting a rotating background to match the total number of detectable sites for each gene.
Co-editing network analysis
To identify sites that are co-edited across SCZ and control samples, we applied unsupervised weighted gene co-expression network analysis (WGCNA)62. Signed networks were constructed for the CMC-derived ACC and DLPFC samples separately, and then again using HBCC-derived DLPFC samples, thus totaling three separate networks. To construct a network, the absolute values of Pearson correlation coefficients were calculated for all the possible editing site pairs and resulting values were transformed using a β-power of 8 for each network so that the final correlation matrix followed an approximate scale-free topology. The WGCNA cut-tree hybrid algorithm was used to detect sub-networks, or co-editing modules, within the global network with the following optimizations: minimum module size of 30 sites, tree-cut height of 0.999 and a deep-split option of 2. For each identified module, we ran singular value decomposition of each module’s editing matrix and used the resulting module eigengene (ME), equivalent to the first principal component, to represent the overall editing profiles for each module. Subsequently, modules with similar editing profiles were merged if ME values were highly correlated (R>0.9). Co-editing modules were interrogated for containing an over-representation of significantly differentially edited sites in SCZ using a one-sided Fisher’s Exact Test and an estimated odds-ratio in comparison to a background of all detected sites for each brain region and cohort. All pairwise tests were corrected using the BH method to control the FDR. To test whether ME values were significantly associated with SCZ, a linear model was applied covarying for individual age, RIN, PMI, sample site and sex using the limma package in R and all statistical tests were BH adjusted.
Gene set and cell type enrichment analyses
All differentially edited sites passing a multiple test corrected P-value <0.05 and all co-editing network modules were subjected to functional annotation. The ToppFunn module of ToppGene Suite software63 (https://toppgene.cchmc.org/) was used to assess enrichment of GO ontology terms relevant to cellular components, molecular factors, biological processes and metabolic pathways using a one-tailed hyper-geometric distribution with a Bonferroni correction. This is a proportion test that assumes a binomial distribution and independence for probability of any gene belonging to any set. We use a one-sided test because we are explicitly testing for over-representation of genes that harbor editing sites across hundreds of GO categories, without any a priori selection of candidate gene sets. A minimum of a three-gene overlap per gene set was necessary to be allowed for testing. Subsequently, modules were tested for over-representation of CNS cell type specific markers collected from a previously conducted single cell RNA-sequencing study64. In order for a gene to be labeled cell type specific, each marker required a minimum log2 expression of 1.4 units and a difference of 0.8 units above the next most abundance cell type measurement, as previously shown. Over-representation of cell type markers within co-editing modules was analyzed using a one-sided Fisher exact test to assess the statistical significance. All P-values, from all gene sets and modules, were adjusted for multiple testing using the BH procedure. We required an adjusted P-value <0.05 to claim that a cell type is enriched within a module.
BrainSpan developmental gene set enrichment analysis
BrainSpan developmental RNA-seq data (www.brainspan.org) were summarized to GENCODE10 and gene-level RPKMs were used across 528 samples. From here, only the neocortical regions were used in our analysis -- dorsolateral prefrontal cortex (DFC), ventrolateral prefrontal cortex (VFC), medial prefrontal cortex (MFC), orbitofrontal cortex (OFC), primary motor cortex (M1C), primary somatosensory cortex (S1C), primary association cortex (A1C), inferior parietal cortex (IPC), superior temporal cortex (STC), inferior temporal cortex (ITC), and primary visual cortex (V1C). Samples with RIN <= 7 were filtered and removed from subsequent analysis. Genes were defined as expressed if they were present at an RPKM of 0.5 in 80% of the samples from at least one neocortical region at one major temporal epoch, resulting in 22,141 transcripts across 299 high-quality samples ranging from post-conception weeks (PCW) 8 to 40 years of age. Finally, expression values were log-transformed (log2[RPKM+1]).
Linear regression was performed at each of 22,141 transcripts, modeling gene expression as a continuous dependent variable, as a function of a binary ‘developmental stage’ variable. A total of 11 developmental stages were analyzed. A moderated t-test, computed using the limma R package, was used to determine which genes were uniquely over-expressed and under-expressed for each specific developmental stage against all other developmental stages. Models included gender, individual as a repeated measure and ethnicity as adjustment variables. Significance values were adjusted for multiple testing using the Benjamini and Hochberg (BH) method to control the false discovery rate (FDR). After the BH correction, genes with Q-value < 0.05 and log2 fold change > 0.5 are defined as genes highly expressed in a given developmental stage, whereas genes with Q-value < 0.05 and an log2 fold change < 0.5 are defined as genes lowly expressed in a given developmental stage. These curated data formed the basis of our developmental stage gene set enrichment analysis. All processed data are available upon request. To test for over-representation of genes with differentially edited sites within a given gene set, a modified version of the GeneOverlap function in R was used so that all pairwise tests were multiple test corrected using the BH method. The Fisher’s exact test function also provides an estimated odds-ratio in comparison to a genome-wide background set to 27,546 transcripts.
cis-edQTL analysis
A total of 11,242 high confidence sites in the ACC and 7,594 sites in the DLPFC edQTL (editing quantitative trait loci) were derived using genetically inferred European samples (ACC=368, DLPFC=426) across the 6.4 million genotyped and imputed markers with imputation score ≥ 0.8 and estimated minor allele frequency ≥ 0.05. For each of the RNA editing sites, we normalized editing levels by centering and scaling each measurement through subtracting out the mean editing level value and dividing by the standard deviation. Quantile normalization was then used to fit the distribution to a standard normal distribution. Subsequently, in order to map genome-wide edQTLs, we used a linear model on the imputed genotype dosages and standardized RNA editing levels using MatrixEQT65. The RNA editing levels were covaried for sample site, sex, individual age, PMI, RIN and clinical diagnosis. In order to control for multiple tests, the FDR was estimated for all cis-edQTLs (defined as 100 KB between SNP marker and editing position), controlling for FDR across all chromosomes. We identified significant cis-edQTLs using a genome-wide significance threshold (q< 0.05). Max cis-edQTL (defined as the most significant eSNP per site, if any) were annotated for genomic regulatory elements according to ENCODE annotations implemented with the SNP nexus annotation tool (http://snp-nexus.org/index.html). To assess whether cis-edQTLs relate to known enhancer sequences, we tested for overlap between edQTLs and tissue-specific enhancer sequences from the FANTOM project covering 40 different tissues. We leveraged the SlideBase database66 (http://slidebase.binf.ku.dk/), which has well curated lists of enhancers found to be exclusively expressed across different tissues in humans. A permutation-based approach with 1,000 random permutations was used to determine statistical significance of the overlap between edSNP coordinates and enhancer regions using the R package regioneR67. A matching permutation analysis was used to assess edQTL overlap with previously generated expression QTL (eQTLs) in the ACC and DLPFC, which are publically available from synapse (syn7188631, syn7254151).
GWAS-edQTL co-localization analysis
A total of 108 genome-wide significant (P<5.0×10−8) SCZ GWAS loci68, as defined by linkage disequilibrium r2 > 0.6 start and end positions, and edQTL sites overlapping those loci were considered for analysis. For those edQTL sites overlapping these GWAS loci, extended edQTL calling was performed using an increased window size in order to obtain edQTL statistics for the entire GWAS locus. GWAS and edQTL summary statistics (beta, standard error) for SNPs within each GWAS locus were used as input to coloc269, and posterior probabilities for five hypotheses (H0, no GWAS or edQTL signal; H1, GWAS signal only; H2, edQTL signal only; H3, GWAS and edQTL signal but not co-localized; H4, co-localized GWAS and edQTL signals) were estimated for each locus. Loci with posterior probability for hypothesis H4 (PPH4) greater than 0.5 were considered to have co-localized GWAS and edQTL signals. While PPH4 ≥ 0.8 has previously been shown to demonstrate strong Bayesian evidence for co-localization, our previous work has found that many loci with PPH4 ≥ 0.5 appear qualitatively consistent with co-localization69.
Data availability.
The CommonMind investigators are committed to the release of data and analysis results, with the anticipation that data sharing in a rapid and transparent manner will speed the pace of research to the benefit of the greater research community. Data and analytical results generated through the CommonMind Consortium are available through the CommonMind Consortium Knowledge Portal: http://dx.doi.org/10.7303/syn2759792.
URLs.
Human Brain Collection Core (HBCC): https://www.nimh.nih.gov/research/research-conducted-at-nimh/research-areas/research-support-services/hbcc/index.shtml
CommonMind Consortium (CMC): http://www.synapse.org/CMC
Code availability.
Code for identifying RNA editing sites and quantifying RNA editing ratios are provided in the public repository: https://github.com/BreenMS/RNAediting
Differential RNA editing, co-editing network analyses and edQTL analysis used standard software packages.
Supplementary Material
ACKNOWLEDGEMENTS
Data were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffman-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881, AG02219, AG05138, MH06692, R01MH110921, R01MH109677, R01MH109897, U01MH103392, and contract HHSN271201300031C through IRP NIMH. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories, and the NIMH Human Brain Collection Core. CMC Leadership: Panos Roussos, Joseph D. Buxbaum, Andrew Chess, Schahram Akbarian, Vahram Haroutunian (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Enrico Domenici (University of Trento), Mette A. Peters, Solveig Sieberts (Sage Bionetworks), Thomas Lehner, Geetha Senthil, Stefano Marenco, Barbara K. Lipska (NIMH). DLPFC RNA-sequencing data, which formed the basis of the validation cohort, was provided by the National Institute of Mental Health Human Brain Collection Core (HBCC). Rhesus Macaque tissue was provided by Scott Hemby through the Stanley Medical Research Institute for Funding for Non-Human Primate Research; and funded by NIMH grant R01MH074313.
Footnotes
CommonMind Consortium members
Schahram Akbarian1,15, Jaroslav Bendl1,7, Kristen Brennand1,7, Leanne Brown1,15, Andrew Browne3, Alexander Charney1,7, Lizette Couto1,15, Greg Crawford16, Olivia Devillers1,15, Enrico Domenici17, Michele Filosi17, Elie Flatow1,15, Nancy Francoeur7, John F Fullard1,2,15, Sergio Espeso Gil1,15, Kiran Girdhar1,7, Attila Gulyás-Kovács11, Raquel Gur19, Chang-Gyu Hahn20, Vahram Haroutunian1,7,8,21, Mads Engel Hauberg9,22, Laura Huckins1,7, Rivky Jacobov1,15, Yan Jiang1,15, Jessica S Johnson1,7, Bibi Kassim1,15, Yungil Kim1,7, Lambertus Klei15, Robin Kramer23, Mario Lauria24, Thomas Lehner25, David A Lewis12, Barbara K Lipska23, Stefano Marenco23, Lara M Mangravite25, Kelsey Montgomery25, Royce Park1,15, Thanneer Malai Perumal25, Mette A Peters25, Chaggai Rosenbluh1, Douglas M Ruderfer26,27, Geetha Senthil25, Hardik R Shah2,4, Solveig K Sieberts25, Laura Sloofman1,7, Lingyun Song28, Patrick Sullivan29, Roberto Visintainer17, Jiebiao Wang30, Ying-Chih Wang2,4, Jennifer Wiseman1,15, Eva Xia11, Wen Zhang1,7, Elizabeth Zharovsky1,15
15Division of Psychiatric Epigenomics, Icahn School of Medicine at Mount Sinai, New York, New York, USA; 16Center for Genomic & Computational Biology, Duke University, Durham, North Carolina, USA; 17Laboratory of Neurogenomic Biomarkers, Centre for Integrative Biology (CIBIO), University of Trento, Trento, Italy; 18Department of Cell, Developmental and Regenerative Biology, Icahn School of Medicine at Mount Sinai, New York, New York, USA; 19Neuropsychiatry Section, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 20Neuropsychiatric Signaling Program, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 21Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York, USA; 22Department of Biomedicine, Aarhus University, Aarhus, Denmark; 23Human Brain Collection Core, National Institutes of Health, NIMH, Bethesda, Maryland, USA; 24Department of Mathematics, University of Trento, Trento, Italy; 25National Institute of Mental Health, Bethesda, MD, USA; 25Sage Bionetworks, Seattle, Washington, USA; 26Department of Medicine, Psychiatry and Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee, USA; 27Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, Tennessee, USA; 28Center for Genomic and Computational Biology, Duke University, Durham, North Carolina, USA; 29Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA; 30Department of Statistics and Data Science, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA.
COMPETING INTERESTS
None to declare.
REFERENCES
- 1.McGrath J, Saha S, Chant D & Welham J Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiologic Reviews 30, 67–76 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Owen M, Sawa A & Mortensen P Schizophrenia. The Lancet 388, 86–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirov G CNVs in neuropsychiatric disorders. Human Molecular Genetics 24, R45–R49 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Xu B et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nature Genetics 44, 1365–1369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takata A et al. Loss-of-Function Variants in Schizophrenia Risk and SETD1A as a Candidate Susceptibility Gene. Neuron 82, 773–780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ripke S et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature neuroscience 19, 1442–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier SM et al. High loading of polygenic risk in cases with chronic schizophrenia. Molecular psychiatry 21, 969–74 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Behm M, & Öhman M RNA editing: a contributor to neuronal dynamics in the mammalian brain. Trends in Genetics 32, 165–175 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Tan M et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nature reviews Molecular cell biology 17, 83–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenthal JJ, Seeburg PH A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74, 432–439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rula EY et al. Developmental modulation of GABAA receptor function by RNA editing. Journal of Neuroscience 28, 6196–6201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krestel HE, et al. A genetic switch for epilepsy in adult mice. Journal of Neuroscience 24, 10568–10578 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahlstedt H, Daniel C, Ensterö M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome research 19, 978–86 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyddon R, Dwork AJ, Keddache M, Siever LJ, Dracheva S. Serotonin 2c receptor RNA editing in major depression and suicide. The world journal of biological psychiatry 14, 590–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaisler-Salomon I, et al. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer’s disease. Neurobiology of aging 35, 1785–1791 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Kwak S, & Kawahara Y Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. Journal of molecular medicine 83, 110–120 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Herrick-Davis K, Grinde E, Niswender CM. Serotonin 5‐HT2C receptor RNA editing alters receptor basal activity: implications for serotonergic signal transduction. Journal of neurochemistry 73, 1711–1717 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Marion S, Weiner DM, Caron MG. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. Journal of Biological Chemistry 279, 2945–2954 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Sodhi MS, Burnet PW, Makoff AJ, Kerwin RW, Harrison PJ. RNA editing of the 5-HT 2C receptor is reduced in schizophrenia. Molecular psychiatry 6, 373–9 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Dracheva S et al. Increased serotonin 2C receptor mRNA editing: a possible risk factor for suicide. Molecular psychiatry 13, 1001–10 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Kubota-Sakashita M, Iwamoto K, Bundo M, Kato T1. A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Molecular brain 7, 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lomeli H, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266, 1709–1713 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Morohashi Y, et al. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. Journal of Biological Chemistry 277, 14965–14975 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Kitagawa H, et al. A regulatory circuit mediating convergence between Nurr1 transcriptional regulation and Wnt signaling. Molecular and cellular biology 27, 7486–7496 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Sullivan PF, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Molecular Psychiatry 14, 1144 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perroud N, et al. Genome-wide association study of increasing suicidal ideation during antidepressant treatment in the GENDEP project. The pharmacogenomics journal 12, 68–88 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Weißflog L, et al. KCNIP4 as a candidate gene for personality disorders and adult ADHD. European Neuropsychopharmacology 23, 436–447 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Ge X, Frank CL, Calderon de Anda F, Tsai LH. Hook3 interacts with PCM1 to regulate pericentriolar material assembly and the timing of neurogenesis. Neuron 65, 191–203 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mistry M, Gillis J, Pavlidis P. Genome-wide expression profiling of schizophrenia using a large combined cohort. Molecular psychiatry 18, 215–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irimia M, et al. Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA biology 9, 12–21 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Murphy FV 4th, Ramakrishnan V, Malkiewicz A, Agris PF. The role of modifications in codon discrimination by tRNA Lys UUU.” Nature Structural and Molecular Biology 11, 1186–91 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Javitt DC Glutamatergic theories of schizophrenia. The Israel journal of psychiatry and related sciences 47, 4–16 (2010). [PubMed] [Google Scholar]
- 36.Ching MS, et al. Deletions of NRXN1 (neurexin‐1) predispose to a wide spectrum of developmental disorders. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 153, 937–947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gauthier J et al. , Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Human genetics 130, 563–573 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kushima I, et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophrenia bulletin 38, 552–560 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glantz LA, & Lewis DA Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives of general psychiatry 57, 65–73 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Massone S, et al. RNA polymerase III drives alternative splicing of the potassium channel–interacting protein contributing to brain complexity and neurodegeneration. The Journal of Cell Biology 193, 851–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata R, et al. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4. 2 potassium channels. Journal of Biological Chemistry 278, 36445–36454 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Marshall CR, et al. CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.C., Yuen RK, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature neuroscience 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hjelm BE, et al. Evidence of mitochondrial dysfunction within the complex genetic etiology of schizophrenia Molecular neuropsychiatry 1, 201–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ben-Shachar D, & Laifenfeld D Mitochondria, synaptic plasticity, and schizophrenia. International review of neurobiology, 273–296 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Rodrigues NR, et al. Characterization of Ngef, a novel member of the Dbl family of genes expressed predominantly in the caudate nucleus. Genomics 65, 53–61 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Tinker A, Qadeer A, Alison. T. The role of ATP‐sensitive potassium channels in cellular function and protection in the cardiovascular system. British journal of pharmacology 171, 12–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akrouh A, Halcomb SE, Nichols CG, Sala-Rabanal M. Molecular biology of KATP channels and implications for health and disease. IUBMB life 61, 971–978 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES (materials and methods)
- 49.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramaswami G & Li JB RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic acids research 42, D109–D113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buuren SV & Groothuis-Oudshoorn K, mice: Multivariate imputation by chained equations in R. Journal of statistical software 45, 1–68 (2010). [Google Scholar]
- 53.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature neuroscience 19, 1442–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lawrence M, Gentleman R Carey, V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan M et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffman GE & Schadt EE, variancePartition: interpreting drivers of variation in complex gene expression studies. BMC bioinformatics 17, 483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research, 43, e47–e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedman J, Hastie T, Tibshirani R, Regularization paths for generalized linear models via coordinate descent. Journal of statistical software 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 59.Robin X, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics, 12, 12–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailey TL, Williams N, Misleh C, Li W.W., MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic acids research 34, W369–W373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paz I, Kosti I, Ares M Jr, Cline M, Mandel-Gutfreund Y, RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic acids research 42, W361–W367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Jing, et al. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research 37, W305–W311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zeisel A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Shabalin AA Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lizio M, et al. Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic acids research 4, D737–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gel B, et al. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ripke S et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobbyn A, et al. Landscape of Conditional eQTL in Dorsolateral Prefrontal Cortex and Co-localization with Schizophrenia GWAS. The American Journal of Human Genetics 102, 1169–1184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CommonMind investigators are committed to the release of data and analysis results, with the anticipation that data sharing in a rapid and transparent manner will speed the pace of research to the benefit of the greater research community. Data and analytical results generated through the CommonMind Consortium are available through the CommonMind Consortium Knowledge Portal: http://dx.doi.org/10.7303/syn2759792.