

Abstract
Background and Purpose
Recent development in drug discovery have shown benzimidazole to be an important pharmacophore,. Benzimidazole derivatives exhibit broad‐spectrum pharmacological properties including anti‐microbial, anti‐diabetic and anti‐tumour activity. However, whether benzimidazole derivatives are effective in suppressing angiogenesis and its underlying mechanisms remain incompletely understood. In this study, we aim to characterize the anti‐angiogenic mechanisms of a novel 2‐aminobenzimidazole‐based compound, Jzu 17, in an effort to develop novel angiogenesis inhibitor.
Experimental Approach
Effects of Jzu 17 on endothelial cell proliferation, migration, invasion, and activation of signalling molecules induced by VEGF‐A, were analysed by immunoblotting, MTT, BrdU, migration, and invasion assays. We performed tube formation assay, aorta ring sprouting assay, matrigel plug assay, and a mouse model of metastasis to evaluate ex vivo and in vivo anti‐angiogenic effects of Jzu 17.
Key Results
Jzu 17 inhibited VEGF‐A‐induced cell proliferation, migration, invasion, and endothelial tube formation of HUVECs. Jzu 17 suppressed VEGF‐A‐induced microvessel sprouting ex vivo and attenuated VEGF‐A‐ or tumour cell‐induced neovascularization in vivo. Jzu 17 also reduced B16F10 melanoma lung metastasis. In addition, Jzu 17 inhibited the phosphorylation of VEGFR‐2 and its downstream signalling molecules in VEGF‐A‐stimulated HUVECs. Results from computer modelling further showed that Jzu 17 binds to VEGFR‐2 with high affinity.
Conclusions and Implications
Jzu 17 may inhibit endothelial remodelling and suppress angiogenesis through targeting VEGF‐A‐VEGFR‐2 signalling. These results also suggest Jzu 17 as a potential lead compound and warrant the clinical development of similar agents in the treatment of cancer and angiogenesis‐related diseases.
Abbreviations
- FAK
focal adhesion kinase
- MTT
3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide
- RTK
receptor TK
What is already known
VEGF‐A‐VEGFR‐2 signalling represents an attractive therapeutic target for intervention of angiogenesis.
Benzimidazole‐based compounds may exhibit pharmacological activities capable of clinical application
What this study adds
Jzu 17, a novel 2‐aminobenzimidazole derivative, suppresses endothelial remodelling and angiogenesis through targeting VEGF‐A‐VEGFR‐2 signalling.
Jzu 17 suppressed neovascularization induced by VEGF‐A or tumour cells in in vivo models.
What is the clinical significance
Jzu 17 is a potential lead in developing novel anti‐angiogenesis agents for treatment of cancer.
1. INTRODUCTION
Angiogenesis is a complicated process of forming new capillary blood vessels from pre‐existing vascular network. It primarily occurs in embryogenesis, wound healing, and reproduction (Ucuzian, Gassman, East, & Greisler, 2010). However, it also participates in various pathological states such as diabetic retinopathy, psoriasis, arthritis, and cancer (Prager, Poettler, Unseld, & Zielinski, 2012). Angiogenesis is mandatory for tumour progression since avascular tumours are restricted in their growth due to lacking blood supply. Angiogenesis is also the most common route for metastatic spread of tumour cells, the main cause of cancer morbidity and mortality (Bielenberg & Zetter, 2015). Therefore, modulating tumour angiogenesis represents a rational and promising strategy for developing anti‐cancer agents (Weis & Cheresh, 2011).
The balance between pro‐angiogenic and anti‐angiogenic stimuli regulates angiogenesis. Numerous pro‐angiogenic factors including https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=963 (Ferrara, 2002), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4916 (Javerzat, Auguste, & Bikfalvi, 2002; Wang et al., 2012), and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4867 (Holopainen et al., 2012) have been implicated in tumour angiogenesis. Of these, https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5085, the member of the VEGF family, is the most critical pro‐angiogenic factor (Carmeliet, 2005). VEGF‐A is expressed in various types of cancers, and its overexpression in tumours is associated with poor prognosis in cancer patients (Goel & Mercurio, 2013). VEGF‐A augments most steps of angiogenesis through binding to the receptor TK VEGF receptor‐2 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1813, also known as Flk‐1) on the endothelial cells (Yancopoulos et al., 2000). VEGFR‐2 is a receptor TK (RTK), which undergoes ligand‐induced dimerization and autophosphorylation upon VEGF‐A binding. Phosphorylated VEGFR‐2 initiates downstream signalling cascades including focal adhesion kinase (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2180), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2206, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285, and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 (Olsson, Dimberg, Kreuger, & Claesson‐Welsh, 2006). These signalling cascades contribute to the regulation of endothelial cell survival, migration, proliferation, and tube formation (Brunton & Frame, 2008). Therefore, VEGF‐A‐VEGFR‐2 signalling represents an attractive therapeutic target for intervention of angiogenesis‐associated diseases including cancer (Ferrara & Kerbel, 2005). A variety of strategies to interfere with VEGF‐A‐VEGFR signalling have been assessed in clinical trials (Jain, Duda, Clark, & Loeffler, 2006). These include soluble decoy receptors (VEGF‐Trap) that sequester VEGF‐A (Papadopoulos et al., 2012), antisense oligonucleotides that target VEGF (Levine et al., 2006), and neutralizing monoclonal antibodies against VEGF‐A or VEGFR (Ferrara, 2004). Inhibition of VEGFR‐2 kinase activity using low MW inhibitors represents another approach to suppress VEGF‐A signalling (Noble, Endicott, & Johnson, 2004). To date, monoclonal antibodies https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6771 (Avastin®) (Summers, Cohen, Keegan, & Pazdur, 2010) and https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7390 (Cyramzar®; Calvetti et al., 2015) and low MW inhibitors such as https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5711 (Nexavar®), https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5713 (Sutent®), or https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5698 (Votrient®) have been already approved by the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMEA) for the treatment of certain types of cancer (Meadows & Hurwitz, 2012).
Benzimidazole, a key pharmacophore with diverse biological and pharmacological properties, has attracted considerable attention in the field of drug discovery. Therefore, synthesis of novel benzimidazole derivatives remains a major focus for synthetic chemistry communities. Most studies have focused on the synthesis of 2‐arylbenzimidazoles and their biological activities. In contrast, there are few studies reporting the synthesis of 2‐aminobenzimidazole derivatives. A variety of benzimidazole‐based compounds have shown their potential use as antidiabetic, anti‐ulcer, anti‐convulsant (Shingalapur, Hosamani, Keri, & Hugar, 2010), anti‐infectious (Ozkay, Tunali, Karaca, & Isikdag, 2011), or anti‐inflammatory (Bukhari et al., 2016) agents. Recent studies demonstrated that benzimidazole derivatives also exhibit anti‐angiogenesis (Huang, Lien, Kuo, & Huang, 2012) and anti‐tumour (Chu et al., 2015) properties. The underlying mechanisms, however, remain incompletely understood. These observations suggest that additional benzimidazole‐based compounds may exhibit pharmacological activities capable of clinical application. Given their potential as lead compound for developing novel anti‐angiogenesis agents, we synthesized a series of compounds with the core structure of 2‐aminobenzimidazole, the Jzu compounds (Figure S1A) and evaluated their anti‐angiogenesis properties. Of these compounds, Jzu 17 was selected for its potent inhibitory activities to reduce cell viability in VEGF‐A‐stimulated HUVECs (Figure S1B). The aim of this study was to characterize the underlying mechanisms by which Jzu 17 suppresses angiogenesis in endothelial cells.
2. METHODS
2.1. Cell culture
MDA‐MB‐231 (BCRC Cat# 60425, RRID:CVCL_0062) breast cancer and B16F10 (BCRC Cat# 60031, RRID:CVCL_0159) melanoma cell lines and HUVECs were obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). HUVECs were maintained in M199 medium containing vascular endothelial cell growth supplement (ECGS; Millipore), 10% FBS, 5 U·ml−1 of heparin, 20‐mM HEPES, 100 U·ml−1 of penicillin G, and 100 μg·ml−1 of streptomycin in a humidified 37°C incubator. MDA‐MB‐231 and B16F10 cells were maintained in DMEM medium containing 10% FCS, 100 μg·ml−1 of streptomycin, and 100 U·ml−1 of penicillin G in a humidified 37°C incubator.
2.2. MTT assay
Cell viability was determined by the colorimetric MTT assay as described previously (Huang et al., 2012).
2.3. LDH release assay
The CytoTox96 non‐radioactive cytotoxicity assay kit (Promega, Madison, WI, USA) was employed to measure LDH leakage to quantify cytotoxicity as described previously (Huang, Lien, Kuo, & Huang, 2014).
2.4. Cell proliferation assay
HUVECs (2 × 104 per well) seeded in 96‐well tissue culture plates were incubated for 24 hr. Cells were starved in M199 medium containing 2% FBS in the absence of endothelial cell growth supplements for another 18 hr. After starvation, cells were treated with Jzu 17 at different concentrations for 30 min, followed by the stimulation with VEGF‐A (25 ng·ml−1) for another 24 hr. Cell proliferation was determined using a BrdU Cell Proliferation kit (Millipore) based on the colorimetric detection of the incorporation of BrdU, following the manufacturer's instructions.
2.5. Cell migration assay
HUVECs were allowed to grow to confluence in 12‐well tissue culture plates coated with 0.1% gelatin (Sigma‐Aldrich). Cells were starved with M199 medium containing 2% FBS for 18 hr. After starvation, monolayer HUVECs were wounded by scratching with pipette tips. Cells were washed with PBS, followed by the treatment with Jzu 17 at different concentrations in the presence or absence of 25 ng·ml−1 of VEGF‐A for another 24 hr. After treatment, cells were fixed with cold 4% paraformaldehyde. Cells were stained with 0.5% toluidine blue. Microscope images were taken at 40× magnification by an OLYMPUS Biological Microscope digital camera (Yuan Li Instrument Co., Taipei, Taiwan). The rate of cell migration was determined by comparing the sizes of scratch area as a percentage of the values obtained with their respective controls at the beginning of experiments (Time 0) using an Image J program (http://rsbweb.nih.gov/ij/index.html; https://imagej.net/, RRID:SCR_003070).
2.6. Transwell invasion assay
The cell invasion assays were performed using transwell plates (Corning, NY, USA). The bottom face of the filter in the transwell plate was coated with 0.2% gelatin. The bottom chambers were filled with M199 medium containing 2% FBS in the presence of VEGF‐A (25 ng·ml−1). HUVECs (104 cells per well) in 200‐μl M199 medium (with 2% FBS) with or without Jzu 17 at different concentrations were seeded in the top chambers. Cells were allowed to invade for 18 hr. Non‐invaded cells (on the top side of filter) were scraped with a cotton swab, and invaded cells were fixed and stained with 0.5% toluidine blue in 4% paraformaldehyde. The cells were photographed under an inverted contrast phase light microscope (×40, Nikon, Japan). Stained HUVECs that invaded through the membrane were quantified by counting in three random fields.
2.7. Matrigel tube formation assay
The tube formation assay was performed as described previously (Huang et al., 2014). Matrigel, a basement membrane matrix (Becton Dickinson, Mountain View, CA, USA), was polymerized at 37°C for 30 min. HUVECs suspended in M199 medium containing 2% FBS in the presence or absence of VEGF‐A (25 ng·ml−1) were seeded onto the Matrigel. They were then treated with vehicle or Jzu 17 at indicated concentrations. After 18 hr, cells were photographed using phase‐contrast microscopy. The cells were photographed under an inverted contrast phase light microscope (×40, Nikon, Japan).
2.8. Immunoblotting
Cells were harvested in an extraction buffer containing 140‐mM NaCl, 10‐mM Tris (pH 7.0), 0.5% NP‐40, 0.2‐mM leupeptin, 0.05‐mM pepstatin A, and 2‐mM PMSF. Equal amounts of protein samples were subjected to SDS‐PAGE and transferred onto an NC membrane (Pall Corporation, Washington, NY, USA). After blocking in a 5% non‐fat milk‐containing blocking buffer for 1 hr, proteins were recognized using specific primary antibodies followed by HRP‐conjugated secondary antibodies. To detect immunoreactivity, enhanced chemiluminescence was employed as per the manufacturer's instructions. Quantitative data were obtained using a computing densitometer with a scientific imaging system (Biospectrum AC System, UVP). The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.9. Animals
All animal care and experimental procedures complied with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH publication No. 85‐23, revised 1996) and were approved by the Taipei Medical University Laboratory Animal Care and Use Committee (Permit Number: LAC‐2017‐0241). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altmans, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
2.10. Aortic ring sprouting assay
Aorta ring sprouting assay was performed as described previously (Huang et al., 2014); 8‐ to 10‐week‐old male Sprague–Dawley rats were obtained from National Laboratory Animal Center (Taipei, Taiwan) and used for the experiment presented in Figure 2c. Rats were killed by CO2 asphyxiation and the aortic arch dissected. After removing the surrounding fibro‐adipose tissues and thoroughly rinsing with M199 culture medium, the aorta was cut into about 1‐mm ring segments. The aortic rings were immersed in Matrigel in the wells of 48‐well plate. VEGF‐A (25 ng·ml−1) with or without Jzu 17 was added to the wells. The aortic rings were cultured in 37°C with 5% CO2, and the cultured medium was changed every 3 days. Growing sprouts of endothelial cells were observed and photographed on Day 7. The images were photographed under microscope, and sprouting area was determined on the computer‐digitized images with Image‐Pro Plus software (Media Cybernetics, Inc., Rockville, MD, USA; Image‐Pro Plus, RRID:SCR_007369). An observer who was unaware of the treatment group assessed the sprouting area.
Figure 2.
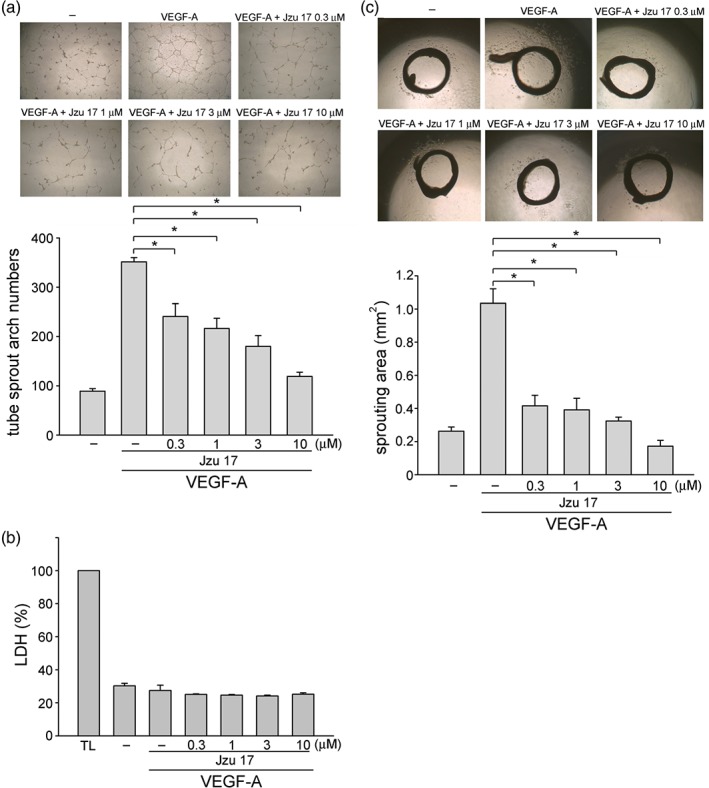
Jzu 17 inhibited VEGF‐A‐induced tube formation in vitro and aorta ring sprouting ex vivo. (a) HUVECs were seeded on Matrigel in the presence of VEGF‐A (200 ng·ml−1) with or without Jzu 17 at indicated concentrations. Cells were photographed under phase contrast after 16 hr as described in Section 2. Bar graphs show compiled data of average sprout arch numbers (n = 6). *P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test. (b) Cells were stimulated with VEGF‐A (25 ng·ml−1) with or without Jzu 17 at indicated concentrations for 16 hr. Jzu 17's cytotoxicity was determined by LDH assay. Cells were also treated with cell lysis buffer (total lysis, TL) to serve as positive control. Each column represents the mean ± SEM of six independent experiments performed in duplicate. Technical replicates were used to ensure the reliability of single values for each experiment. *P < .05, significantly different from VEGF‐A alone; Kruskal–Wallis test. (c) Rat aortic rings were placed in Matrigel and treated with VEGF‐A (25 ng·ml−1) in the presence or absence of Jzu 17 at indicated concentrations. The effects of Jzu 17 on formation of vessel sprout from various aorta samples was determined on Day 8. Bar graphs show compiled data of average microvessels area (n = 7). *P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test
2.11. VEGF‐A‐ or tumour cells‐induced angiogenesis Matrigel plug assay
The angiogenesis matrigel plug assay with nudenu/nu mice as described previously (Huang et al., 2012) was used to determine in vivo anti‐angiogenic effects of Jzu 17; 3‐ to 5‐week‐old male nudenu/nu mice with body weight about 20 g were obtained from BioLasco (Taipei, Taiwan) and used for the experiment presented in Figure 3. All the mice were housed (three mice per cage) in clean specific pathogen‐free (SPF) rooms (standard 12‐hr light/12‐hr dark cycle at 22°C) in Laboratory Animal Center of Taipei Medical University and maintained on standard chow and autoclaved water. The cage floor was covered with Bed O'Cobs animal bedding (The Andersons, Maumee, OH, USA). All mice were randomly allocated to individually ventilated cage (IVC) by vivarium staff, upon transfer from BioLASCO into the animal housing room. All mice purchased from BioLASCO were acclimatized in the animal housing room for 7 days prior to starting experiments. Mice were anaesthetized with intraperitoneal pentobarbital (50 mg·kg−1). Once anaesthesia was induced, an aliquot (500 μl) of Matrigel containing VEGF‐A (200 ng·ml−1) with heparin (20 U) was injected subcutaneously into the right flank of each mouse (VEGF‐A‐induced angiogenesis model). In the other set of experiments, MDA‐MB‐231 cells were harvested and re‐suspended in PBS. Cells (5 × 106 cells) in a volume of 150 μl in the presence of heparin (20 U) were mixed with Matrigel (150 μl) and injected subcutaneously into the right flank of each mouse (tumour cells‐induced angiogenesis model). After implantation, animals were randomized to either the vehicle‐treated control group or the treatment group, which received Jzu 17 at indicated concentrations. The treatment was administered intraperitoneally once daily for 7 (VEGF‐A‐induced angiogenesis model) or 10 (tumour cells‐induced angiogenesis model) days. At the end of treatment, animals were killed using CO2 asphyxiation, Matrigel plugs were removed, and the surrounding tissues trimmed. Haemoglobin levels of the Matrigel plugs were evaluated with Drabkin's reagent kit (Sigma‐Aldrich) according to the manufacturer's instructions. The concentration of haemoglobin was calculated based on a set of haemoglobin standards.
Figure 3.
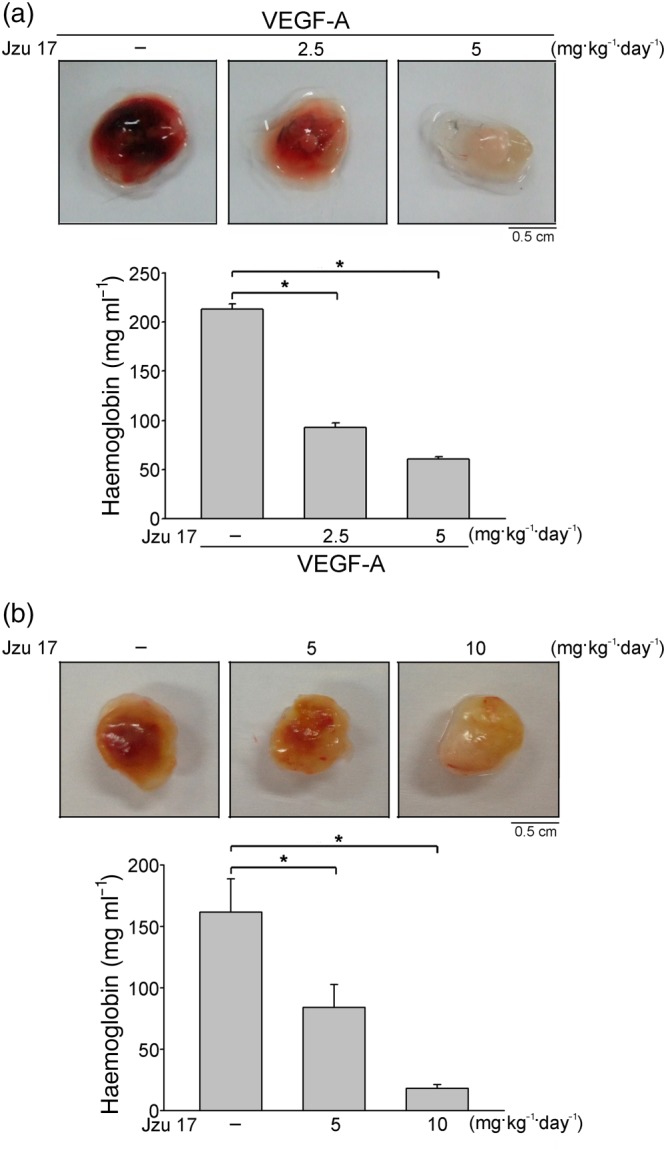
Jzu 17 suppressed VEGF‐A‐ or tumour cells‐induced angiogenesis in vivo. (a) Matrigel mixed with VEGF‐A was injected subcutaneously into the right flank of nude mice. After implantation, animals were treated intraperitoneally with vehicle or Jzu 17 (2.5 or 5 mg·kg−1·day−1) for 7 days. Matrigel plugs removed from the mice injected intraperitoneally with vehicle or Jzu 17 were shown in the upper of the chart. Haemoglobin levels in the Matrigel plug were also quantified. Each column represents the mean ± SEM of six plugs in each group. *P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test.. (b) Matrigel mix with MDA‐MB‐231 breast cancer cells was injected subcutaneously into the right flank of nude mice. After implantation, animals were treated intraperitoneally with vehicle or Jzu 17 (5 or 10 mg·kg−1·day−1) for 10 days. Matrigel plugs removed from the mice treated with vehicle or Jzu 17 were shown in the upper of the chart. Haemoglobin levels in the Matrigel plug were quantified. Each column represents the mean ± SEM of six plugs in each group. *P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test
2.12. Melanoma lung metastatic mouse model
The melanoma lung metastatic model with C57BL/6 mice as described previously (Lin, Chen, Chou, & Wang, 2011) was used to determine anti‐metastatic effects of Jzu 17. 6‐ to 8‐week‐old male C57BL/6 mice with body weight about 35 g were obtained from National Laboratory Animal Center (Taipei, Taiwan) and used for the experiment presented in Figure 4. All the mice were housed (five mice per conventional cage) in clean conventional animal housing rooms (standard 12‐hr light/12‐hr dark cycle at 22°C) in Laboratory Animal Center of Taipei Medical University and maintained on standard chow and autoclaved water. The cage floor was covered with Bed O'Cobs animal bedding (The Andersons). All mice were randomly allocated to conventional cage by vivarium staff, upon transfer from National Laboratory Animal Center into the animal housing room. Mice were anaesthetized with intraperitoneal pentobarbital (50 mg·kg−1). Once anaesthesia was induced, mice were inoculated at the tail vein with B16F10 melanoma cells (106 cells per mouse) suspended in 150 μl of sterile saline. Mice were randomized to either the vehicle‐treated control group or the treatment group, which received Jzu 17 (10 mg·kg−1·day−1). The treatment was given intraperitoneally once daily for 18 days. At the end of treatment, mice were killed by CO2 asphyxiation and dissected. The lungs were collected and fixed in 10% formalin. Metastatic melanoma nodules were scored by counting. Paraffin wax‐embedded sections (5 μm) of lung tissue were stained with haematoxylin and eosin (H&E) staining and photographed under a microscope to assess metastatic nodules area. The metastatic nodules area was determined on the computer‐digitized images with Image‐Pro Plus software (Media Cybernetics, Inc., Rockville, MD, USA; Image‐Pro Plus, RRID:SCR_007369).
Figure 4.
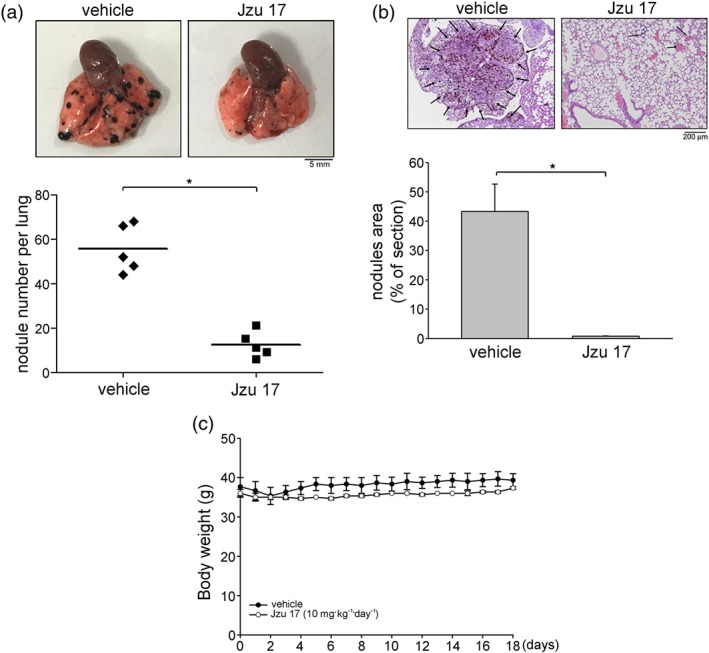
Jzu 17 inhibited murine melanoma B16F10 cell lung metastasis. (a) B16F10 melanoma cells were injected into the tail vein of the C57BL/6 mice. These mice were injected intraperitoneally with vehicle or Jzu 17 (10 mg·kg−1·day−1) for 18 consecutive days. Representative photograph of pulmonary metastatic foci produced 18 days after intravenous injection of B16F10 cells. Each symbol represents the average number of metastasis nodules in a lung from an individual mouse (n = 5 for each group). *P < .05, significantly different from vehicle‐treated control group; Student's t‐test. (b) Representative lung haematoxylin and eosin (H&E) stained sections of metastases from vehicle‐ and Jzu 17‐treated mice. Arrows point to metastatic foci. Bar graphs show compiled data of average area of metastasis nodules per section (n = 5 for each group). *P < .05, significantly different from vehicle‐treated control group; Student's t‐test. (c) The body weights of the nude mice were examined daily within 18 days treatment of vehicle or Jzu 17. Values represent the mean ± SEM (n = 5 for each group)
2.13. VEGF‐A binding analysis
We performed VEGF‐A binding analysis using human VEGF‐A biotinylated Fluorokine Kit (R&D Systems). Briefly, HUVECs were deattached using Accutase™ cell detachment solution (BD Biosciences). Cells were washed with PBS and incubated with biotinylated recombinant VEGF‐A or negative control reagent (biotinylated soybean trypsin inhibitor) in the absence or presence of Jzu 17 for 1 hr. Cells were incubated subsequently with avidin‐conjugated fluorescein for another 30 min. Cells were washed with PBS, and fluorescence of labelled cells was determined using flow cytometer (FACScan; BD Biosciences, San Jose, CA, USA) and analysed with CellQuest software (BD Biosciences; BD CellQuest Pro, RRID:SCR_014489).
2.14. Molecular docking simulation
For docking simulation, the X‐ray crystallography structure for VEGF‐A (PDB ID: 3V2A; Brozzo et al., 2012) and VEGFR‐2 (PDB ID: 5EW3; Bold et al., 2016) was downloaded from RCSB Protein Data Bank. The preparation of protein was performed by Prepare Protein module in Discovery Studio 2.5 (DS2.5) to remove crystal water in crystallography structure, insert missing atoms in incomplete residues, protonate the structure of both protein with Chemistry at HARvard Macromolecular Mechanics (CHARMM) force field (Brooks et al., 1983), and optimize side‐chain conformation for residues with inserted atoms. For VEGF‐A, there are two small receptor cavities between VEGF‐A and VEGFR‐2. We combine these two small receptor cavities to define the binding site with the volume of 341.375 Å for VEGF‐A (Figure S3A). The binding site of VEGFR‐2 was defined as volume of the co‐crystallized compound in the X‐ray crystallography (Figure S3B). The docking poses of compound was performed by Ligand Fit module in DS2.5 using a shape filter and Monte‐Carlo ligand conformation generation and optionally minimized with CHARMM force field (Brooks et al., 1983).
2.15. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). To provide randomization and blinding in our experiments, in each experiment, the same cell was used to evaluate the effects of Jzu 17 versus the related control. Formal randomization was therefore not employed. Mice used in this study were randomly allocated to cages by vivarium staff and randomized into vehicle‐treated or Jzu 17‐treated group before the treatment. For blinding, we have different people conducting experiments (operator) and analysing data (analyst).
The exact group size (n) was the same for each experiment in this study. Results are expressed as mean ± SEM; n ≥ 5, where “n” refers to independent values, and not replicates. To control for unwanted sources of variation and to reveal relevant trends, normalization was performed to compare the differences after the treatment. For MTT or BrdU assay, the viability or BrdU incorporation was expressed as fold changes over that of the vehicle‐treated cells, whose expression was set to 1 (100%). For LDH assay, LDH release from lysis buffer‐treated cells (TL group) was considered to be 100% and LDH release from the VEGF‐A‐treated cells in the presence or absence of Jzu 17 was expressed as a percentage of the control. For immunoblotting, the levels of protein modification (e.g., VEGFR‐2 or FAK phosphorylation) were normalized to that of un‐modified protein (e.g., VEGFR‐2 or FAK). The status of protein modification was expressed as fold changes over that of the vehicle‐treated cells, whose expression was set to 1 (100%). The SEM was normalized appropriately. The status of protein modification was expressed by normalization that generates control values with no variance (SEM = 0) to reduce the effect of variation from different exposure of blotting, and such data are subjected to non‐parametric statistical analysis. The group data subjected to statistical analysis have a minimum of n = 5 independent samples per group in this study. Statistical analysis was performed using SigmaPlot 10 (Build 10.0.0.54; Systat Software, San Jose, CA, USA; http://www.sigmaplot.com/products/sigmaplot/, RRID:SCR_003210). Statistical comparisons between two groups were evaluated by the unpaired Student's t‐test for parametric analysis or Mann–Whitney test for non‐parametric analysis. Statistical comparisons among more than two groups were evaluated by one‐way ANOVA with Tukey's post hoc test for parametric analysis or Kruskal–Wallis test followed by Dunn's multiple comparison for non‐parametric analysis. Post hoc tests were run only if F achieved P < .05 and there was no significant inhomogeneity. A P value <.05 was defined as statistically significant.
2.16. Materials
The 2‐aminobenzimidazole‐based Jzu compounds, including Jzu17, were synthesized as described in the http://bph14813.docx/#F8. Other compounds and materials were obtained as follows: 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide (MTT) was from Sigma‐Aldrich (St Louis, MO, USA). Medium 199 (M199), DMEM, TrypLE™, FBS, and all cell culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). Recombinant VEGF‐A was purchased from PeproTech (Rocky Hill, NJ, USA). Antibodies against VEGFR2 (Cell Signaling Technology Cat# 2479, RRID:AB_2212507), VEGFR2 phosphorylated at tyrosine 1175 (Y1175; Cell Signaling Technology Cat# 3770, RRID:AB_1642326), FAK (Cell Signaling Technology Cat# 3285, RRID:AB_2269034), FAK phosphorylated at tyrosine 397 (Y397; Cell Signaling Technology Cat# 3283, RRID:AB_2173659), Src (Cell Signaling Technology Cat# 2108, RRID:AB_331137), Src phosphorylated at tyrosine 416 (Y416; Cell Signaling Technology Cat# 6943, RRID:AB_10013641), Akt (Cell Signaling Technology Cat# 9272, RRID:AB_329827), Akt phosphorylated at serine 473 (S473; Cell Signaling Technology Cat# 9271, RRID:AB_329825), ERK1/2 (Cell Signaling Technology Cat# 4695, RRID:AB_390779) and ERK1/2 phosphorylated at threonine 202/tyrosine 204 (T202/Y204; Cell Signaling Technology Cat# 4370, RRID:AB_2315112) were purchased from Cell Signaling (Danvers, MA, USA). Anti‐mouse and anti‐rabbit IgG conjugated HRP antibodies, as well as antibody against α‐tubulin (GeneTex Cat# GTX628802, RRID:AB_2716636), were obtained from GeneTex Inc (Irvine, CA, USA). Cell Proliferation ELISA, BrdU assay kit was from Roche (Indianapolis, IN, USA). All materials for immunoblotting were from Bio‐Rad (Hercules, CA, USA). The enhanced chemiluminescence detection kit was from Millipore (Billerica, MA, USA). All other chemicals were obtained from Sigma‐Aldrich.
2.17. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro, et al., 2017; Alexander, Kelly, et al., 2017).
3. RESULTS
A novel 2‐aminobenzimidazole derivative Jzu 17 inhibited VEGF‐A‐induced HUVEC proliferation, migration, and invasion.
Vascular endothelial cell proliferation, migration, invasion, and tube formation are critical steps in angiogenesis (Solinas et al., 2012). To investigate whether the novel 2‐aminobenzimidazole‐based compounds with benzyl, methylbenzyl, fluorobenzyl, or chlorobenzyl structure, the Jzu compounds (Jzu 01–24; Figure S1A), exhibit anti‐angiogenic activities, we evaluated their effects on cell proliferation in VEGF‐A‐stimulated HUVECs. HUVECs were synchronized by starvation medium (2% FBS‐containing M199 medium) for 18 hr. After starvation, cells were stimulated by VEGF‐A (25 ng·ml−1) in the absence or presence of the Jzu compounds (1, 3, or 10 μM) for another 24 hr. As shown in Figure S1B, Jzu 01 (2‐aminobenzimidazole, the core structure) and its derivatives (Jzu 04–Jzu 13, Jzu 17, Jzu 20–Jzu 22, and Jzu 24) significantly reduced cell viability in VEGF‐A‐stimulated HUVECs as determined by MTT assay. Among these 16 effective compounds, Jzu 17 exhibited the most prominent effects in reducing cell viability in VEGF‐A‐stimulated HUVECs (Figure S1B). Therefore, we sought to explore the underlying mechanisms by which Jzu 17 suppresses VEGF‐A‐induced angiogenesis in the following experiments. Figure 1a illustrates the chemical structure of the novel 2‐aminobenzimidazole‐based compound, Jzu 17. Results from BrdU labelling analysis demonstrated that Jzu 17 concentration dependently suppressed VEGF‐A‐induced HUVEC proliferation (Figure 1b). As shown in Figure 1c, Jzu 17 also significantly inhibited VEGF‐A‐induced HUVEC migration as determined by wound‐healing migration assay. We also used the transwell invasion assay and, as shown in Figure 1d, Jzu 17 at 0.3 to 10 μM significantly reduced the number of invading cells, passing through the gelatin‐coated transwell membrane barrier, using VEGF‐A as the chemoattractant.
Figure 1.
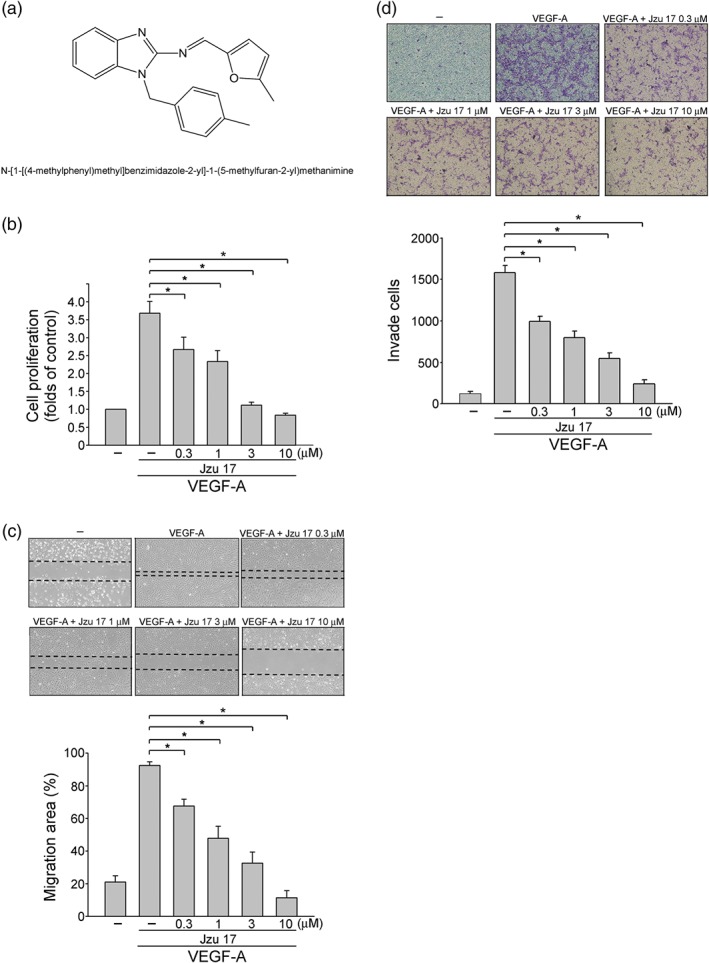
Jzu 17 inhibited VEGF‐A‐induced cell proliferation, migration, and invasion. (a) Chemical structures of Jzu 17. (b) HUVECs were starved in 2% FBS containing medium without ECGS for 16 hr. After starvation, cells were treated with indicated concentrations of Jzu 17 followed by the stimulation with VEGF‐A (25 ng·ml−1) for another 24 hr. Cell proliferation was determined as described in Section 2. Each column represents the mean ± SEM of eight independent experiments performed in duplicate. Technical replicates were used to ensure the reliability of single values for each experiment. *P < .05, significantly different from VEGF‐A alone; Kruskal–Wallis test.. (c) After starvation as described in (b), cells were scratched and treated with vehicle or JZU 17 at indicated concentrations in the presence of VEGF‐A for another 24 hr. The rate of cell migration was determined as described in Section 2. Each column represents the mean ± SEM of five independent experiments. * P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test. (d) After starvation as described in (b), a total of 105 HUVECs were seeded in the top gelatin‐coated chamber and treated with vehicle or indicated concentrations of Jzu 17 using VEGF‐A as chemoattractant. After 16 hr, the HUVECs that invaded through the gelatin‐coated membrane were stained and quantified as described in the Section 2. Each column represents the mean ± SEM of six independent experiments *P < .05, significantly different from VEGF‐A alone; one‐way ANOVA, with Tukey's post‐hoc test
3.1. Jzu 17 inhibited VEGF‐A‐induced tube formation of HUVECs and suppressed microvessel sprouting ex vivo
We next examined Jzu 17 on tubular formation of endothelial cells, a key step in angiogenesis. HUVECs seeded on top of Matrigel, were stimulated by VEGF‐A (25 ng·ml−1) with or without Jzu 17 (0.3–10 μM). As shown in Figure 2a, HUVECs became elongated and formed capillary‐like structure after 24‐hr exposure to VEGF‐A. However, Jzu 17 significantly inhibited the formation of capillary‐like network in a concentration‐dependent manner. Results from the LDH assay further showed that Jzu 17 at 0.3 to 10 μM did not increase LDH release in HUVECs exposed to VEGF‐A for 24 hr (Figure 2b). Together, these results indicate that Jzu 17 exhibits anti‐angiogenesis property through suppression of cell proliferation, migration, invasion, and tube formation of endothelial cells. It also suggests that Jzu 17's inhibitory actions in VEGF‐A‐stimulated HUVECs may not attribute to its cytotoxic effects. Moreover, we also performed an ex vivo rat aortic ring sprouting assay. As shown in Figure 2c, VEGF‐A (25 ng·ml−1) induced significant increases in sprouting microvessels to form complex network around the aortic rings. However, treatment with Jzu 17 (0.3–10 μM) significantly reduced VEGF‐A's effects on microvessel sprouting (Figure 2c). It indicates that Jzu 17 is effective in suppressing VEGF‐A‐induced angiogenesis ex vivo.
3.2. Jzu 17 suppressed angiogenesis induced by VEGF‐A or tumour cells in vivo
We employed murine Matrigel plug models to explore whether Jzu 17 exhibits anti‐angiogenesis effects in vivo. As shown in Figure 3a, VEGF‐A significantly induced microvessel formation in the Matrigel plug. The pale colour of the plugs removed from the Jzu 17‐treated mice indicates that VEGF‐A induced less neovascularization after 7‐day treatment with Jzu 17 (2.5 or 5 mg·kg−1·day−1; Figure 3a, upper panel). We also measured the haemoglobin content of the plugs to quantify angiogenesis level. A significant reduction in neovascularization was observed in plugs from Jzu 17‐treated mice when compared with those from vehicle‐treated mice (Figure 3a, bottom panel). Moreover, we used a tumour cells‐induced angiogenesis model to investigate whether Jzu 17 suppresses tumour angiogenesis. MDA‐MB‐231 breast cancer cells mixed with Matrigel were injected into the flanks of mice. After implantation for 10 days, Matrigel plugs were harvested. As shown in Figure 3b, MDA‐MB‐231 cells markedly increased neovascularization in the plug while Jzu 17 reduced this effect (Figure 3b, upper panel). The angiogenesis level was also quantified. As compared with the vehicle‐treated control group, Jzu 17 significantly reduced tumour cells‐elicited angiogenesis in vivo (Figure 3b, bottom panel). We further examined whether Jzu 17 affects MDA‐MB‐231 cell viability. As shown in Figure S2, Jzu 17 slightly decreased MDA‐MB‐231 cell viability. Jzu 17 at 10 μM caused about 10% reduction in cell viability in MDA‐MB‐231 cells (Figure S2). These results suggest that Jzu 17 inhibits MDA‐MB231 tumour cells‐induced angiogenesis by targeting the proliferating endothelial cells, rather than tumour cells. It also indicates that systemic administration with Jzu 17 is capable of suppressing angiogenesis in vivo.
3.3. Jzu 17 reduced lung metastasis of B16F10 melanoma cells
We exploited the well‐described lung metastasis assay to evaluate the anti‐metastatic effects of Jzu 17 in vivo. C57BL/6 mice were inoculated via the tail vein, with B16F10 mouse melanoma cells exhibiting high metastasis activity from small inoculums. After inoculation for 18 days, lungs from the vehicle‐treated mice exhibited multiple metastasis nodules of different sizes (Figure 4a, upper panel). In contrast, the lungs from mice receiving daily intraperitoneal injections of Jzu 17 (10 mg·kg−1·day−1) displayed fewer and smaller melanoma nodules (Figure 4a, upper panel). We also quantified the level of lung metastasis by determining the number of nodules. A significant reduction in the number of lung metastatic nodules was shown in lungs from Jzu 17‐treated mice when compared with those from vehicle‐treated control mice (Figure 4a, bottom panel). The lung tissues were also paraffin embedded for histological analysis with H&E staining (Figure 4b, upper panel). Jzu 17 markedly reduced lung metastatic nodular areas, compared with the vehicle‐treated control group (Figure 4b, bottom panel). In addition, treatment with Jzu 17 at 10 mg·kg−1·day−1 had no significant effects on mouse body weight, compared to the control group within 18 days (Figure 4c). These findings suggest that Jzu 17 also exhibits inhibitory effects on tumour metastasis in vivo.
3.4. Jzu 17 inhibited VEGFR‐2 signalling in VEGF‐A‐stimulated HUVECs
VEGF‐A signalling via VEGFR‐2 plays a prominent role in regulating angiogenesis (Shibuya, 2011). Following exposure to VEGF‐A, VEGFR‐2 becomes phosphorylated, resulting in the activation of many downstream signalling molecules such as FAK (Farhan, Azad, Touret, & Murray, 2017), Src (Sun et al., 2012), Akt, and ERK1/2 (Huang et al., 2014). VEGF‐A triggers auto‐phosphorylation of VEGFR‐2 on several tyrosine residues. Of these, VEGFR‐2 Tyr1175 phosphorylation is believed to be a key determinant of angiogenic signalling in VEGF‐A‐stimulated HUVECs (Olsson et al., 2006). As shown in Figure 5, Jzu 17 significantly inhibited VEGF‐A‐induced VEGFR‐2 Tyr1175 phosphorylation (Figure 5b). Moreover, Jzu 17 also reduced the phosphorylation of FAK (Figure 5c), Src (Figure 5d), Akt (Figure 5e), and ERK1/2 (Figure 5f) in HUVECs exposed to VEGF‐A. These results suggest that Jzu 17 exhibits its anti‐angiogenesis activities through suppressing VEGF‐A‐VEGFR‐2 signalling.
Figure 5.
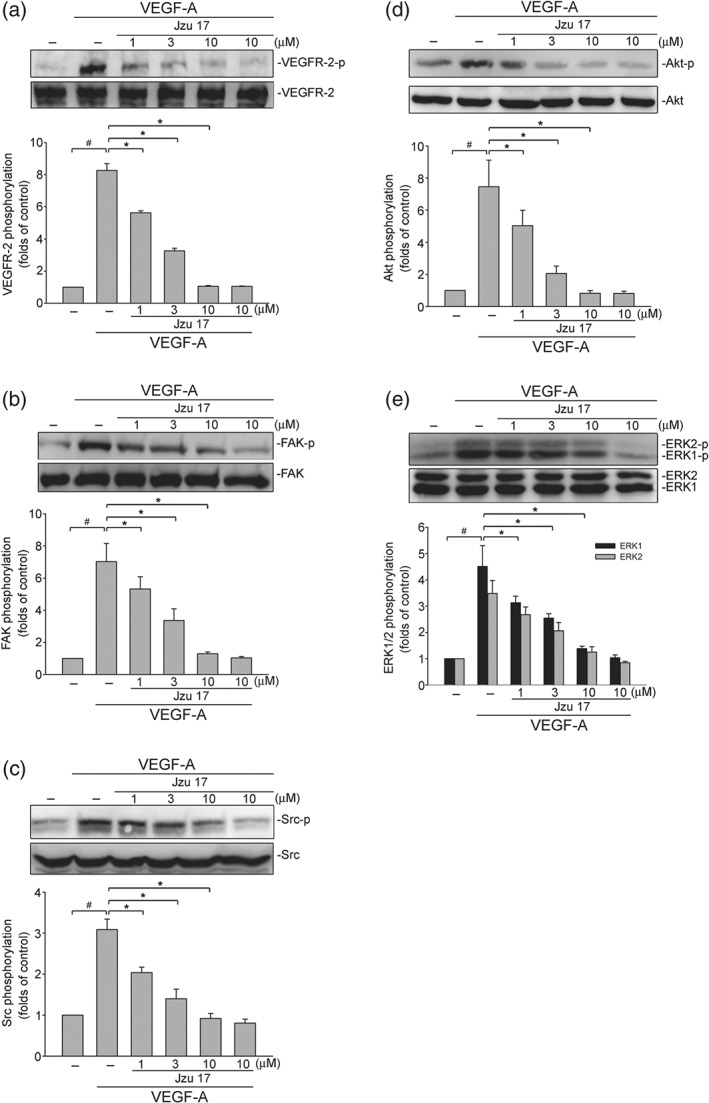
Jzu 17 inhibits VEGFR‐2‐mediated signalling in HUVECs. HUVECs were treated with indicated concentrations of Jzu 17 for 30 min, followed by the stimulation with VEGF‐A (25 ng·ml−1) for another 5 (VEGFR‐2) or 20 (FAK, Src, Akt and ERK1/2) min. Phosphorylation status of VEGFR‐2 (a), FAK (b), Src (c), Akt (d), and ERK1/2 (e) were determined by immunoblotting. The compiled results of VEGFR‐2 Tyr1175 (a), Src Tyr416 (b), FAK Tyr397 (c), Akt Ser473 (d), and ERK1/2 Thr202/Tyr204 (e) phosphorylations are shown. Each column represents the mean ± SEM of six independent experiments. # P < .05, compared with the control group; *P < .05, significantly different from VEGF‐A alone; Kruskal–Wallis test
3.5. Jzu 17, sunitinib, or sorafenib inhibited VEGFR‐2 TK activity in vitro
We next compared the inhibitory effects of Jzu 17, sunitinib, and sorafenib on VEGFR‐2 phosphorylation in HUVECs exposed to VEGF‐A. As shown in Figure S3, Jzu 17 (1–10 μM) suppressed VEGF‐A‐induced VEGFR‐2 phosphorylation with similar potency to sunitinib (Figure S3A) or sorafenib (Figure S3B). The potency of angiogenesis inhibitors might vary in cell‐based systems and in cell‐free systems (Kankanala et al., 2012; Latham et al., 2012). We thus used an in vitro kinase assay to examine the effects of Jzu 17, sunitinib, and sorafenib on the intrinsic TK activity of VEGFR‐2. Sunitinib (Latham et al., 2012) and sorafenib (Pattarozzi et al., 2017) are inhibitors of many RTKs, which also suppress the kinase activity of bFGFR. The effects of these compounds on bFGFR1 kinase activity were also determined. As shown in Figure S4A, sunitinib and sorafenib at concentrations ranging from 0.1 to 3 μM markedly inhibited purified recombinant VEGFR‐2 TK activity. Both drugs at 1 μM elicited approximately 90% inhibition of VEGFR‐2 kinase activity in vitro. To our surprise, Jzu 17 exhibited only about 30% inhibition of kinase activity at a high concentration of 10 μM (Figure S4A). The suppressive effects of these compounds on bFGFR1 kinase activity were less pronounced. Sunitinib and sorafenib at a concentration of 1 μM displayed about 50% inhibition of bFGFR1 kinase activity. However, Jzu 17 is a much weaker inhibitor of this receptor (Figure S4B).
3.6. Computational modelling of the interactions between Jzu 17 and VEGF‐A or VEGFR‐2
We next examined whether Jzu 17 affects VEGF‐A binding to VEGFR in HUVECs. As shown in Figure 6a, Jzu 17 suppressed VEGF‐A binding to VEGFR in HUVECs as determined by flow cytometry. It is likely that Jzu 17 may antagonize VEGF‐A‐VEGFR‐2 signalling through binding to VEGFR‐2 or VEGF‐A. We thus conducted molecular docking simulations to analyse the possible interactions between Jzu 17 and VEGF‐A or VEGFR‐2. The binding site of VEGF‐A was defined by two small receptor cavities between VEGF‐A and VEGFR‐2 (Figure S5A). In addition, the binding site of VEGFR‐2 was defined as the volume of the co‐crystallized compound (Figure S5B). Table 1 shows the docking score and scoring results (Table 1). The docking pose of Jzu 17 has π–cation interactions with Lys868 and His1026 residues of VEGFR‐2 (Figure 6b). It forms a stable docking pose with VEGFR‐2, which is similar to the docking pose of the co‐crystallized compound in the X‐ray crystallography. Figure 6c further illustrated the 2D ligand–protein interaction diagram between Jzu 17 and VEGFR‐2. In contrast, there are three different possible docking poses of Jzu 17 for VEGF‐A (Figure S6). The Poses 1 and 3 have π–cation interactions with Lys84 and Phe36 residues of VEGF‐A. Whereas the Pose 1, 2, or 3 lay in different position of binding site. It appears that Jzu 17 does not have a stable docking pose for VEGF‐A. Taken together, these observations suggest that Jzu 17 may bind to VEGFR‐2 rather than VEGF‐A to interrupt VEGF‐A‐VEGFR2 signalling.
Figure 6.
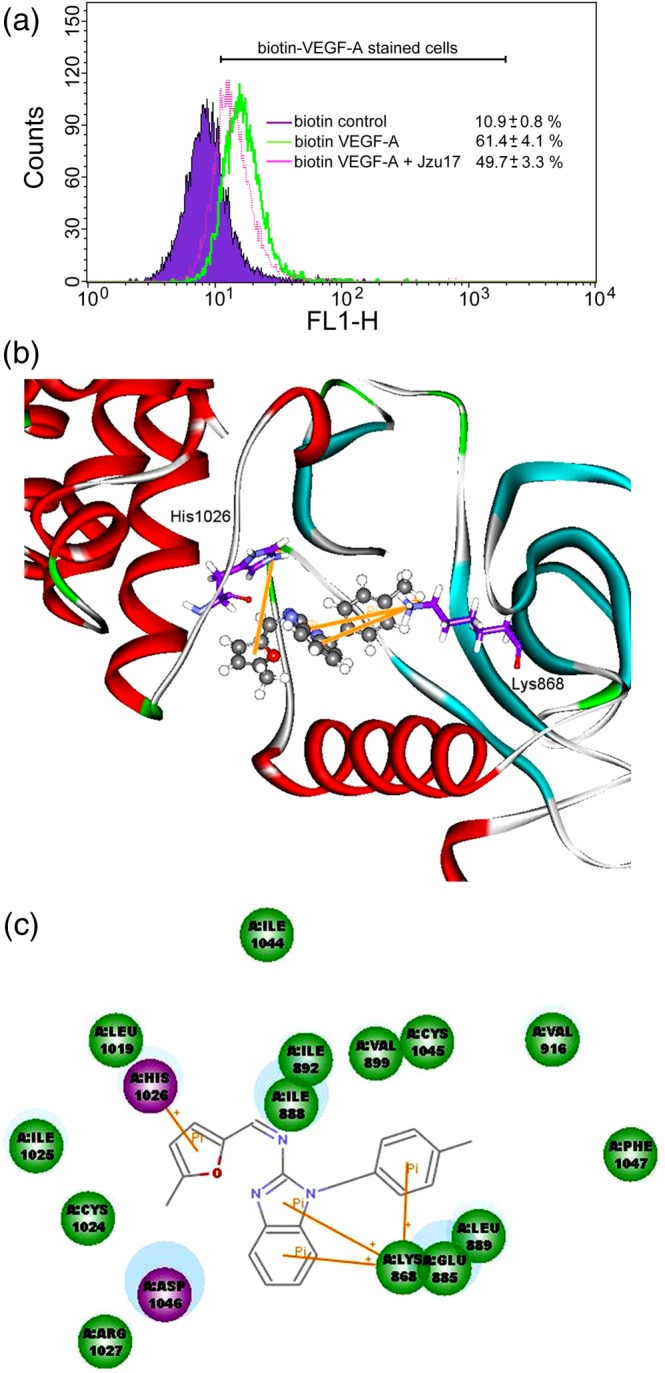
Molecular docking simulation analysis of Jzu 17. (a)HUVECs were detached, suspended in PBS, and treated with biotin control or biotin VEGF‐A in the absence or presence of Jzu 17. Cells were then treated with avidin‐fluorescein, and the fluorescence derived from biotin VEGF‐A‐stained cells was measured by flow‐cytometry. Results shown are representative of seven independent experiments. (b) Molecular modelling of the interactions between VEGFR‐2 and Jzu 17. The graph shows the docking pose of VEGFR‐2 with Jzu 17. (c) Docking pose of VEGFR‐2 with Jzu 17 using 2D ligand–protein interaction diagrams
Table 1.
Scoring functions of compound 17 with VEGF‐A and VEGFR‐2
| Protein | Compound | Pose | ‐PLP1 | ‐PLP2 | ‐PMF |
|---|---|---|---|---|---|
| VEGFR‐2 | Jzu 17 | 1 | 70.61 | 71.31 | 78.35 |
| VEGF‐A | Jzu 17 | 1 | 56.81 | 51.74 | 44.71 |
| Jzu 17 | 2 | 62.32 | 54.81 | 43.99 | |
| Jzu 17 | 3 | 72.09 | 68.08 | 48.22 |
Note. Triple consensus scoring: PLP1, PLP2, and PMF.
Abbreviations: PLP, piecewise linear potential; PMF, potential of mean force.
4. DISCUSSION
In the last decades, cancer remains the leading cause of death globally with increasing incidence. Tumour metastasis is the primary cause of cancer mortality. It is estimated that tumour metastasis is responsible for about 90% of cancer‐related deaths. Inhibition of angiogenesis potentially prevents tumours from growing and metastatic spread to other organs (Fischer, Mazzone, Jonckx, & Carmeliet, 2008). It may also improve chemotherapy targeting tumours through vascular remodelling. Therefore, blockade of angiogenesis has been considered an essential modality for normalizing the tumour‐associated vasculature and serves as a complementary therapeutic paradigm for cancer therapy (Heath & Bicknell, 2009). Clinical efforts developing novel anti‐angiogenesis agents have largely focused on targeting VEGF‐A‐VEGFR‐2 signalling due to its central role in angiogenesis (Welti, Loges, Dimmeler, & Carmeliet, 2013). There is increasing evidence highlighting the beneficial effects of benzimidazole derivatives in the treatment of cancer (El Rashedy & Aboul‐Enein, 2013). In the present study, we identified a low MW compound Jzu 17 from a series of 2‐aminobenzimidazole derivatives as a potent angiogenesis inhibitor. We demonstrated that Jzu 17 suppressed VEGF‐A‐ or tumour cells‐induced angiogenesis in in vivo models. We also noted that Jzu 17 significantly reduced the number and size of metastatic melanoma nodules in the lungs in a murine model of metastasis. Moreover, we demonstrated that suppression of VEGFR‐2‐mediated signalling might contribute to Jzu 17's inhibitory actions on angiogenesis.
VEGFR‐2 is a prototypic RTK that plays a pivotal role in physiological and pathological angiogenesis. Auto‐phosphorylation of VEGFR‐2 upon VEGF‐A binding leads to the activation of its downstream signalling cascades including Src, FAK, Akt, and ERK (Liu & Agarwal, 2010). Several VEGFR‐2 tyrosine residues become phosphorylated in response to VEGF‐A. VEGFR‐2 Tyr1175 phosphorylation is believed to be a key determinant of angiogenic signalling upon VEGF‐A stimulation (Olsson et al., 2006). In addition, recent studies indicated that inhibition of Src, FAK, Akt, or ERK downstream of VEGFR‐2 may be beneficial for cancer therapy (Zhu & Zhou, 2015). In agreement with these observations, we noted that Jzu 17 reduced VEGFR‐2 Tyr1175 phosphorylation and the subsequent phosphorylation of Src, FAK, Akt, and ERK in VEGF‐A‐stimulated HUVECs. Jzu 17 also reduced VEGF‐A‐ or breast cancer cells‐induced angiogenesis in in vivo animal models. Moreover, Jzu 17 suppressed VEGF‐A binding to VEGFR in HUVECs. Molecular docking results further indicate that Jzu 17 may bind to VEGFR‐2 rather than VEGF‐A to interrupt VEGF‐A‐VEGFR2 signalling. However, Jzu 17 may also exhibit inhibitory effects against other RTKs, because of the structural similarities within this family. We noted in this study that Jzu 17 is a weak inhibitor of bFGFR1. Whether Jzu 17 affects other RTKs remains to be clarified. Together, these findings suggest that Jzu 17, at least in part, may represent as a VEGFR‐2 antagonist to interfere with VEGF‐A‐VEGFR‐2 signalling and inhibit tumour angiogenesis.
Besides inhibiting VEGF‐A's angiogenic actions, we also noted that Jzu 17 significantly suppressed MDA‐MB‐231 breast cancer cells‐induced angiogenesis in vivo. However, Jzu 17 only slightly altered MDA‐MB‐231 cell viability. It suggests that the suppression, by Jzu 17, of angiogenesis induced by MDA‐MB‐231 cells may not contribute to the reduction of cancer cell viability. Jzu 17 may target the proliferating endothelial cells, rather than tumour cells. In addition, benzimidazole has emerged as a key pharmacophore in the development of chemotherapeutic drugs (El Rashedy & Aboul‐Enein, 2013; Nofal et al., 2011). Benzimidazole‐based compounds caused tumour cell cycle arrest by blocking EGFR and HER2 signalling cascades (Chu et al., 2015). Apoptotic mechanisms may also contribute to the anti‐tumour properties of benzimidazole derivatives (Chu et al., 2015; Nayak et al., 2017). We noted in this study that Jzu 17, a novel 2‐aminobenzimidazole derivative, only slightly suppressed cell viability with 10% reduction in MDA‐MB‐231 breast cancer cells (Figure S2). In contrast, Jzu 17 caused a marked reduction in cell viability in HCT116 and SW480 colorectal cancer cells. However, Jzu 17 did not alter cell viability in non‐tumour HaCaT keratinocytes (Figure S2). It is likely that Jzu 17's effects in reducing tumour cell viability may vary among different cancer types. The precise mechanisms underlying Jzu 17‐reduced tumour cell viability needs to be established.
The critical role of angiogenic factors and its receptors in tumour cells has been recently described. Knizetova et al (2008) demonstrated that VEGF‐mediated autocrine promotes glioblastoma aggressiveness. VEGF‐VEGFR signalling also contributes to tumour growth or survival in other type of cancers (Barr et al., 2015). http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=963 and VEGFRs are highly expressed in melanoma cells, and high circulating serum VEGF levels are associated with poor pro+gnosis in patients with melanoma (Tas et al., 2006). We noted in this study that Jzu 17 also markedly reduced B16F10 melanoma cell viability (Figure S2). Therefore, it also worth to further investigate whether Jzu 17‐reduced melanoma cell viability involves VEGFR‐2 signalling blockade.
It is expected that low MW inhibitors of angiogenesis are capable of suppressing tumour metastasis without causing severe adverse effects, in contrast to widely used chemotherapeutic drugs (Ivy, Wick, & Kaufman, 2009). However, most angiogenesis inhibitors possess adverse effects such as bleeding complications (Elice & Rodeghiero, 2010). The mechanisms underlying these adverse effects remain incompletely understood. It appears that these angiogenesis inhibitors exhibited poor target selectivity (Verheul & Pinedo, 2007). Understanding the mechanisms involved in causing toxicity may help to develop more specific and potent angiogenesis inhibitors. We performed a tail‐bleeding assay to examine Jzu 17's in vivo effect on haemostasis or the risk of haemorrhage. As shown in Figure S7, systemic administration of mice with Jzu 17 for 10 days did not alter tail‐bleeding time. It suggests that Jzu 17 might be safer in posing less bleeding risk. In addition, administration of Jzu 17 (10 mg·kg−1·day−1) for 10 days did not alter the body weight of the mice but showed significant inhibitory effects on VEGF‐A or tumour cells‐induced angiogenesis. Together, these observations suggest that Jzu 17 may be a novel anti‐angiogenesis agent with limited off‐target toxicity.
It is crucial to efficiently design and evaluate novel agents by applying current knowledge. Based on the studies showing that benzimidazole, a key pharmacophore, exhibits diverse pharmacological properties, we used cell‐based and in vivo models to evaluate and identify a low MW compound, Jzu 17, from a series of 2‐aminobenzimidazole derivatives, as a potent angiogenesis inhibitor. Targeted RTK inhibitors designed to disrupt tumour vascularization or inhibit tumour proliferation remain a major focus in anti‐cancer drug development. Recent studies showed that in silico structure‐based design technology might help to rapidly identify potent low MW angiogenesis inhibitors and provide an important basis for the development of multi‐TKs inhibitors (Kankanala et al., 2012; Latham et al., 2012). A rationally designed multi‐kinase inhibitor based on comprehensive structure–function analyses exhibits a synergistic effect targeting not only angiogenesis but also tumour cell proliferation (Latham et al., 2014). Utilizing the structure‐based methods to design and synthesize additional novel 2‐aminobenzimidazole‐based compounds may help to develop multi‐kinase inhibitors in the future.
In conclusion, we have shown in the present study that a novel 2‐aminobenzimidazole derivative Jzu 17 exhibits anti‐angiogenic properties via targeting the VEGFR‐2 signalling in vascular endothelial cells. Moreover, Jzu 17 may also have additional properties with anti‐tumour effects. The exact mechanisms of these activities remain to be characterized, but together, these observations support the potential of Jzu 17 as a valuable lead compound in developing anti‐angiogenesis and/or anti‐cancer agents for future cancer therapy.
AUTHOR CONTRIBUTIONS
C.L.C., T.F.H., M.J.H., and S.W.H designed the experiments. M.J.H. and S.W.H. performed the experiments. C.L.C., M.J.H., and S.W.H. analysed the data. T.C.C., G.Y.G., and J.C.L. contributed reagents/synthesized Jzu compounds. K.C.C. and J.C.L were responsible for the molecular docking simulation. M.J.H and S.W.H wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, https://doi.org/10.1111/bph.14208 and https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14206, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1.
Effects of 2 aminobenzimidazole derivatives, Jzu compounds, on cell viability in VEGF‐A‐stimulated HUVECs (A) Chemical structures of 2‐aminobenzimidazole derivatives, Jzu compounds. (B) HUVECs were starved in 2% FBS‐containing M199 without ECGS for 16 h. After starvation, cells were treated with Jzu compounds (Jzu 1–24) at the concentrations of 1, 3, or 10 μM followed by the stimulation with VEGF‐A (25 ng ml‐1) for another 24 h. Cell viability was determined by MTT assay. Each column represents the mean ± S.E.M. of six independent experiments performed in duplicate (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the group treated with VEGF‐A alone). Technical replicates were used to ensure the reliability of singe values for each experiment.
Figure S2. Effects of Jzu 17 on cell viability in non‐tumor HaCaT keratinocytes or tumor cells. Cells were treated with Jzu 17 at indicated concentrations for 24 h. Cell viability was determined by MTT assay. Each column represents the mean ± S.E.M. of six independent experiments performed in duplicate (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the control group). Technical replicates were used to ensure the reliability of singe values for each experiment.
Figure S3. Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 phosphorylation in VEGF‐A‐stimulated HUVECs HUVECs were treated with indicated concentrations of sunitinib (A) or sorafenib (B) and Jzu 17 for 30 min, followed by the stimulation with VEGF‐A (25 ng ml‐1) for another 5 min. Phosphorylation status of VEGFR‐2 was determined by immunoblotting. The compiled results of VEGFR‐2 Tyr1175 phosphorylations are shown. Each column represents the mean ± S.E.M. of six independent experiments. (Statistically significant differences were determined using the Kruskal‐Wallis test. #p < 0.05, compared with the control group; *p < 0.05, compared with the group treated with VEGF‐A alone.).
Figure S4. Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 or FGFR1 kinase activities Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 (A) or FGFR1 (B) kinase activity was determined by in vitro kinase assay using Kinase‐Glo Plus luminescence kinase assay kit (Promega, Madison, WI, U.S.A.) as described in “Supplement Methods” section. Each column represents the mean ± S.E.M. of seven independent experiments. (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the control group).
Figure S5. Putative binding sites of VEGF‐A and VEGFR‐2. (A) The binding site of VEGF‐A was defined by two small receptor cavities between VEGF‐A and VEGFR‐2. Green, the putative binding site; Blue and Red, VEGF‐A protein. (B) The binding site of VEGFR‐2 was defined as the volume of the co‐crystallized compound. Green, the putative binding site; Blue and Red, VEGFR‐2 protein.
Figure S6 The predicted binding models of Jzu 17 and VEGF‐A. Three possible docking poses of VEGF‐A with Jzu 17 (A, Pose 1; B, Pose 2; C, Pose 3) and their 2D ligand‐protein interaction diagrams (D, Pose 1; E, Pose 2; F, Pose 3).
Figure S7. Effects of Jzu 17 on tail bleeding times of mice Mice were administered intraperitoneally with vehicle or Jzu 17 (5 or 10 mg kg‐1 day‐1) for 10 days. Tail bleeding time was determined. Each column represents the mean ± S.E.M. (N = 6 for each group).
ACKNOWLEDGEMENTS
This work was supported by grants (MOST 106‐2320‐B‐038‐013‐MY2, MOST 107‐2320‐B‐038‐007, MOST 107‐2320‐B‐039‐058, and MOST 108‐2314‐B‐038‐019) from the Ministry of Science and Technology of Taiwan, grant (CMU107‐S‐33) from the China Medical University, Taichung, Taiwan, grant (107TMU‐TMUH‐12) from the Taipei Medical University Hospital, and grant (TMU107‐AE1‐B01) from the Taipei Medical University, Taipei, Taiwan.
Lien J‐C, Chung C‐L, Huang T‐F, et al. A novel 2‐aminobenzimidazole‐based compound Jzu 17 exhibits anti‐angiogenesis effects by targeting VEGFR‐2 signalling. Br J Pharmacol. 2019;176:4034–4049. 10.1111/bph.14813
Shiu‐Wen Huang will communicate with the editorial office and, if necessary, the production office.
Contributor Information
Ming‐Jen Hsu, Email: aspirin@tmu.edu.tw.
Shiu‐Wen Huang, Email: shiuwen@tmu.edu.tw.
REFERENCES
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Other protein targets. British Journal of Pharmacology, 174(Suppl 1), S1–S16. 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr, M. P. , Gray, S. G. , Gately, K. , Hams, E. , Fallon, P. G. , Davies, A. M. , … O'Byrne, K. J. (2015). Vascular endothelial growth factor is an autocrine growth factor, signaling through neuropilin‐1 in non‐small cell lung cancer. Molecular Cancer, 14, 45 10.1186/s12943-015-0310-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielenberg, D. R. , & Zetter, B. R. (2015). The contribution of angiogenesis to the process of metastasis. Cancer Journal, 21, 267–273. 10.1097/PPO.0000000000000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bold, G. , Schnell, C. , Furet, P. , McSheehy, P. , Bruggen, J. , Mestan, J. , … Littlewood‐Evans, A. (2016). A novel potent oral series of VEGFR2 inhibitors abrogate tumor growth by inhibiting angiogenesis. Journal of Medicinal Chemistry, 59, 132–146. 10.1021/acs.jmedchem.5b01582 [DOI] [PubMed] [Google Scholar]
- Brooks, B. R. , Bruccoleri, R. E. , Olafson, B. D. , States, D. J. , Swaminathan, S. , & Karplus, M. (1983). CHARMM: A program for macromolecular energy minimization and dynamics calculations. Journal of Computational Chemistry, 4, 187–217. 10.1002/jcc.540040211 [DOI] [Google Scholar]
- Brozzo, M. S. , Bjelic, S. , Kisko, K. , Schleier, T. , Leppanen, V. M. , Alitalo, K. , … Ballmer‐Hofer, K. (2012). Thermodynamic and structural description of allosterically regulated VEGFR‐2 dimerization. Blood, 119, 1781–1788. 10.1182/blood-2011-11-390922 [DOI] [PubMed] [Google Scholar]
- Brunton, V. G. , & Frame, M. C. (2008). Src and focal adhesion kinase as therapeutic targets in cancer. Current Opinion in Pharmacology, 8, 427–432. 10.1016/j.coph.2008.06.012 [DOI] [PubMed] [Google Scholar]
- Bukhari, S. N. , Lauro, G. , Jantan, I. , Fei Chee, C. , Amjad, M. W. , Bifulco, G. , … Rahman, N. A. (2016). Anti‐inflammatory trends of new benzimidazole derivatives. Future Medicinal Chemistry, 8, 1953–1967. 10.4155/fmc-2016-0062 [DOI] [PubMed] [Google Scholar]
- Calvetti, L. , Pilotto, S. , Carbognin, L. , Ferrara, R. , Caccese, M. , Tortora, G. , & Bria, E. (2015). The coming of ramucirumab in the landscape of anti‐angiogenic drugs: Potential clinical and translational perspectives. Expert Opinion on Biological Therapy, 15, 1359–1370. 10.1517/14712598.2015.1071350 [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. (2005). VEGF as a key mediator of angiogenesis in cancer. Oncology, 69(Suppl 3), 4–10. 10.1159/000088478 [DOI] [PubMed] [Google Scholar]
- Chu, B. , Liu, F. , Li, L. , Ding, C. , Chen, K. , Sun, Q. , … Jiang, Y. (2015). A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell Death & Disease, 6, e1686 10.1038/cddis.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Rashedy, A. A. , & Aboul‐Enein, H. Y. (2013). Benzimidazole derivatives as potential anticancer agents. Mini Reviews in Medicinal Chemistry, 13, 399–407. [DOI] [PubMed] [Google Scholar]
- Elice, F. , & Rodeghiero, F. (2010). Bleeding complications of antiangiogenic therapy: pathogenetic mechanisms and clinical impact. Thrombosis Research, 125(Suppl 2), S55–S57. 10.1016/S0049-3848(10)70014-1 [DOI] [PubMed] [Google Scholar]
- Farhan, M. A. , Azad, A. K. , Touret, N. , & Murray, A. G. (2017). FGD5 regulates VEGF receptor‐2 coupling to PI3 kinase and receptor recycling. Arteriosclerosis, Thrombosis, and Vascular Biology, 37, 2301–2310. 10.1161/ATVBAHA.117.309978 [DOI] [PubMed] [Google Scholar]
- Ferrara, N. (2002). VEGF and the quest for tumour angiogenesis factors. Nature Reviews. Cancer, 2, 795–803. 10.1038/nrc909 [DOI] [PubMed] [Google Scholar]
- Ferrara, N. (2004). Vascular endothelial growth factor as a target for anticancer therapy. The Oncologist, 9(Suppl 1), 2–10. 10.1634/theoncologist.9-suppl_1-2 [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , & Kerbel, R. S. (2005). Angiogenesis as a therapeutic target. Nature, 438, 967–974. 10.1038/nature04483 [DOI] [PubMed] [Google Scholar]
- Fischer, C. , Mazzone, M. , Jonckx, B. , & Carmeliet, P. (2008). FLT1 and its ligands VEGFB and PlGF: Drug targets for anti‐angiogenic therapy? Nature Reviews. Cancer, 8, 942–956. 10.1038/nrc2524 [DOI] [PubMed] [Google Scholar]
- Goel, H. L. , & Mercurio, A. M. (2013). VEGF targets the tumour cell. Nature Reviews. Cancer, 13, 871–882. 10.1038/nrc3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, V. L. , & Bicknell, R. (2009). Anticancer strategies involving the vasculature. Nature Reviews. Clinical Oncology, 6, 395–404. 10.1038/nrclinonc.2009.52 [DOI] [PubMed] [Google Scholar]
- Holopainen, T. , Saharinen, P. , D'Amico, G. , Lampinen, A. , Eklund, L. , Sormunen, R. , … Alitalo, K. (2012). Effects of angiopoietin‐2‐blocking antibody on endothelial cell‐cell junctions and lung metastasis. Journal of the National Cancer Institute, 104, 461–475. 10.1093/jnci/djs009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. W. , Lien, J. C. , Kuo, S. C. , & Huang, T. F. (2012). Antiangiogenic mechanisms of PJ‐8, a novel inhibitor of vascular endothelial growth factor receptor signaling. Carcinogenesis, 33, 1022–1030. 10.1093/carcin/bgs127 [DOI] [PubMed] [Google Scholar]
- Huang, S. W. , Lien, J. C. , Kuo, S. C. , & Huang, T. F. (2014). PPemd26, an anthraquinone derivative, suppresses angiogenesis via inhibiting VEGFR2 signalling. British Journal of Pharmacology, 171, 5728–5742. 10.1111/bph.12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy, S. P. , Wick, J. Y. , & Kaufman, B. M. (2009). An overview of small‐molecule inhibitors of VEGFR signaling. Nature Reviews. Clinical Oncology, 6, 569–579. 10.1038/nrclinonc.2009.130 [DOI] [PubMed] [Google Scholar]
- Jain, R. K. , Duda, D. G. , Clark, J. W. , & Loeffler, J. S. (2006). Lessons from phase III clinical trials on anti‐VEGF therapy for cancer. Nature Clinical Practice. Oncology, 3, 24–40. 10.1038/ncponc0403 [DOI] [PubMed] [Google Scholar]
- Javerzat, S. , Auguste, P. , & Bikfalvi, A. (2002). The role of fibroblast growth factors in vascular development. Trends in Molecular Medicine, 8, 483–489. 10.1016/S1471-4914(02)02394-8 [DOI] [PubMed] [Google Scholar]
- Kankanala, J. , Latham, A. M. , Johnson, A. P. , Homer‐Vanniasinkam, S. , Fishwick, C. W. , & Ponnambalam, S. (2012). A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. British Journal of Pharmacology, 166, 737–748. 10.1111/j.1476-5381.2011.01801.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altmans, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. The Journal of Gene Medicine, 12, 561–563. 10.1002/jgm.1473 [DOI] [PubMed] [Google Scholar]
- Knizetova, P. , Ehrmann, J. , Hlobilkova, A. , Vancova, I. , Kalita, O. , Kolar, Z. , & Bartek, J. (2008). Autocrine regulation of glioblastoma cell cycle progression, viability and radioresistance through the VEGF‐VEGFR2 (KDR) interplay. Cell Cycle, 7, 2553–2561. 10.4161/cc.7.16.6442 [DOI] [PubMed] [Google Scholar]
- Latham, A. M. , Bruns, A. F. , Kankanala, J. , Johnson, A. P. , Fishwick, C. W. , Homer‐Vanniasinkam, S. , & Ponnambalam, S. (2012). Indolinones and anilinophthalazines differentially target VEGF‐A‐ and basic fibroblast growth factor‐mediated responses in primary human endothelial cells. British Journal of Pharmacology, 165, 245–259. 10.1111/j.1476-5381.2011.01545.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham, A. M. , Kankanala, J. , Fearnley, G. W. , Gage, M. C. , Kearney, M. T. , Homer‐Vanniasinkam, S. , … Ponnambalam, S. (2014). In silico design and biological evaluation of a dual specificity kinase inhibitor targeting cell cycle progression and angiogenesis. PLoS ONE, 9, e110997 10.1371/journal.pone.0110997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, A. M. , Tulpule, A. , Quinn, D. I. , Gorospe, G. 3rd , Smith, D. L. , Hornor, L. , … Gill, P. S. (2006). Phase I study of antisense oligonucleotide against vascular endothelial growth factor: Decrease in plasma vascular endothelial growth factor with potential clinical efficacy. Journal of Clinical Oncology, 24, 1712–1719. 10.1200/JCO.2005.03.4801 [DOI] [PubMed] [Google Scholar]
- Lin, H. H. , Chen, J. H. , Chou, F. P. , & Wang, C. J. (2011). Protocatechuic acid inhibits cancer cell metastasis involving the down‐regulation of Ras/Akt/NF‐κB pathway and MMP‐2 production by targeting RhoB activation. British Journal of Pharmacology, 162, 237–254. 10.1111/j.1476-5381.2010.01022.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , & Agarwal, S. (2010). Mechanical signals activate vascular endothelial growth factor receptor‐2 to upregulate endothelial cell proliferation during inflammation. Journal of Immunology, 185, 1215–1221. 10.4049/jimmunol.0903660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows, K. L. , & Hurwitz, H. I. (2012). Anti‐VEGF therapies in the clinic. Cold Spring Harbor Perspectives in Medicine, 2 10.1101/cshperspect.a006577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, V. L. , Nagesh, N. , Ravikumar, A. , Bagul, C. , Vishnuvardhan, M. , Srinivasulu, V. , & Kamal, A. (2017). 2‐aryl benzimidazole conjugate induced apoptosis in human breast cancer MCF‐7 cells through caspase independent pathway. Apoptosis, 22, 118–134. 10.1007/s10495-016-1290-x [DOI] [PubMed] [Google Scholar]
- Noble, M. E. , Endicott, J. A. , & Johnson, L. N. (2004). Protein kinase inhibitors: Insights into drug design from structure. Science, 303, 1800–1805. 10.1126/science.1095920 [DOI] [PubMed] [Google Scholar]
- Nofal, Z. M. , Soliman, E. A. , Abd El‐Karim, S. S. , El Zahar, M. I. , Srour, A. M. , Sethumadhavan, S. , & Maher, T. J. (2011). Novel benzimidazole derivatives as expected anticancer agents. Acta Poloniae Pharmaceutica, 68, 519–534. [PubMed] [Google Scholar]
- Olsson, A. K. , Dimberg, A. , Kreuger, J. , & Claesson‐Welsh, L. (2006). VEGF receptor signalling? In control of vascular function. Nature Reviews. Molecular Cell Biology, 7, 359–371. 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- Ozkay, Y. , Tunali, Y. , Karaca, H. , & Isikdag, I. (2011). Antimicrobial activity of a new series of benzimidazole derivatives. Archives of Pharmacal Research, 34, 1427–1435. 10.1007/s12272-011-0903-8 [DOI] [PubMed] [Google Scholar]
- Papadopoulos, N. , Martin, J. , Ruan, Q. , Rafique, A. , Rosconi, M. P. , Shi, E. , … Wiegand, S. J. (2012). Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis, 15, 171–185. 10.1007/s10456-011-9249-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattarozzi, A. , Carra, E. , Favoni, R. E. , Wurth, R. , Marubbi, D. , Filiberti, R. A. , … Daga, A. (2017). The inhibition of FGF receptor 1 activity mediates sorafenib antiproliferative effects in human malignant pleural mesothelioma tumor‐initiating cells. Stem Cell Research & Therapy, 8, 119 10.1186/s13287-017-0573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager, G. W. , Poettler, M. , Unseld, M. , & Zielinski, C. C. (2012). Angiogenesis in cancer: Anti‐VEGF escape mechanisms. Transl Lung Cancer Res, 1, 14–25. 10.3978/j.issn.2218-6751.2011.11.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya, M. (2011). Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti‐ and pro‐angiogenic therapies. Genes & Cancer, 2, 1097–1105. 10.1177/1947601911423031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingalapur, R. V. , Hosamani, K. M. , Keri, R. S. , & Hugar, M. H. (2010). Derivatives of benzimidazole pharmacophore: Synthesis, anticonvulsant, antidiabetic and DNA cleavage studies. European Journal of Medicinal Chemistry, 45, 1753–1759. 10.1016/j.ejmech.2010.01.007 [DOI] [PubMed] [Google Scholar]
- Solinas, M. , Massi, P. , Cantelmo, A. R. , Cattaneo, M. G. , Cammarota, R. , Bartolini, D. , … Parolaro, D. (2012). Cannabidiol inhibits angiogenesis by multiple mechanisms. British Journal of Pharmacology, 167, 1218–1231. 10.1111/j.1476-5381.2012.02050.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers, J. , Cohen, M. H. , Keegan, P. , & Pazdur, R. (2010). FDA drug approval summary: Bevacizumab plus interferon for advanced renal cell carcinoma. The Oncologist, 15, 104–111. 10.1634/theoncologist.2009-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Li, X. , Massena, S. , Kutschera, S. , Padhan, N. , Gualandi, L. , … Claesson‐Welsh, L. (2012). VEGFR2 induces c‐Src signaling and vascular permeability in vivo via the adaptor protein TSAd. The Journal of Experimental Medicine, 209, 1363–1377. 10.1084/jem.20111343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas, F. , Duranyildiz, D. , Oguz, H. , Camlica, H. , Yasasever, V. , & Topuz, E. (2006). Circulating serum levels of angiogenic factors and vascular endothelial growth factor receptors 1 and 2 in melanoma patients. Melanoma Research, 16, 405–411. 10.1097/01.cmr.0000222598.27438.82 [DOI] [PubMed] [Google Scholar]
- Ucuzian, A. A. , Gassman, A. A. , East, A. T. , & Greisler, H. P. (2010). Molecular mediators of angiogenesis. Journal of Burn Care & Research, 31, 158–175. 10.1097/BCR.0b013e3181c7ed82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheul, H. M. , & Pinedo, H. M. (2007). Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nature Reviews. Cancer, 7, 475–485. 10.1038/nrc2152 [DOI] [PubMed] [Google Scholar]
- Wang, P. , Zhen, H. , Zhang, J. , Zhang, W. , Zhang, R. , Cheng, X. , … Zhang, X. (2012). Survivin promotes glioma angiogenesis through vascular endothelial growth factor and basic fibroblast growth factor in vitro and in vivo. Molecular Carcinogenesis, 51, 586–595. 10.1002/mc.20829 [DOI] [PubMed] [Google Scholar]
- Weis, S. M. , & Cheresh, D. A. (2011). Tumor angiogenesis: Molecular pathways and therapeutic targets. Nature Medicine, 17, 1359–1370. 10.1038/nm.2537 [DOI] [PubMed] [Google Scholar]
- Welti, J. , Loges, S. , Dimmeler, S. , & Carmeliet, P. (2013). Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. The Journal of Clinical Investigation, 123, 3190–3200. 10.1172/JCI70212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos, G. D. , Davis, S. , Gale, N. W. , Rudge, J. S. , Wiegand, S. J. , & Holash, J. (2000). Vascular‐specific growth factors and blood vessel formation. Nature, 407, 242–248. 10.1038/35025215 [DOI] [PubMed] [Google Scholar]
- Zhu, X. , & Zhou, W. (2015). The emerging regulation of VEGFR‐2 in triple‐negative breast cancer. Frontiers in Endocrinology (Lausanne), 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Effects of 2 aminobenzimidazole derivatives, Jzu compounds, on cell viability in VEGF‐A‐stimulated HUVECs (A) Chemical structures of 2‐aminobenzimidazole derivatives, Jzu compounds. (B) HUVECs were starved in 2% FBS‐containing M199 without ECGS for 16 h. After starvation, cells were treated with Jzu compounds (Jzu 1–24) at the concentrations of 1, 3, or 10 μM followed by the stimulation with VEGF‐A (25 ng ml‐1) for another 24 h. Cell viability was determined by MTT assay. Each column represents the mean ± S.E.M. of six independent experiments performed in duplicate (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the group treated with VEGF‐A alone). Technical replicates were used to ensure the reliability of singe values for each experiment.
Figure S2. Effects of Jzu 17 on cell viability in non‐tumor HaCaT keratinocytes or tumor cells. Cells were treated with Jzu 17 at indicated concentrations for 24 h. Cell viability was determined by MTT assay. Each column represents the mean ± S.E.M. of six independent experiments performed in duplicate (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the control group). Technical replicates were used to ensure the reliability of singe values for each experiment.
Figure S3. Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 phosphorylation in VEGF‐A‐stimulated HUVECs HUVECs were treated with indicated concentrations of sunitinib (A) or sorafenib (B) and Jzu 17 for 30 min, followed by the stimulation with VEGF‐A (25 ng ml‐1) for another 5 min. Phosphorylation status of VEGFR‐2 was determined by immunoblotting. The compiled results of VEGFR‐2 Tyr1175 phosphorylations are shown. Each column represents the mean ± S.E.M. of six independent experiments. (Statistically significant differences were determined using the Kruskal‐Wallis test. #p < 0.05, compared with the control group; *p < 0.05, compared with the group treated with VEGF‐A alone.).
Figure S4. Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 or FGFR1 kinase activities Effects of sunitinib, sorafenib and Jzu 17 on VEGFR‐2 (A) or FGFR1 (B) kinase activity was determined by in vitro kinase assay using Kinase‐Glo Plus luminescence kinase assay kit (Promega, Madison, WI, U.S.A.) as described in “Supplement Methods” section. Each column represents the mean ± S.E.M. of seven independent experiments. (Statistically significant differences were determined using the Kruskal‐Wallis test. *p < 0.05, compared with the control group).
Figure S5. Putative binding sites of VEGF‐A and VEGFR‐2. (A) The binding site of VEGF‐A was defined by two small receptor cavities between VEGF‐A and VEGFR‐2. Green, the putative binding site; Blue and Red, VEGF‐A protein. (B) The binding site of VEGFR‐2 was defined as the volume of the co‐crystallized compound. Green, the putative binding site; Blue and Red, VEGFR‐2 protein.
Figure S6 The predicted binding models of Jzu 17 and VEGF‐A. Three possible docking poses of VEGF‐A with Jzu 17 (A, Pose 1; B, Pose 2; C, Pose 3) and their 2D ligand‐protein interaction diagrams (D, Pose 1; E, Pose 2; F, Pose 3).
Figure S7. Effects of Jzu 17 on tail bleeding times of mice Mice were administered intraperitoneally with vehicle or Jzu 17 (5 or 10 mg kg‐1 day‐1) for 10 days. Tail bleeding time was determined. Each column represents the mean ± S.E.M. (N = 6 for each group).