

In this report, we show that IFN-γ is produced by HSV-1 viral miR-H28 and viral replication is blocked in cells exposed to IFN-γ before infection but not during or after infection. The inevitable conclusion is that HSV-1 induces IFN-γ to curtail its spread from infected cells to uninfected cells. In essence, this report supports the hypothesis that HSV-1 encodes functions that restrict the transmission of virus from cell to cell.
KEYWORDS: HSV-1, IFN-γ, miRNA
ABSTRACT
An earlier report showed that herpes simplex virus 1 (HSV-1) expresses two microRNAs (miRNAs), miR-H28 and miR-H29, late in the infectious cycle. The miRNAs are packed in exosomes and, in recipient cells, restrict the transmission of virus from infected cells to uninfected cells. We now report that (i) miR-H28 induced the synthesis of gamma interferon (IFN-γ) in both infected cells and cells transfected with miR-H28, (ii) IFN-γ accumulated concurrently with viral proteins in infected cells, (iii) IFN-γ was produced in HEp-2 cells derived from cancer tissue and in HEK293T cells derived from normal tissue, and (iv) HSV-1 replication was affected by exposure to IFN-γ before infection but not during or after infection. The results presented in this report support the growing body of evidence indicating that HSV-1 encodes functions designed to reduce the spread of infection from infected cells to uninfected cells, possibly in order to maximize the transmission of virus from infected individuals to uninfected individuals.
IMPORTANCE In this report, we show that IFN-γ is produced by HSV-1 viral miR-H28 and viral replication is blocked in cells exposed to IFN-γ before infection but not during or after infection. The inevitable conclusion is that HSV-1 induces IFN-γ to curtail its spread from infected cells to uninfected cells. In essence, this report supports the hypothesis that HSV-1 encodes functions that restrict the transmission of virus from cell to cell.
INTRODUCTION
The genesis of this report stems from an observation made in the course of analyses of the expression of 20 genes potentially associated with innate immunity to herpes simplex virus 1 (HSV-1) infection. In brief, as illustrated below, the mRNAs encoding 18 proteins showed no significant increase or increased in abundance early in infection and declined precipitously by 18 h. In contrast, the mRNAs of the remaining two genes peaked at late time points and were barely detectable at an early time point. The two mRNAs encoded gamma interferon (IFN-γ) and CXCL10, a product of a gene activated by IFN-γ (1–3).
Interest in these observations stemmed from two considerations. First, IFN-γ is a potent inhibitor of viral infections (4–6). The induction of IFN-γ at any time in the course of HSV infection seemed contrary to the accepted idea that the intent of viruses is to replicate and to spread. Second, the countervailing consideration centered on the observation that HSV-1 encodes two microRNAs (miRNAs), miR-H28 and miR-H29, expressed late in infection. The miRNAs are packaged in exosomes and exported to uninfected cells (7). The spread of virus from cell to cell was restricted in cultures exposed to exosomes containing the miRNAs (7).
In this report, we show that IFN-γ is produced in two human cell lines of diverse origins, that IFN-γ is induced by miR-H28 but not by miR-H29, that IFN-γ and infected cell polypeptide 8 (ICP8) accumulate in the same cells, and, finally, that viral replication is blocked in cells exposed to IFN-γ before infection but not during or after infection. The inevitable conclusion is that HSV-1 induces IFN-γ to curtail its spread from infected cells to uninfected cells. In essence, this report supports the hypothesis that HSV-1 encodes functions that restrict the transmission of virus from cell to cell.
RESULTS
Accumulation of mRNAs for selected cellular genes at specific times after HSV-1(F) infection.
Replicate cultures of HEp-2 cells were mock treated or exposed to 5 PFU of HSV-1(F) per cell. At 7, 18, or 36 h after infection, the cells were harvested and total RNAs from cell lysates were extracted and reverse transcribed to cDNA, as described in Materials and Methods. The mRNAs encoding activating transcription factor 3 (ATF3), cGMP-AMP synthase (cGAS), CXCL10, histone deacetylase 1 (HDAC1), HDAC4, IFN-γ-inducible protein 16 (IFI16), IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), IFN-α, IFN-β, IFN-γ, interleukin 6 (IL-6), IFN regulatory factor 3 (IRF3), LGP2, lysine-specific histone demethylase 1 (LSD1), MDA5, MYD88, PUM1, RIG-I, and stimulator of IFN genes (STING) were normalized with respect to 18S RNA and results were shown as fold changes compared with the mRNA levels measured in control HEp-2 cells. The primers used for quantification of the mRNAs are listed in Table 1. The results were as follows (Fig. 1).
TABLE 1.
Sequences of primers used to measure gene expression
| Gene | Forward primer | Reverse primer |
|---|---|---|
| ATF3 | 5′-AAGGAAAAAGAGGCGACGAG-3′ | 5′-TCAGTTCAGCATTCACACTTTCC-3′ |
| cGAS | 5′-ACCCAGAACCCTCAAGAC-3′ | 5′-GAGGCACTGAAGAAAGTATGTC-3′ |
| CXCL10 | 5′-ACGTGTTGAGATCATTGCTAC-3′ | 5′-ATCTTTTAGACCTTTCCTTGC-3′ |
| GADD45γ | 5′-CCCGACAATGTGACCTTCTG-3′ | 5′-AAAGCCTGGATCAGCGTAAA-3′ |
| HDAC1 | 5′-TTCAAGCTCCACATCAGTC-3′ | 5′-GTCAGGGTCGTCTTCGTC-3′ |
| HDAC4 | 5′-TCAAGAACAAGGAGAAGGGCAAAG-3′ | 5′-TCCCGTACCAGTAGCGAGGGTC-3′ |
| IFI16 | 5′-AAAGTTCCGAGGTGATGC-3′ | 5′-TGACAGTGCTGCTTGTGG-3′ |
| IFIT1 | 5′-CAACCAAGCAAATGTGAG-3′ | 5′-AGGGGAAGCAAAGAAAATGG-3′ |
| IFN-α | 5′-AGAGTCACCCATCTCAGCAAG-3′ | 5′-CACCAGGACCATCAGTAAAGC-3′ |
| IFN-β | 5′-TTGTGCTTCTCCACTACAGC-3′ | 5′-CTGTAAGTCTGTTAATGAAG-3′ |
| IFN-γ | 5′-ATGTCCAACGCAAAGCAATAC-3′ | 5′-GCTCTTCGACCTCGAAACAG-3′ |
| IL-6 | 5′-GCCACTCACCTCTTCAGAACG-3′ | 5′-CAGTGCCTCTTTGCTGCTTTC-3′ |
| IRF3 | 5′-GCCGAGGCCACTGGTGCATAT-3′ | 5′-TGGGTCGTGAGGGTCCTTGCT-3′ |
| LGP2 | 5′-GAGCCAGTACCTAGAACTTAAAC-3′ | 5′-ATGGCCCCATCGAGTTTG-3′ |
| LSD1 | 5′-CCGCTCCACGAGTCAAAC-3′ | 5′-ATCCCAGAACACCCGATC-3′ |
| MDA5 | 5′-AGGAGTCAAAGCCCACCATC-3′ | 5′-GTGACGAGACCATAACGGATAAC-3′ |
| MYD88 | 5′-TACAAGGCAATGAAGAAAG-3′ | 5′-CAAGGCGAGTCCAGAA-3′ |
| PUM1 | 5′-CTTGCATTTGGACAAGGTCTG-3′ | 5′-CATTCACTACAAGGGCACCAG-3′ |
| RIG-I | 5′-AGAAGAGTACCACTTAAACCCAG-3′ | 5′-TTGCCACGTCCAGTCAATATG-3′ |
| STING | 5′-TCAGCATTACAACAACCTGCTAC-3′ | 5′-TTATCCAGGAAGCGAATGTTG-3′ |
| 18S | 5′-CTCAACACGGGAAACCTCAC-3′ | 5′-CGCTCCACCAACTAAGAACG-3′ |
FIG 1.

Temporal patterns of accumulation of mRNAs for 20 genes associated with various aspects of innate immunity. Replicate cultures of HEp-2 cells were mock treated (M) or exposed to 5 PFU of HSV-1(F) per cell. The cells were harvested at the indicated times after infection, and total RNAs were extracted and reverse transcribed to cDNA as described in Materials and Methods. Levels of mRNAs for ATF3, cGAS, CXCL10, HDAC1, HDAC4, IFI16, IFIT1, IFN-α, IFN-β, IFN-γ, IL-6, IRF3, LGP2, LSD1, MDA5, MYD88, PUM1, RIG-I, and STING were normalized to those of 18S RNA, and results are shown as fold changes, compared with the mRNA levels measured in uninfected HEp-2 cells.
The genes whose mRNAs were quantified in these assays fell into three groups. The mRNAs of genes in group 1 increased in amount at 7 h and then decreased to basal levels by 18 h after infection. The genes in this group included the ATF3, growth arrest and DNA damage-inducible protein 45γ (GADD45γ), and IL-6 genes.
The genes in group 2 included genes whose mRNA levels remained unchanged or decreased, relative to the levels observed in mock-infected cells, at 7 h after infection. Examples of genes in this group were the cGAS, IFI16, IRF3, and LGP2 genes.
Lastly, group 3 contained two genes, the CXCL10 and IFN-γ genes. The mRNAs of both genes were barely detectable at 7 h after infection but significantly increased in amount by 18 h. CXCL10 is activated by IFN-γ (1–3). The accumulation of mRNAs encoding IFN-γ was unexpected and is the focus of the remainder of this report.
IFN-γ is produced in infected human cells independent of their origin.
There are at least three trivial explanations for the data presented above, as follows. (i) The exposure of HEp-2 cells to 5 PFU of HSV-1 per cell was not sufficient to infect all cells, and IFN-γ was produced by uninfected cells. (ii) Production of IFN-γ is a characteristic of infected HEp-2 cells and not a general characteristic of HSV-1-infected human cells. (iii) Even if all cells become infected, the cells producing IFN-γ do not concurrently express viral gene products. We report two series of experiments to test these hypotheses.
Briefly, replicate cultures of HEp-2 cells or HEK293T cells were mock treated or exposed to 20 PFU of HSV-1(F) per cell, to saturate susceptible cells with virus. At 7 or 18 h after infection, the cells were harvested, total RNAs were extracted, and IFN-γ mRNAs was assayed as described above. The results showed that, at 20 PFU per cell, IFN-γ mRNA accumulated in both HEp-2 and HEK293T cells at 18 h after infection (Fig. 2). The key conclusions are that both HEp-2 and HEK293T cells exposed to 20 PFU of virus per cell produced IFN-γ at late times after HSV-1 infection.
FIG 2.
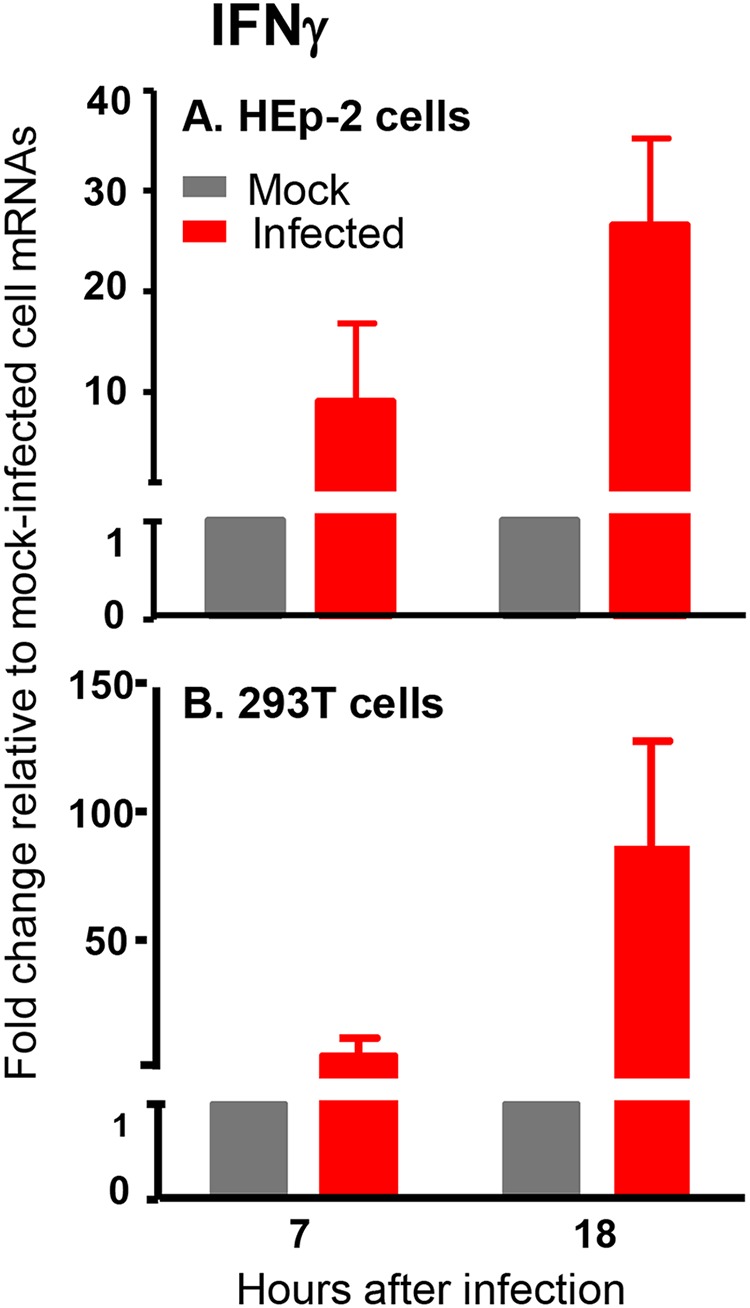
Analyses of accumulation of IFN-γ mRNA in HEp-2 and HEK293T cells after HSV-1(F) infection. Replicate cultures of HEp-2 cells (A) and HEK293T cells (B) were mock treated or exposed to 20 PFU of HSV-1(F) per cell. The cells were harvested at the indicated times after infection, and total RNAs from cell lysates were extracted and reverse transcribed to cDNA as described in Materials and Methods. Levels of mRNA encoding IFN-γ were normalized to those of 18S RNA, and results are shown as fold changes, compared with the mRNA levels measured in uninfected HEp-2 or HEK293T cells.
The objective of the second series of experiments was to test the hypothesis that infected cells accumulated viral gene products or IFN-γ but not both. In this series of experiments, HEp-2 cells seeded on coverslips were mock infected or infected with 5 PFU per cell for 1 h. The inoculum was replaced with fresh culture medium. The cells were fixed at 6 or 20 h after infection and were stained with 4′,6-diamidino-2-phenylindole (DAPI) or reacted with antibodies to ICP8 and or IFN-γ, as indicated in Fig. 3. The images were captured at ×63 magnification with the aid of a laser scanning microscope. As expected, ICP8 was detected in the nuclei of the infected cells, whereas IFN-γ was detected in the cytoplasm; the two were detected in the same cells in cultures stained with both antibodies (Fig. 3). Analyses of the infected cells fixed at 20 h after infection showed that slightly more than 50% of the cells accumulated both ICP8 and IFN-γ. Since ICP8 is made relatively early after infection, whereas IFN-γ is made relatively late, the results suggest that viral gene products and IFN-γ are made sequentially by the same cells.
FIG 3.
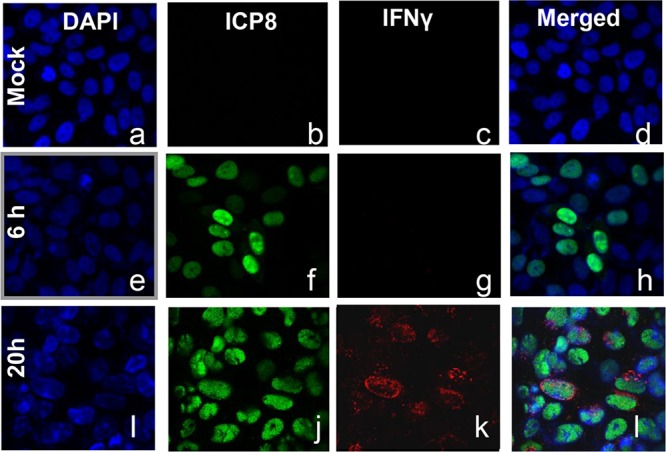
Accumulation of ICP8 (a viral gene product) and IFN-γ in the same cells. HEp-2 cells were mock infected or infected with 5 PFU of HSV-1 per cell. After 1 h of exposure, the inoculum was replaced with fresh culture medium. The cells were fixed at the times shown and were costained with an antibody to ICP8 (green), an antibody to IFN-γ (red), and DAPI (blue). The images were captured using a confocal laser scanning microscope. Magnification, ×63.
HSV-1 miR-H28 transfected into HEp-2 cells induces the synthesis of IFN-γ.
HSV-1 encodes numerous miRNAs made at different times throughout the replicative cycle (8–11). Published studies focused on two miRNAs, miR-H28 and miR-H29, made very late in infection (7). Both miRNAs are packaged into exosomes. Moreover, viral replication was adversely affected in cells transfected with miR28 or miR29 mimics (7). In light of the observation that induction of IFN-γ and the synthesis of the two miRNAs both take place late in the reproductive cycle, the question of whether the miRNAs induce IFN-γ on transfection into uninfected cells arose. To test this hypothesis, replicate cultures of HEp-2 cells were mock treated or transfected with 100 nM nontarget (NT), miR-H5-3p, miR-H6-3p, miR-H1-5p, miR-H26, miR-H27, miR-H28, or miR-H29 mimics. At 7 and 18 h after transfection, the cells were harvested, total RNAs were extracted and reverse transcribed into cDNA, and mRNAs encoding IFN-α, IFN-β, and IFN-γ were normalized with respect to 18S RNA. The results are shown as fold changes, compared with the mRNA levels detected in HEp-2 cells (Fig. 4). The results shown in Fig. 4 indicate that miR-H28 activates significant but relatively low levels of IFN-α and IFN-β and significantly higher levels of IFN-γ. It is noteworthy that IFN-α and IFN-β levels were higher at 7 h after transfection, whereas the levels of IFN-γ were higher at 18 h after transfection than at 7 h. The results suggest that miR-H28 activates the synthesis of IFN-γ. The results obtained in these studies do not shed light on the target of miR-H29. The results do not rule out the possibility that other HSV-1 gene products also induce IFN-γ late in infection.
FIG 4.
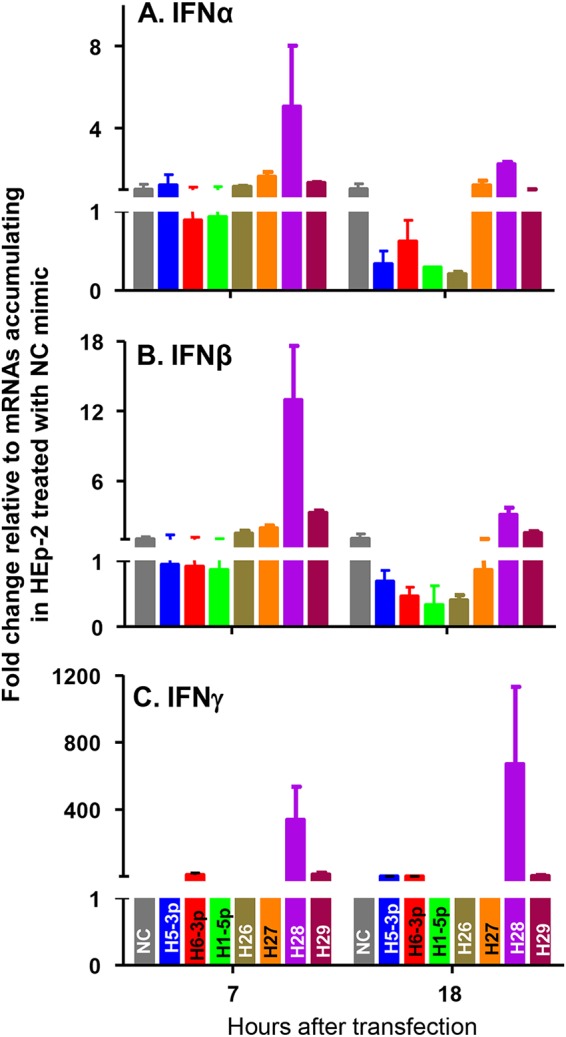
Accumulation of mRNAs for IFN-α, IFN-β, and IFN-γ with HEp-2 transfection of miR-H28 mimics. Replicate cultures of HEp-2 cells were mock treated or transfected with 100 nM NC, miR-H5-3p, miR-H6-3p, miR-H1-5p, miR-H26, miR-H27, miR-H28, or miR-H29 mimic. The cells were harvested at the indicated times after transfection, and total RNAs from cell lysates were extracted and reverse transcribed to cDNA as described in Materials and Methods. Levels of mRNAs for IFN-α (A), IFN-β (B), and IFN-γ (C) were normalized to those of 18S RNA, and results are shown as fold changes, compared with the mRNA levels measured in HEp-2 cells. NC, miRNA mimic negative control.
HSV-1 replication is blocked in cells exposed to IFN-γ before infection but not during or after infection.
A central question posed by the studies described above is whether cells infected with HSV-1 are affected by IFN-γ at any time in the course of its replication. We report two series of experiments.
In the first series of experiments, replicate cultures of HEp-2 cells were mock treated, pretreated with IFN-γ (250 ng/ml) for 24 h before infection, or treated concurrently with 1 PFU of HSV-1(F) per cell. The results shown in Fig. 5A indicated that viral replication was inhibited in cells exposed to IFN-γ before infection but not in cells exposed to IFN-γ during and presumably after infection.
FIG 5.
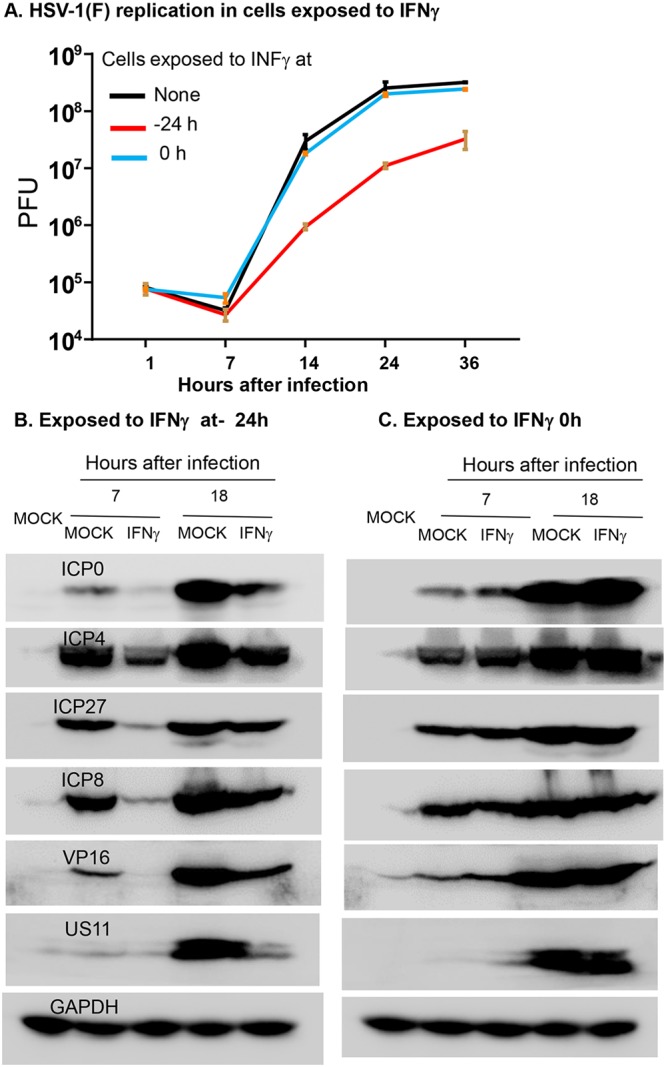
Replication of HSV-1(F) and accumulation of viral proteins in HEp-2 cells exposed to IFN-γ. (A) Replicate cultures of HEp-2 cells were mock treated or exposed to 250 ng/ml of IFN-γ for 24 h before infection or concurrently with 1 PFU of HSV-1(F) per cell. After 1 h, the inoculum was replaced with fresh medium. (B and C). The HEp-2 cells were mock treated or exposed to IFN-γ 24 h before infection or concurrently with 5 PFU of HSV-1(F) per cell. The cells were harvested at 7 or 18 h after infection, solubilized, subjected to electrophoresis in denaturing gels, and probed with specific antibodies to ICP0, ICP4, ICP8, ICP27, VP16, and US11. GAPDH served as a loading control.
In the second series of experiments, we examined the accumulation of viral proteins. The design of the experiment was similar to that described above except that HEp-2 cells exposed to IFN-γ and 5 PFU of HSV-1(F) per cell were harvested at 7 or 18 h after infection, solubilized, subjected to electrophoresis in denaturing gels, and probed with antibodies to ICP0, ICP4, ICP27, ICP8, VP16, and US11. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. The results shown in Fig. 5B indicated that IFN-γ affected the accumulation of viral proteins in cells exposed to IFN-γ before infection but not after or during infection.
DISCUSSION
HSV-1 infects and establishes lifetime residence, in the form of latent infections, in nearly 100% of the human adult population (12–14). The most frequent form of transmission is by physical contact between small recurrent lesions induced by reactivation of latent virus and mucous membranes of uninfected individuals (15–17). The lesions caused by reactivated virus are usually small and associated with minimal discomfort (18–20). Transmission of virus to uninfected individuals would likely be greatly reduced if the lesions were large and associated with significant discomfort or were repulsive in appearance. The central question posed in numerous studies is what limits the spread of virus from cell to cell.
In principle, the mission of viruses is to replicate and to spread. Control of virus replication and spread are generally the functions of innate and adaptive immunity (21–30). What makes HSV-1 particularly interesting is that it has evolved functions that specifically trigger an innate immune response designed to restrict the spread of virus from infected cells to uninfected cells in order to maximize the transmission of HSV-1 from infected individuals to uninfected individuals (15, 31–33).
The evidence that HSV evolved functions to control its replication and spread emerged from several studies (7, 31, 34–37). Studies by Kalamvoki et al. (31) demonstrated that STING is exported in exosomes from infected cells to uninfected cells and the recipients of STING yield less virus than cells exposed to exosomes lacking STING. These studies were followed by the aforementioned observation that HSV encodes two miRNAs, miR-H28 and miR-H29, that are expressed very late in infection (7). The miRNAs are packaged in exosomes and reduce virus yields and cell-to-cell spread in recipient cells. In this report, we show that miR-H28 induces IFN-γ expression; cells exposed to IFN-γ before infection become resistant to HSV-1. Our results do not rule out the possibility of the existence of other viral functions designed to control viral yields and the spread of virus from cell to cell.
The evolution of viral genes designed to limit pathogenicity is not unexpected (36, 38, 39). Viruses with high mortality rates in human populations do not survive unless they are maintained in another host in which the spread of infection and virulence are compatible with survival of the host and maintenance of the infectious agent (29, 39, 40). To remain stably associated with human populations, viruses must shed functions that disable effective innate immune responses or, as in the case of HSV-1, evolve functions designed to trigger a potent appropriate response.
MATERIALS AND METHODS
Cell lines and virus strains.
HEp-2 cells, HEK293T cells, and Vero cells were obtained from the American Type Culture Collection. HEp-2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (high glucose) supplemented with 5% (vol/vol) fetal calf serum, and HEK293T cells were cultured in DMEM supplemented with 10% (vol/vol) fetal calf serum. Vero cells were cultured in DMEM (high glucose) supplemented with 5% (vol/vol) newborn calf serum. HSV-1(F) is a limited-passage prototype HSV-1 strain used in our laboratories, as reported previously (41); it was amplified and titrated on Vero cells.
RNA isolation and real-time quantitative PCR.
Replicate cultures of HEp-2 cells in 6-well plates were mock treated or exposed to 5 PFU of HSV-1(F) per cell for 7, 18, or 36 h (Fig. 1) or HEp-2 cells or HEK293T cells in 6-well plates were mock treated or exposed to 20 PFU of HSV-1(F) per cell for 7 or 18 h (Fig. 2). The cells were then harvested at the indicated times after infection. Total RNA was isolated using TRI Reagent solution (Invitrogen) and treated with DNase I (TaKaRa). cDNA was synthesized from 0.5 μg RNA with the aid of the ReverTra Ace qPCR RT kit (Toyobo), in accordance with instructions provided by the suppliers. Gene expression was measured by real-time quantitative PCR analysis using SYBR green Realtime PCR Master Mix (Toyobo) in step 1 and a real-time PCR system (AB Applied Biosystems), using the primers shown in Table 1. Transcript expression was normalized to that of 18S RNA, and relative expression changes were determined using the 2−ΔΔCT method.
IFN-γ protein and antibodies.
Recombinant human IFN-γ protein was purchased from Sino Biological (product no. 11725-HNAS). Antibodies against ICP0, ICP4, ICP8 (Rumbaugh-Goodwin Institute for Cancer Research, Inc.), ICP27 (42), VP16 (43), and US11 (44) have been described elsewhere. The anti-GAPDH antibody (product no. 2118) and anti-IFN-γ antibody (product no. AF-285-SP) were purchased from Cell Signaling Technology and R&D Systems, respectively.
Immunofluorescence assays.
Ep-2 cells (5 × 104) seeded on slides and incubated for 16 h were mock infected or exposed to 5 PFU of HSV-1(F) per cell for 1 h. The inoculum was replaced with fresh culture medium. At the indicated times after infection, the cells were rinsed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 30 min at room temperature, and permeabilized with 0.1% Triton X-100. The cells either were reacted overnight at 4°C with anti-ICP8 antibody and then for 1 h at room temperature with anti-mouse IgG secondary antibody conjugated to Alexa Fluor Plus 488 (product no. A32766; Invitrogen) or were reacted overnight at 4°C with anti-IFN-γ antibody and then for 1 h at room temperature with Cy3-labeled anti-goat IgG (H+L) secondary antibody (product no. A0502; Beyotime). The cells were then washed with PBS and embedded in DAPI-containing mounting medium (product no. 18961S; Cell Signaling Technology). The images were captured and processed using a confocal laser scanning microscope, at a magnification of ×63.
Transfection of miRNA mimics.
miRNA mimics were purchased from GenePharma. The sequences of miRNA mimics are shown in Table 2. The NT mimic was used as a negative control. For transfection of miRNA mimics, HEp-2 cells (5 × 105 cells per well) seeded in 6-well plates were transfected with miRNA mimics at a final concentration of 100 nM. At 7 or 18 h after transfection, the cells were harvested for real-time PCR analyses. All transfections were carried out using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions.
TABLE 2.
Sequences of miRNA mimics
| miRNA mimic | Sense | Antisense |
|---|---|---|
| NT | 5′-UUCUCCGAACGUGUCACGUUU-3′ | 5′-ACGUGACACGUUCGGAGAAUU-3′ |
| miR-H1-5p | 5′-GAUGGAAGGACGGGAAGUGGA-3′ | 5′-CACUUCCCGUCCUUCCAUCUU-3′ |
| miR-H5-3p | 5′-GUCAGAGAUCCAAACCCUCCGG-3′ | 5′-GGAGGGUUUGGAUCUCUGACUU-3′ |
| miR-H6-3p | 5′-CACUUCCCGUCCUUCCAUCCC-3′ | 5′-GAUGGAAGGACGGGAAGUGUU-3′ |
| miR-H26 | 5′-UGGCUCGGUGAGCGACGGUC-3′ | 5′-CCGUCGCUCACCGAGCCAUU-3′ |
| miR-H27 | 5′-CAGACCCCUUUCUCCCCCCUCUU-3′ | 5′-GAGGGGGGAGAAAGGGGUCUGUU-3′ |
| miR-H28 | 5′-CGAUGGUCGUCUGUGGAU-3′ | 5′-CCACAGACGACCAUCGUU-3′ |
| miR-H29 | 5′-CUGGAGGCGGGCAAGGACUACC-3′ | 5′-UAGUCCUUGCCCGCCUCCAGUU-3′ |
Immunoblotting.
Replicate cultures of HEp-2 cells in 12-well plates were mock treated, pretreated with 250 ng/ml of recombinant IFN-γ for 24 h before infection, or posttreated with 250 ng/ml of recombinant IFN-γ at 0 h after infection and then were exposed to 1 PFU of HSV-1(F) per cell. Cells were harvested at the indicated times after processing in experiments and were lysed with a RIPA lysis buffer (Beyotime) supplemented with 1 mM protease inhibitor phenylmethyl sulfonyl fluoride (PMSF) (Beyotime). Cell lysates were heat denatured, separated by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Millipore). The proteins were detected by incubation with appropriate primary antibody, followed by horseradish peroxidase-conjugated secondary antibody (Pierce) and the enhanced chemiluminescence (ECL) reagent (Pierce), and exposed to a film.
Virus titration.
HEp-2 cells (7 × 105 cells per well) seeded in 6-well plates were mock treated, pretreated with 250 ng/ml of IFN-γ for 24 h before infection, or posttreated with 250 ng/ml of IFN-γ at 0 h after infection and then were exposed to 1 PFU of HSV-1(F) per cell. The cells were harvested at 1, 7, 14, 24, and 36 h postinfection. Viral progenies were titrated on Vero cells after three freeze-thaw cycles and brief sonication.
ACKNOWLEDGMENTS
These studies were supported by the Shenzhen Overseas High-Caliber Peacock Foundation under grant KQTD2015071414385495, Shenzhen Science and Innovation Commission Project grants JCYJ20160229153541081 and JCYJ20170411094933148, Dapeng Project grant KY20180101 to the Shenzhen International Institute for Biomedical Research, and Guangdong Nature Science Foundation grant 2016A030308007 to Guangzhou Medical University.
We declare no conflicts of interest.
REFERENCES
- 1.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. 2018. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation: a target for novel cancer therapy. Cancer Treat Rev 63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamassia N, Calzetti F, Ear T, Cloutier A, Gasperini S, Bazzoni F, McDonald PP, Cassatella MA. 2007. Molecular mechanisms underlying the synergistic induction of CXCL10 by LPS and IFN-γ in human neutrophils. Eur J Immunol 37:2627–2634. doi: 10.1002/eji.200737340. [DOI] [PubMed] [Google Scholar]
- 3.Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. 2014. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev 13:272–280. doi: 10.1016/j.autrev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Kang S, Brown HM, Hwang S. 2018. Direct antiviral mechanisms of interferon-gamma. Immune Netw 18:e33. doi: 10.4110/in.2018.18.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spalluto CM, Singhania A, Cellura D, Woelk CH, Sanchez-Elsner T, Staples KJ, Wilkinson T. 2017. IFN-γ influences epithelial antiviral responses via histone methylation of the RIG-I promoter. Am J Respir Cell Mol Biol 57:428–438. doi: 10.1165/rcmb.2016-0392OC. [DOI] [PubMed] [Google Scholar]
- 6.McTavish H, Zerebiec KW, Zeller JC, Shekels LL, Matson MA, Kren BT. 2019. Immune characteristics correlating with HSV-1 immune control and effect of squaric acid dibutyl ester on immune characteristics of subjects with frequent herpes labialis episodes. Immun Inflamm Dis 7:22. doi: 10.1002/iid3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han Z, Liu X, Chen X, Zhou X, Du T, Roizman B, Zhou G. 2016. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. Proc Natl Acad Sci U S A 113:E894–E901. doi: 10.1073/pnas.1525674113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du T, Han Z, Zhou G, Zhou G, Roizman B. 2015. Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc Natl Acad Sci U S A 112:E49–E55. doi: 10.1073/pnas.1422657112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Li Q. 2012. The miRNAs of herpes simplex virus (HSV). Virol Sin 27:333–338. doi: 10.1007/s12250-012-3266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurak I, Hackenberg M, Kim JY, Pesola JM, Everett RD, Preston CM, Wilson AC, Coen DM. 2014. Expression of herpes simplex virus 1 microRNAs in cell culture models of quiescent and latent infection. J Virol 88:2337–2339. doi: 10.1128/JVI.03486-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 14.Antoine TE, Park PJ, Shukla D. 2013. Glycoprotein targeted therapeutics: a new era of anti-herpes simplex virus-1 therapeutics. Rev Med Virol 23:194–208. doi: 10.1002/rmv.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmichael JC, Yokota H, Craven RC, Schmitt A, Wills JW. 2018. The HSV-1 mechanisms of cell-to-cell spread and fusion are critically dependent on host PTP1B. PLoS Pathog 14:e1007054. doi: 10.1371/journal.ppat.1007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. doi: 10.3390/v5020678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tovaru S, Parlatescu I, Tovaru M, Cionca L, Arduino PG. 2011. Recurrent intraoral HSV-1 infection: a retrospective study of 58 immunocompetent patients from Eastern Europe. Med Oral Patol Oral Cir Bucal 16:e163–e169. doi: 10.4317/medoral.16.e163. [DOI] [PubMed] [Google Scholar]
- 19.Ramchandani M, Kong M, Tronstein E, Selke S, Mikhaylova A, Magaret A, Huang ML, Johnston C, Corey L, Wald A. 2016. Herpes simplex virus type 1 shedding in tears and nasal and oral mucosa of healthy adults. Sex Transm Dis 43:756–760. doi: 10.1097/OLQ.0000000000000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goade DE, Nofchissey RA, Kusewitt DF, Hjelle B, Kreisel J, Moore J, Lyons CR. 2001. Ultraviolet light induces reactivation in a murine model of cutaneous herpes simplex virus-1 infection. Photochem Photobiol 74:108–114. doi:. [DOI] [PubMed] [Google Scholar]
- 21.Luecke S, Paludan SR. 2015. Innate recognition of alphaherpesvirus DNA. Adv Virus Res 92:63–100. doi: 10.1016/bs.aivir.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 22.She M, Jiang H, Chen X, Chen X, Liu X, Zhang X, Roizman B, Zhou GG. 2019. GADD45γ activated early in the course of herpes simplex virus 1 infection suppresses the activation of a network of innate immunity genes. J Virol 93:e02201-18. doi: 10.1128/JVI.02201-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Liu Y, Wu J, Roizman B, Zhou GG. 2018. Innate responses to gene knockouts impact overlapping gene networks and vary with respect to resistance to viral infection. Proc Natl Acad Sci U S A 115:E3230–E3237. doi: 10.1073/pnas.1720464115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Qu L, Liu Y, Roizman B, Zhou GG. 2017. PUM1 is a biphasic negative regulator of innate immunity genes by suppressing LGP2. Proc Natl Acad Sci U S A 114:E6902–E6911. doi: 10.1073/pnas.1708713114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouwendijk WJ, Laing KJ, Verjans GM, Koelle DM. 2013. T-cell immunity to human alphaherpesviruses. Curr Opin Virol 3:452–460. doi: 10.1016/j.coviro.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lafaille FG, Ciancanelli MJ, Studer L, Smith G, Notarangelo L, Casanova JL, Zhang SY. 2015. Deciphering human cell-autonomous anti-HSV-1 immunity in the central nervous system. Front Immunol 6:208. doi: 10.3389/fimmu.2015.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai HS, Caligiuri MA. 2018. Molecular basis for the recognition of herpes simplex virus type 1 infection by human natural killer cells. Front Immunol 9:183. doi: 10.3389/fimmu.2018.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melchjorsen J, Matikainen S, Paludan SR. 2009. Activation and evasion of innate antiviral immunity by herpes simplex virus. Viruses 1:737–759. doi: 10.3390/v1030737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chahar HS, Bao X, Casola A. 2015. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses 7:3204–3225. doi: 10.3390/v7062770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collins SE, Noyce RS, Mossman KL. 2004. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 78:1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalamvoki M, Du T, Roizman B. 2014. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc Natl Acad Sci U S A 111:E4991–E4996. doi: 10.1073/pnas.1419338111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turan A, Grosche L, Krawczyk A, Muhl-Zurbes P, Drassner C, Duthorn A, Kummer M, Hasenberg M, Voortmann S, Jastrow H, Dorrie J, Schaft N, Kraner M, Dohner K, Sodeik B, Steinkasserer A, Heilingloh CS. 2019. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J Cell Biol 218:508–523. doi: 10.1083/jcb.201801151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.da Silva LF, Jones C. 2013. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res 175:101–109. doi: 10.1016/j.virusres.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, Zhong CQ, Xia W, Zhou R, Zheng C, Han J. 2015. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17:229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Huang R, Zhou X, Ren S, Liu X, Han Z, Zhou GG. 2019. Effect of loss-of-function of the herpes simplex virus-1 microRNA H6-5p on virus replication. Virol Sin 34:386. doi: 10.1007/s12250-019-00111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Diao C, Yang X, Yang Z, Liu M, Li X, Tang H. 2016. ICP4-induced miR-101 attenuates HSV-1 replication. Sci Rep 6:23205. doi: 10.1038/srep23205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu B, Rivera-Gonzalez R, Smith CA, DeLuca NA. 1993. Herpes simplex virus infected cell polypeptide 4 preferentially represses Sp1-activated over basal transcription from its own promoter. Proc Natl Acad Sci U S A 90:9528–9532. doi: 10.1073/pnas.90.20.9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgado-Eckert E, Ojosnegros S, Beerenwinkel N. 2011. The evolution of virulence in RNA viruses under a competition-colonization trade-off. Bull Math Biol 73:1881–1908. doi: 10.1007/s11538-010-9596-2. [DOI] [PubMed] [Google Scholar]
- 39.Alizon S, Hurford A, Mideo N, Van Baalen M. 2009. Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J Evol Biol 22:245–259. doi: 10.1111/j.1420-9101.2008.01658.x. [DOI] [PubMed] [Google Scholar]
- 40.Neumann S, El Maadidi S, Faletti L, Haun F, Labib S, Schejtman A, Maurer U, Borner C. 2015. How do viruses control mitochondria-mediated apoptosis? Virus Res 209:45–55. doi: 10.1016/j.virusres.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 42.Ackermann M, Braun DK, Pereira L, Roizman B. 1984. Characterization of herpes simplex virus 1 α proteins 0, 4, and 27 with monoclonal antibodies. J Virol 52:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKnight JL, Kristie TM, Roizman B. 1987. Binding of the virion protein mediating α gene induction in herpes simplex virus 1-infected cells to its cis site requires cellular proteins. Proc Natl Acad Sci U S A 84:7061–7065. doi: 10.1073/pnas.84.20.7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roller RJ, Roizman B. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J Virol 66:3624–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]