

Abstract
Motivation
Flow and mass cytometry are experimental techniques used to measure the level of proteins expressed by cells at the single-cell resolution. Several algorithms were developed in flow cytometry to increase the number of simultaneously measurable markers. These approaches aim to combine phenotypic information of different cytometric profiles obtained from different cytometry panels.
Results
We present here a new algorithm, called CytoBackBone, which can merge phenotypic information from different cytometric profiles. This algorithm is based on nearest-neighbor imputation, but introduces the notion of acceptable and non-ambiguous nearest neighbors. We used mass cytometry data to illustrate the merging of cytometric profiles obtained by the CytoBackBone algorithm.
Availability and implementation
CytoBackBone is implemented in R and the source code is available at https://github.com/tchitchek-lab/CytoBackBone.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Flow and mass cytometry are experimental techniques used to measure the level of proteins expressed by cells at single-cell resolution. Flow cytometry is currently limited to the measurement of approximately 15–22 cell markers. Mass cytometry, derived from this technique and mass spectrometry, increased the number of available measurements per single cell to more than 50 cell markers. However, the study of immune responses would be further improved by increasing the number of simultaneously measurable markers.
Computational approaches were designed in flow cytometry for simultaneously studying cell markers from different cytometric profiles. In these approaches, algorithms overlap phenotypic information in different profiles using a set of common markers. An approach based on nearest neighbor (NN) imputation was first proposed (Pedreira et al., 2008), and subsequent developments were later provided (Lee et al., 2011; O’Neill et al., 2015).
We designed a new algorithm, called CytoBackBone, which allows combining phenotypic information of cells from different cytometric profiles obtained from different cytometry panels. Importantly, profiles to combine must be obtained from the same sample or same tissue type. CytoBackBone is based on NN imputation, but introduces the notion of acceptable and non-ambiguous NNs. This notion is key to produce symmetrical and noise-free results. We used mass cytometry data to illustrate the merging performed by CytoBackBone.
2 Algorithm details
CytoBackBone is an NN-based algorithm that merges phenotypic information obtained from different cytometry panels.
In our approach, CytoBackBone combines marker expression information of two cells from different cytometric profiles if, and only if, these two cells are acceptable and non-ambiguous NNs. To find these NN cells, CytoBackBone uses the expression levels of the set of markers shared by the two cytometric profiles. We define hereafter this set of common markers as the backbone. This NN imputation is based on a k NNs algorithm (with k = 1) based on k-d tree space partitioning.
The merging of two cytometric profiles performed by the CytoBackBone algorithm is shown in a three-dimensional space (Fig. 1). In details, CytoBackBone works as follows: (i) cells of each cytometric profile with no acceptable neighbors are first excluded from the two input profiles; (ii) all acceptable and non-ambiguous NN cells remaining in the two profiles are merged into a new profile and discarded from the two input profiles; (iii) the second step is repeated until no more acceptable and non-ambiguous neighbor cells can be found in the two input profiles and (iv) finally, all excluded and remaining cells are isolated in a supplementary cytometric profile for information purposes. In the merged profile, the phenotypes of cells correspond to the average marker expressions for the set of backbone markers and to the specific marker expressions for non-backbone markers.
Fig. 1.
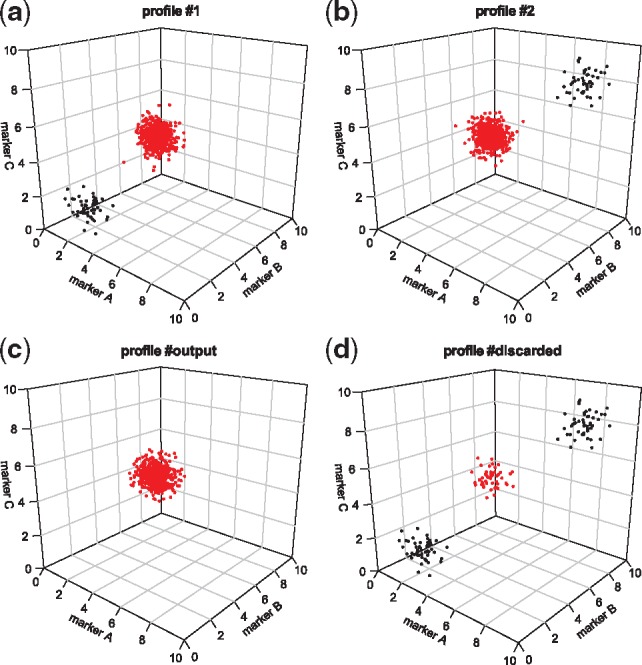
3D representation of the merging process. Axes correspond to backbone markers and cells are positioned based on their marker expressions. (a) and (b) Profiles #1 and #2 correspond to the cytometric profiles to merge. Red dots correspond to cells with acceptable neighbors between the two cytometric profiles. Black dots correspond to cells without acceptable NNs. (c) Profile #output represents the merged cytometric profile. Red dots correspond to the combined phenotypes of cells from profiles #1 and #2 with non-ambiguous and acceptable NNs. (d) Profile #discarded corresponds to cells without acceptable NNs. The presence of red dots (i.e. cells with potential acceptable neighbors) in the profile #discarded can be explained by a higher number of cells in one of the two cytometric profiles
These successive iterations ensure that the algorithm finds a new set of acceptable and non-ambiguous neighbor cells at each step. In the best situation (i.e. if the two cytometric profiles to merge are highly similar based on their backbone markers), the resulting merged profile will contain as many cells as the smallest profile.
To be acceptable neighbors, the phenotypic distance between two cells must be lower than a specific distance threshold (defined by the user). This phenotypic distance corresponds to the Euclidean distance computed as the square root of the sum of the squared expression differences for each pair of common markers. More precisely, we defined this phenotypic distance between two cells as
where c1 and c2 correspond to two cells, n to the number of backbone markers, and Median Signal Intensity (MSI) to the transformed arcs in expression intensities for the backbone marker i of the cell c.
To be defined as non-ambiguous NNs, two cells of the two different profiles must reciprocally be the closest neighbors. To identify these non-ambiguous NNs, the algorithm identifies the closest cells in cytometric profile #2 for each cell from cytometric profile #1. Then, the algorithm identifies the closest cells in cytometric profile #1 for each cell from cytometric profile #2. The merging of phenotypic information is possible only if the two cells from the two different cytometric profiles are identified as mutual non-ambiguous NNs.
A distance threshold, defining acceptable NNs, needs to be specified to avoid merging two cells with large differences in the expression levels of backbone markers: the lower the threshold, the more stringent is the merging. Distribution expressions of backbone markers can be quantile-normalized to ensure similar intensity levels across the different cytometric profiles (Bolstad et al., 2003). Such a strategy is based on the assumption that the differences of backbone marker distributions between the cytometric profiles to merged are only due to experimental variabilities.
3 Merging illustration
The efficiency of the CytoBackBone algorithm was illustrated using whole blood samples from a healthy patient. Samples were stained either with a complete mass cytometry panel of 35 markers, or with one of the four incomplete mass cytometry panels. Incomplete antibody panels were derived by omitting several markers from the complete panel, and were used to generate combined cytometric profiles (Supplementary Appendix S1 and Supplementary Table 1). As shown in Supplementary Appendix S2 and Supplementary Figure S1, the distributions of cells present in the combined Flow Cytometry Standard (FCS) files were similar to those from reference FCS file.
Merging produced by CytoBackBone are symmetrical and noise-free thanks to the notion of acceptable and non-ambiguous NNs (Supplementary Appendix S3 and Supplementary Fig. S2). Without the notion of non-ambiguous neighbors, multiple cells from one given profile can be mapped to the same cell from the other profile. The concept of acceptable neighbors avoids the merging of very distinct cells.
A benchmarking of merging settings revealed that both the length and the content of the backbone impact the merging quality (Supplementary Appendix S4 and Supplementary Fig. S3). The normalization increased the number of merged cells but only slightly the merging quality.
4 Conclusion
In principle, there is no limit to the number of cytometric profiles that can be merged by the CytoBackBone algorithm. Merging results produced by CytoBackBone are symmetrical and more-stringent compared to other approaches.
Funding
This work was supported by the IDMIT infrastructure and funded by ANR Grant No. ANR-11-INBS-0008. Nicolas Tchitchek held fellowships from the ANRS (France Recherche Nord & Sud Sida-HIV Hépatites).
Conflict of Interest: none declared.
Supplementary Material
References
- Bolstad B.M. et al. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics, 19, 185–193. [DOI] [PubMed] [Google Scholar]
- Lee G. et al. (2011) Statistical file matching of flow cytometry data. J. Biomed. Inform., 44, 663–676. [DOI] [PubMed] [Google Scholar]
- O’Neill K. et al. (2015) Deep profiling of multitube flow cytometry data. Bioinformatics, 31, 1623–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira C.E. et al. (2008) Generation of flow cytometry data files with a potentially infinite number of dimensions. Cytometry A, 73, 834–846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.