

Abstract
Background
Current routine tests for premarital screening of β‐thalassemia carriers are not applicable for diagnosis of rare atypical minor β‐thalassemia cases. A more specialized laboratory evaluation for them is the measurement of β/α chain synthesis ratio with the assistance of radioactive amino acids. This method is also no longer routinely accessible. Consequently it is required to establish a rapid, trouble‐free, and reliable method that encompasses all the cases of β‐thalassemia carriers. Therefore we have determined β/α‐globin mRNA ratio by applying relative qRT‐PCR in various β‐thalassemia patients.
Methods
Reticulocytes RNA extraction and subsequent cDNA synthesis were performed, followed by relative qRT‐PCR for α‐ and β‐globin chain genes and ‐actin gene as an endogenous reference. ‐Globin gene ratio was then evaluated with the Pfaffl method.
Results
The mean of β/α ratio was 0.99, 0.81, 0.69, and 0.69 for normal population, minor, intermediate, and major β‐thalassemia, respectively. Approximately 6% of cases with minor thalassemia RBC index and normal HbA2 and having a decreased β/α ratio were located in the minor β‐thalassemia group. The mean of β/α mRNA ratio in normal individuals and minor β‐thalassemia was significantly different with all other groups (P‐value < 0.05). Nevertheless, there was no such association between β/α mRNA ratio in major and intermediate β‐thalassemia.
Conclusion
According to the significant differences achieved, no overlapping between minor β‐thalassemia and normal group, capability of diagnosing atypical minor β‐thalassemia, and accessibility of this technique, we can declare that this method could be suggested as a routine premarital screening test for β‐thalassemia carriers.
Keywords: thalassemia, premarital screening, relative qRT‐PCR
INTRODUCTION
Thalassemia, a genetic defect that is inherited as autosomal recessive, is one of the most frequent single gene diseases worldwide that is the result of abnormal quantity of hemoglobin chains synthesis, due to mutations in the globin chains 1. Two major clinical forms are the α‐ and β‐thalassemia. α‐Thalassemia with absence or reduced α‐globin chain takes place as a result of affected ‐globin genes with two copies, located on the short arm of chromosome 16, while β‐thalassemia results in reduced synthesis of β‐globin chain as a consequence of point mutations on ‐globin gene located as a cluster on the short arm of chromosome 11 2. As it was declared, in the β‐thalassemia there is reduction (β+) or lacking (β0) synthesis of β‐globin chains 3. Accordingly, an imbalanced amount of β‐globin chain results in relative excess of alpha chain, precipitating in erythrocytes, since they are not stable enough to form soluble tetramers. Therefore notable variability exists in the clinical manifestations of β‐thalassemia due to the degree of alpha chain excess, ranging from asymptomatic phenotype (minor thalassemia) to severe anemia (major thalassemia). Major thalassemia is a transfusion‐dependent and life‐threatening form of thalassemia appearing in infancy or childhood in which normal hemoglobin is absent, characterized by severe anemia, enlargement of the heart, liver, spleen, and skeletal deformation 4.
One of the most significant ways to prevent the birth of major β‐thalassemia children is premarital screening of the carriers, since elective abortion of the affected fetus has many restrictions, such as religious boundaries on abortion in Islamic countries; therefore, prenatal diagnosis is not an appropriate method to avoid β‐thalassemia birth. Nowadays premarital screening is performed in most at‐risk populations, such as Iranian in which the preventive strategies started in 1995. Although this strategy has extremely lessened the birth prevalence of affected homozygotes, unfortunately it has not eliminated it completely 5.
One of the main reasons is that the current routinely used tests for premarital screening of β‐thalassemia carriers are CBC test, HbA2 measurement, and Hb electrophoresis that are not applicable for diagnosis of rare atypical minor β‐thalassemia cases (minor β‐thalassemia with normal HbA2 level such as heterozygote delta β‐thalassemia, minor β‐thalassemia with iron deficiency, simultaneous defect in α‐ and β‐globin, and other cases with unknown reasons; 5).
Consequently it is required to establish a rapid, trouble‐free, and reliable method that encompasses all the cases of β‐thalassemia carriers. In the current study, we have determined β/α‐globin mRNA ratios in β‐thalassemia by applying relative quantitative real‐time reverse transcription polymerase chain reaction (relative qRT‐PCR) to quantify in vitro synthesis of β‐ and α‐globin chains in various β‐thalassemia subjects and normal population.
MATERIALS AND METHODS
Sample Collection
Blood samples of 2 ml each, were collected in K3EDTA vials from 45 major β‐thalassemia, 18 intermediate β‐thalassemia, 84 minor β‐thalassemia, and 29 healthy individuals with normal Hb profile. Patients with major thalassemia were screened according to clinical and laboratory findings, such as Hb electrophoresis, and minor thalassemia cases were identified by RBC index and HbA2 level. The samples were achieved at Dastgheib and Motahari hospital, during December 2011 to May 2012, and stored at 4°C for less than 6 hours prior to RNA extraction.
Reticulocyte RNA Preparation and cDNA Synthesis
Blood samples were centrifuged, followed by removing Plasma and buffy coat layer. The packed RBCs were then diluted 20 times with 1× PBS and passed through the flexible in‐line red cell filtration system Leucoflex LCR Diamond (Macopharma, France). After centrifuging for 15 min at 150 × g (low spin centrifugation), platelets were separated from erythroid cells. Then the erythroid cells RNA were extracted with RNX‐Plus (Cinnagen, Iran) and dissolved in 50 μl Tris EDTA buffer. RNA concentration was measured at 260 and 280 nm by using a spectrophotometer (Biochrom, UK) to estimate its quantity and quality for cDNA synthesis. Subsequently, 1 μg of isolated RNA was reverse transcribed in a final volume of 20 μl using the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas, EU) based on the supplier's instruction.
Relative qRT‐PCR
Relative quantitative real‐time PCR was performed for α‐ and β‐globin chain as target genes and β‐actin as an endogenous reference gene using QIAGEN Rotor Gene Q Real time PCR detector system (QIAGEN, Germany) with Syber Green PCR Master Mix (Maxima SYBR Green/RoxqPCR Master Mix). Table 1 demonstrates the sequences of primer pairs for each gene. They were designed to obtain the PCR products of between 60 and 150 base pairs in length, as short products tend to amplify most efficiently.
Table 1.
Forward and Reverse Primer Sequences Used for Relative qRT‐PCR Reactions
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Alpha globin | AACTTCAAGCTCCTAAGCCACTGC | CGAGGCTCCAGCTTAACGGTATTTG |
| Beta globin | ATCCTGAGAACTTCAGGCTCCTGGG | GAGCTTAGTGATACTTGTGGGCCAG |
| Beta actin | CCACACTGTGCCCATCTACG | CCGTGGTGGTGAAGCTGTAG |
The relative qRT‐PCR reactions were carried out in 25 μl volume including 50 ng of cDNA product, 0.1 μmol of each primer and 12.5 μl of 2× reaction mix containing FastStart DNA polymerase reaction buffer, dNTPs, and SYBR green I (Fermentas).
The thermal cycling conditions comprised an Optional UDG pretreatment at 50°C for 2 min, an initial denaturation step at 95°C for 10 min, followed by 40 cycles (denaturation at 95°C for 20 sec, annealing at 60°C for 30 sec, and extension at 72°C for 30 sec). Melting curve analysis verified the qRT‐PCR amplification and electrophoresis on 2% agarose gel validated it as well.
PCR efficiencies for each set of primers were determined using serial dilutions of PCR products and resulting plots of CT versus the logarithmic PCR product input, using the equation E (PCR efficiency = 10(−1/slope)). The relative quantity of β‐ and α‐globin were determined from ΔΔCt and (efficiency) −ΔΔCt formulas. ‐ to ‐globin gene ratio was evaluated with the Pfaffl method 6 as follows:
Statistical Analysis
The comparison between four groups (major β‐thalassemia, intermediate β‐thalassemia, minor β‐thalassemia, and healthy individuals) was performed with one‐way analysis of variance (ANOVA) statistical method using SPSS software v.17 (Spss Inc, New York, USA). Graphs were plotted and analyzed applying SPSS as well. A P‐value of less than 0.05 was considered as statistically significant.
RESULTS
‐ and ‐globin gene expression were computed applying relative quantitative real‐time PCR and were compared with ‐actin gene expression as a reference gene. Subsequently the ratio of β/α‐globin mRNA in each sample was measured using Pfaffl method 6 based on real‐time PCR efficiency of both target and reference gene along with CT difference (Δ) of unknown sample versus the mean ΔCT of the control individuals.
The mean ΔCT of controls from 29 normal samples were −15.2 for β‐globin and −15.5 for α‐globin. The efficiencies of the β‐globin, α‐globin, and β‐actin PCR reactions were calculated as 1.02, 0.93, and 0.97, respectively. β‐Globin mRNA expression was measured by 1.02−ΔΔCT and α‐globin was calculated with 0.93−ΔΔCT. Therefore β/α ratio was achieved by dividing β‐ to α‐globin expression and the mean of β/α mRNA ratio for them was 0.99 (Min = 0.92, Max = 1.11).
Nevertheless, 84 samples were elected as minor β‐thalassemia regarding to their RBC index. All of the chosen minor cases had mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) less than 72 fl and 21.5 pg, respectively, and RBC count of more than 5.5 million/μl. Individuals with minor β‐thalassemia were verified by HbA2 measuring kit with the microcolumn ion exchange chromatography method (Biosystem, Spain). Among them 60 samples had HbA2 > 4% in which the mean ratio of β/α mRNA was 0.8 (Min = 0.63, Max = 0.89). This group shows a noticeable reduction in β‐globin expression with respect to the normal group. The other 24 cases had the normal HbA2 level (related to reference value of 1.3–3.7 given by the HbA2 measuring kit) of whom five show a decrease in β/α mRNA ratio and located in minor β‐thalassemia group, 16 demonstrate normal β/α mRNA ratio and placed in normal group, and 3 of 24 cases have shown β/α ratio of more than normal, which, according to RBC indices, can be categorized into α minor thalassemia.
It is noted that the β/α mRNA ratio in some minor β‐thalassemia were overlapped with major and intermediate β‐thalassemia patients.
In this study, 63 samples of β‐thalassemia patients were also considered, which were divided to two categories (major β‐thalassemia and intermediate β‐thalassemia) in accordance to clinical symptoms and the severity of anemia according to phenotypic scoring system offered by Nadkarni et al. (Table 2; 7).
Table 2.
Characteristics of the Minor, Intermediate, and Major Groups of β‐Thalassemia and Normal Population
| Characteristics | Major | Intermediate | Minor | Normal |
|---|---|---|---|---|
| No. (%) | 45 | 18 | 84 | 29 |
| Age at presentation (years) | 2.1 ± 2 | 8.4 ± 6.05 | ‐ | ‐ |
| Liver size <4 cm (no. /%) | 23 (51.1) | 6 (33.3) | 84 (100) | 29 (100) |
| Spleen size <4 cm (no. /%) | 1 (2.22) | 8 (44.4) | 84 (100) | 29 (100) |
| Hemoglobin level (g/dl) | 8.1 ± 1.2 | 9.7 ± 1 | 12.2 ± 2.1 | 14.5 ± 1.9 |
The average expression of β and α mRNA of both groups represent a substantial decrease of ‐globin gene in comparison with both control group and heterozygote β‐thalassemia (minor β‐thalassemia). The relating results for the groups 1 and 2 were as follows: In group 1 (major β‐thalassemia) the mean of β/α mRNA ratio was 0.69 (Min = 0.23, Max = 0.89). In group 2 (intermediate β‐thalassemia), the mean of β/α mRNA ratio was 0.69 (Min = 0.62, Max = 0.80).
Evaluation of the variation between the results was performed by using one‐way ANOVA test. The mean of β/α mRNA ratio in normal individuals was significantly different from all the other groups (P‐value = 0). The minor β‐thalassemia group was also significantly different with the other three groups (P‐value < 0.05). Nevertheless, there was no such association between β/α mRNA expression ratio in major and intermediate β‐thalassemia (Fig. 1).
Figure 1.
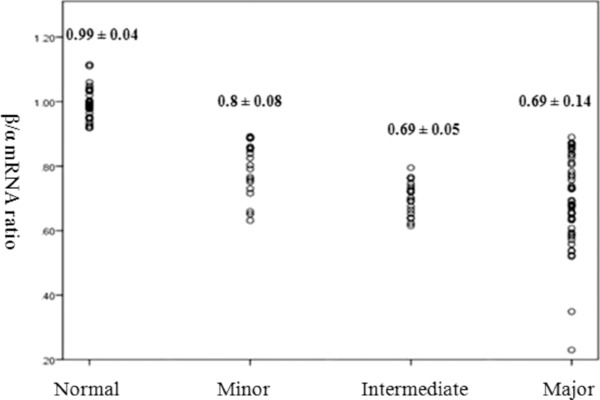
β/α mRNA ratio in various β‐thalassemia subjects and normal population.
DISCUSSION
Definitive diagnosis of suspected minor β‐thalassemia cases that was done in a more specialized laboratory is based on the measurement of β/α chain synthesis ratio with the assistance of radioactive amino acids such as 3H‐leucine or 14C‐leucin with which reticulocytes were incubated 1. Fresh radionuclides necessity and an excess step, chromatography application for separation of two types of globin chains, which were hazardous and laborious, caused this method to be no longer routinely accessible 8. Also some diagnostic centers apply amplification refractory mutation system (ARMS)‐PCR for detecting these suspected cases 9. However, this method may not be effective enough to identify all possible cases, as there is a large spectrum of common and uncommon mutations that complicate the population screening 10, 11. In this study, we apply relative quantitative PCR to investigate mRNA expression of α‐ and β‐globin. As a result, the reduction of β‐globin mRNA is measured without the necessity to know the exact type of mutation of ‐globin gene. Moreover, less amount of blood, up to 2 ml, was needed for the test, because the remaining RNA included a high amount of reticulocyte RNA due to the removing of WBCs and the platelets by employing red cell filtration system and centrifuging, respectively 12, 13, whereas in research work handling radionuclides, the amount of necessary blood reached up to 10 ml 14.
The mean of β/α mRNA ratio in normal individuals was 0.99 in our study that was consistent with prior reports 14, 15, which employed specific radioactive activity and that of Watanapokasin et al. and Tubsuwan et al., who applied one‐step qRT‐PCR 16, 17. Nevertheless, our results were not in agreement with the previous studies that utilized quantitative RT‐PCR, with the β/α mRNA ratio result of 0.76 8 and 0.21 18. This is the risk in which the relative method does not contain at all rather than the quantitative because internal control was used for each sample and target gene expression was determined toward it, instead of external control, it causes elimination of efficiency variations resulted from PCR inhibitors and RNA/cDNA distribution errors 6.
The mean of β/α mRNA ratio in minor β‐thalassemia was 0.8 (Min = 0.65, Max = 0.89), which was significantly different from that of intermediate and major β‐thalassemia groups. However this result was rather higher than earlier reports of Shchory and Chalevelakis 14, 19 that minor thalassemia results were somewhat close to that of intermediates. In the Shchory experience, only one minor β‐thalassemia was analyzed. Accordingly, in our experience among the minor β‐thalassemia, some had reduced β/α mRNA ratio with overlap to some of intermediate thalassemia ratios. This may be due to the mutations that result in lower stability of β‐globin mRNA.
Approximately 6% of cases, which were suspected of minor β‐thalassemia and had normal HbA2, were considered as β minor thalassemia by this method.
All the five cases of minor β‐thalassemia who had normal HbA2 and reduced β‐chain synthesis demonstrated drastic decline in β/α mRNA ratio compared with other minor thalassemia (mean = 0.67 ± 0.05); however, in overlapped with intermediate and major thalassemia we have no description for this finding.
Since the segregation of minor β‐thalassemia from intermediates and major β‐thalassemia is clearly applicable by using peripheral blood smear, Hb electrophoresis, and clinical symptoms, we have focused on making a distinction between minor β‐thalassemia, normal population, and other microcytic hypochromic anemia.
The average ratio of and in the normal group are equal to 1.03 and 1.00, respectively. The ratio reduces in β‐thalassemia cases parallel to increase in clinical symptoms. However, the ratio was approximately constant in all groups. Therefore, it can be concluded that in the presence of defect in globin gene the ratio of affected gene to β‐actin is reduced rather than this ratio in normal cases.
In 3 of 24 cases of minor thalassemia with normal HbA2, which was considered as α‐thalassemia, this ratio has shown reduction and was equal to 0.83, 0.79, and 0.75.
In this study, the cases that represent both α‐ and β‐globin deficiency at the same time have not been considered. Nevertheless, it seems that these cases can be detected by use of these ratios and considering the simultaneous decline of and ratios in each case in comparison to the normal range. For the definitive approval of this hypothesis, we should study more cases.
According to the significant differences achieved, no overlapping between minor β‐thalassemia and normal group, capability of diagnosing atypical minor β‐thalassemia, and accessibility of this technique, we can declare that this method could be suggested as a routine premarital screening test for β‐thalassemia carriers.
Grant sponsor: Shiraz University of Medical Sciences, Iran.
REFERENCES
- 1. Weatherall DJ, Clegg JB. Frontmatter The Thalassaemia Syndromes. Blackwell Science Ltd, Oxford; 2008. p i–xiv. [Google Scholar]
- 2. Fucharoen S, Winichagoon P. Thalassemia in South East Asia: Problems and strategy for prevention and control. Southeast Asian J Trop Med Public Health 1992;23(4):647–655. [PubMed] [Google Scholar]
- 3. Cao A, Moi P, Galanello R. Recent advances in beta‐thalassemias. Pediatr Rep 2011;3(2):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peters M, Heijboer H, Smiers F, Giordano PC. Diagnosis and management of thalassaemia. Br Med J 2012;344:e228. [DOI] [PubMed] [Google Scholar]
- 5. Karimi M, Jamalian N, Yarmohammadi H, Askarnejad A, Afrasiabi A, Hashemi A. Premarital screening for β‐thalassaemia in Southern Iran: Options for improving the programme. J Med Screen 2007;14(2):62–66. [DOI] [PubMed] [Google Scholar]
- 6. Pfaffl MW. Quantification strategies in real‐time PCR In: Bustin SA, editor. A‐Z of Quantitative PCR, La Jolla, CA: International University Line (IUL); 2004. p 87–112. [Google Scholar]
- 7. Nadkarni A, Gorakshakar A, Colah R, Mohanty D, Ghosh K. Evaluation of the clinical severity of β‐thalassemia homozygous patients using a phenotypic scoring system. J Chinese Clin Med 2007;2(8):439–447. [Google Scholar]
- 8. Chaisue C, Kitcharoen S, Wilairat P, Jetsrisuparb A, Fucharoen G, Fucharoen S. α/β‐Globin mRNA ratio determination by multiplex quantitative real‐time reverse transcription‐polymerase chain reaction as an indicator of globin gene function. Clin Biochem 2007;40(18):1373–1377. [DOI] [PubMed] [Google Scholar]
- 9. Rahimi Z, Muniz A, Parsian A. Detection of responsible mutations for beta thalassemia in the Kermanshah Province of Iran using PCR‐based techniques. Mol Biol Rep 2010;37:149–154. [DOI] [PubMed] [Google Scholar]
- 10. Tamhankar PM, Agarwal S, Arya V, Kumar R, Gupta UR, Agarwal SS. Prevention of homozygous beta thalassemia by premarital screening and prenatal diagnosis in India. Prenatal Diag 2009;29(1):83–88. [DOI] [PubMed] [Google Scholar]
- 11. Kotila TR. Guidelines for the diagnosis of the haemoglobinopathies in Nigeria. Ann Ibadan Postgrad Med 2010;8(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goh S‐H, Josleyn M, Lee YT, et al. The human reticulocyte transcriptome. Physiol Genom 2007;30(2):172–178. [DOI] [PubMed] [Google Scholar]
- 13. Goh S‐H, Lee YT, Bhanu NV, et al. A newly discovered human α‐globin gene. Blood 2005;106(4):1466–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shchory M, Ramot B. Globin chain synthesis in the marrow and reticulocytes of beta thalassemia, hemoglobin H disease, and beta delta thalassemia. Blood 1972;40(1):105–111. [PubMed] [Google Scholar]
- 15. Hassan K, Vijayasilan T, Mahmood Z, Abdul Hamid H, Chin YM. Investigations on the relative synthesis of globin chains in thalassaemia: A preliminary study in Malaysian subjects. Singapore Med J 1988;29(5):462–468. [PubMed] [Google Scholar]
- 16. Watanapokasin Y, Winichagoon P, Fuchareon S, Wilairat P. Relative quantitation of mRNA in beta‐thalassemia/Hb E using real‐time polymerase chain reaction. Hemoglobin 2000;24(2):105–116. [DOI] [PubMed] [Google Scholar]
- 17. Tubsuwan A, Munkongdee T, Jearawiriyapaisarn N, et al. Molecular analysis of globin gene expression in different thalassaemia disorders: Individual variation of βE pre‐mRNA splicing determine disease severity. Br J Haematol 2011;154(5):635–643. [DOI] [PubMed] [Google Scholar]
- 18. Han JY, Zeng RP, Cheng G, Hu B, Li H, Lai YR. Quantitative analysis of human globin gene expression in beta‐thalassemia using real‐time RT‐PCR. Yi Chuan 2005;27(1):57–64. [PubMed] [Google Scholar]
- 19. Chalevelakis G, Clegg JB, Weatherall DJ. Imbalanced globin chain synthesis in heterozygous beta‐thalassemic bone marrow. Proc Natl Acad Sci U S A 1975;72(10):3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]