

Abstract
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disorder, yet the genetic cause of up to 50% of cases remains unknown. Here, we show that mutations in KLHL24 cause HCM in humans. Using genome-wide linkage analysis and exome sequencing, we identified homozygous mutations in KLHL24 in two consanguineous families with HCM. Of the 11 young affected adults identified, 3 died suddenly and 1 had a cardiac transplant due to heart failure. KLHL24 is a member of the Kelch-like protein family, which acts as substrate-specific adaptors to Cullin E3 ubiquitin ligases. Endomyocardial and skeletal muscle biopsies from affected individuals of both families demonstrated characteristic alterations, including accumulation of desmin intermediate filaments. Knock-down of the zebrafish homologue klhl24a results in heart defects similar to that described for other HCM-linked genes providing additional support for KLHL24 as a HCM-associated gene. Our findings reveal a crucial role for KLHL24 in cardiac development and function.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disorder, and the prevalence is approximately 1 in 500 individuals (1). In most cases, HCM is caused by autosomal dominant mutations in genes encoding proteins of the sarcomere; among these, the most common genes are slow/beta cardiac myosin heavy chain (MYH7) and the cardiac myosin-binding protein C (MYBPC3) located within the thick filament and the thin filament-associated troponin T (TNNT2) and troponin I (TNNI3). Pathogenic variants of genes associated with metabolic disorders may also cause a cardiomyopathy that mimics HCM but usually with recessive inheritance. HCM is diagnosed on cardiac imaging, showing unexplained hypertrophy together with typical electrocardiographic abnormalities (1). Next-generation sequencing panels for clinical genetic testing for HCM provide a definitive molecular diagnosis in up to 50% of patients (2). Still, many patients remain without a clear genetic diagnosis, suggesting that additional causes of HCM remain to be identified (1).
Here, we report a novel cardiomyopathy that mimics HCM with recessive inheritance identified in two unrelated families. Several individuals died of sudden cardiac arrest in young adulthood. Endomyocardial and skeletal muscle biopsy demonstrated characteristic alterations, and we provide evidence that the disease is caused by inactivation of the Kelch-like (KLHL) protein 24 by molecular genetic investigations and functional studies in zebrafish.
Results
A novel autosomal recessive cardiomyopathy causing lethal arrhythmias and heart failure
We describe two families with autosomal recessive cardiomyopathy originating from Iraq (family A) and Iran (family B) (Fig. 1). In summary, affected members of both families presented with symptoms characteristic of HCM including recurrent syncope, dyspnea on exertion and palpitations (Table 1). No signs of muscular atrophy or weakness were found, and nerve conduction studies were normal. Importantly, the clinical phenotype is associated with a poor prognosis due to lethal arrhythmias and cardiac failure.
Figure 1.
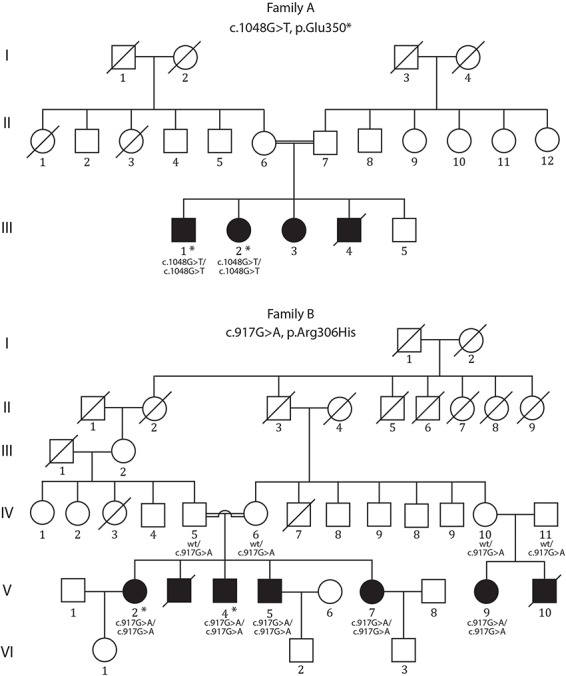
Pedigrees of two consanguineous families with segregation of KLHL24 mutations. Filled squares and circles indicate individuals with cardiomyopathy. Asterisks indicate the individuals whose DNA was analysed by exome sequencing. wt = wild type. Individual III:4 in family A died suddenly at the age of 20. In family B, individual V:3 died suddenly at 26 years of age, and V:10 died of sudden cardiac arrest at the age of 26.
Table 1.
Clinical findings
| Family A | Family B | |||||||
|---|---|---|---|---|---|---|---|---|
| III:1 | III:2 | V:2 | V:3 | V:4 | V:5 | V:7 | V:9 | |
| Gender | M | F | F | M | M | M | F | F |
| Descent | Iraqi | Iraqi | Iranian | Iranian | Iranian | Iranian | Iranian | Iranian |
| Age, years | 32 | 27 | 36 | 27 | 17 | 32 | 29 | 28 |
| Age of onset, (years) | 28 | 19 | nd | nd | 16 | nd | 24 | 21 |
| Initial symptoms | Palpitations, vertigo and shortness of breath | Fatigue, shortness of breath and palpitations | Palpitations, dyspnea on exertion | NYHAIII | Palpitations | Dyspnea on exertion | Palpitations, dyspnea on exertion | nd |
| ICD (years) | 28 | 23 | 35 | — | — | 31 | — | — |
| ECG | Sinus rhythm. General ST-T changes. PR 188 ms. QRS duration 154 ms |
Sinus rhythm. General ST-T changes. PR 194 ms. QRS duration 120 ms. Frequent episodes of non-sustained VT |
Normal sinus rhythm | ST-T change; PR 210 ms | General ST-T changes, PR 186 ms, QRS duration 125 ms |
nd | nd | nd |
| Echocardiogram (age, years) | 28 | 25 | 32 | 27 | 16 | 31 | 28 | nd |
| Echocardiogram results | Left ventricular outflow tract obstruction, left atrium slightly dilated, no valve abnormalities | Moderately dilated left ventricle with regions of akinesia, left atrium slightly dilated, no valve abnormalities | Small left ventricular cavity, severe concentric left ventricular hypertrophy, no SAM, mild mitral regurgitation | ASH | ASH, left ventricular outflow tract obstruction = 48 mmHg, moderate SAM | Small left ventricular cavity, severe SAM, left ventricular outflow tract obstruction = 112 mmHg, moderate mitral regurgitation | Small left ventricular cavity, normal left ventricular function, stage-2 diastolic dysfunction, no left ventricular outflow tract obstruction | Reduced LV systolic, severe left ventricle hypertrophy, mild to moderate mitral regurgitation, no left ventricular outflow tract obstruction, severe ASH, no SAM |
| Left ventricular (LV) end-diastolic volume (mL) | 83 | 146 | 37 | 40 | 37 | 30 | 47 | nd |
| Septal wall thickness (cm) | 2.0 | 0.9 | 2.2 | 3.3 | 2.2 | 2.8 | 4 | nd |
| Posterior wall thickness (cm) | 2.8 | 1.5 | 1.9 | 0.8 | 1 | 1.4 | 1.3 | 2 |
| Ejection fraction (%) | 50 | 25 | 67 | 65 | 70 | 70 | 35 | 40–45 |
| Coronary angiogram | Normal | Normal | nd | nd | nd | nd | nd | nd |
| Heart transplant. (age, years) | — | 26 | — | — | — | — | — | — |
| Dermatological findings | Dark-haired, without freckles, no skin problems | Red-haired with freckles, problems with sun exposure, at the age of 27 no skin abnormality, except pityriasis versicolor | ||||||
| KLHL24 mutation | c.1048G>T p.E350* Homozygous |
c.1048G>T p.E350* Homozygous |
c.917G>A p.R306H Homozygous |
nd | c.917G>A p.R306H Homozygous |
c.917G>A p.R306H Homozygous |
c.917G>A p.R306H Homozygous |
c.917G>A p.R306H Homozygous |
ICD: implantable cardioverter defibrillator; SAM: systolic anterior motion of the mitral, nd: not determined, ASH: Asymmetric Septal Hypertrophy, ECG: electrocardiogram, VT: ventricular tachycardia
Diagnostic histopathological hallmarks in cardiac and skeletal muscle tissue
Individual III:2 in family A had a cardiac transplant at the age of 26. The heart explant showed, similar to a previous endomyocardial biopsy, hypertrophy and scattered cardiomyocytes with polyglucosan. There were also interstitial fibrosis and small macrophage infiltrates that were occasionally associated with polyglucosan bodies (Fig. 2A–G).
Figure 2.
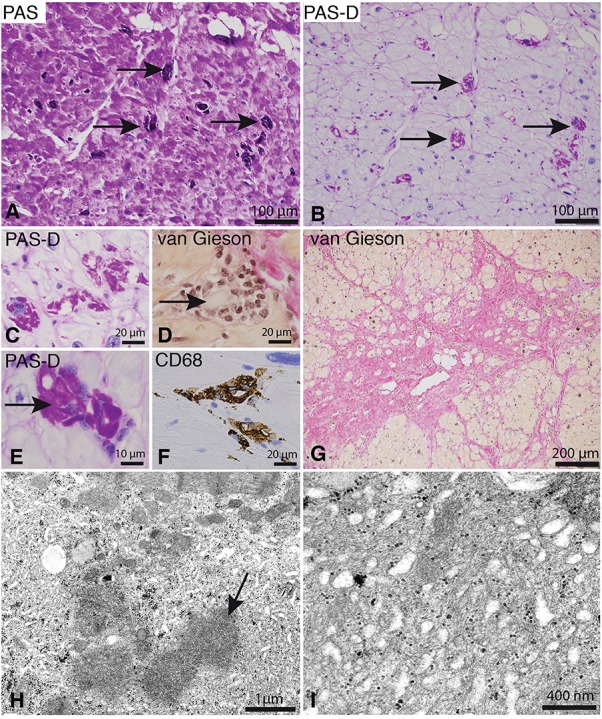
Histopathology of the heart in family A. (A–G) Cardiac explant from individual III:2 after fixation in paraformaldehyde and paraffin embedding. (A and B). There is accumulation of glycogen as revealed by PAS staining (A). Cardiomyocytes have often accumulated PAS-positive material that is alpha-amylase resistant (polyglucosan; arrows) (PAS-D: PAS-diastase). (C) Accumulation of polyglucosan in cardiomyocytes (PAS-Diastase). (D and E) Scattered cardiomyocytes, many of which include polyglucosan (arrows), are associated with inflammatory cells. (F) The inflammatory cells stain positively for CD68, a marker for macrophages. (G) There is patchy fibrosis in the heart that stains red on van Gieson staining, compared to brownish cardiomyocytes. (H and I) Electron microscopy of endomyocardial biopsy material from individual III:1 after glutaraldehyde fixation and embedding in resin. Polyglucosan (arrow in panel H) is associated with intermyofibrillar accumulation of glycogen, filaments and tubular structures, which are seen at higher magnification in panel I.
A myocardial biopsy of individual III:1 from family A was performed at 30 years of age and revealed myocyte hypertrophy and occasional polyglucosan bodies. Electron microscopy showed accumulation of glycogen, tubular structures and irregularly arranged intermediate filaments in the intermyofibrillar regions (Fig. 2H–I).
Skeletal muscle biopsy in individual III:1 and III:2 in family A and V:5 in family B demonstrated characteristic focal subsarcolemmal and intermyofibrillar accumulation of glycogen. The accumulated material was also stained with antibodies against desmin. This accumulation resulted in a jagged appearance at the edges of the majority of both type 1 and type 2 muscle fibres, giving them a characteristic cogwheel-like appearance (Fig. 3A–H and Supplementary Material, Fig. S1). The accumulated material was also stained for nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) but was negative for the mitochondria-specific enzyme succinate dehydrogenase (SDH), indicating presence of tubules derived from the sarcoplasmic reticulum (Supplementary Material, Fig. S2). Electron microscopy confirmed that the accumulated material consisted of intermediate filaments, measuring 8–12 nm in diameter, glycogen and tubular structures (Fig. 3I–J and Supplementary Material, Figs S1 and S2).
Figure 3.
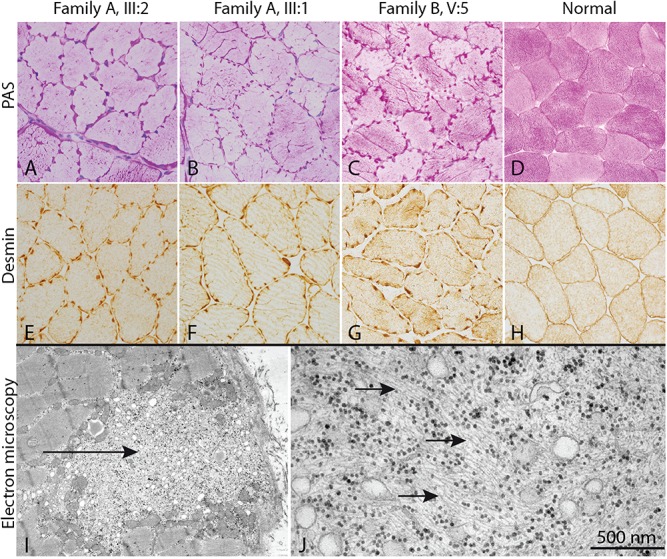
Skeletal muscle biopsy from three individuals from two families with cardiomyopathy and homozygous KLHL24 mutations and a normal control. (A–H) In all three individuals with cardiomyopathy, a characteristic cogwheel appearance of the fibres are present due to jagged accumulation of glycogen (PAS staining) and intermediate filaments (desmin immunostaining). Electron microscopy (I, J) of individual III:2 shows focal subsarcolemmal accumulation of glycogen, tubular structures and intermediate filaments (arrows).
Mutations in KLHL24 are associated with a novel cardiomyopathy
Two affected individuals from each family were investigated by exome sequencing (Fig. 1).
In family A, a homozygous nonsense mutation in the KLHL family member 24 gene (KLHL24) (NM_017644.3) (Fig. 4A–D) was identified in both siblings (III:1 and III:2). The mutation at position c.1048G>T p.Glu350* (Fig. 4B) located in exon 4 of the KLHL24 gene that encodes the functional kelch domains of the protein and is located in an extended 8.7 Mb region of homozygosity. The parents were heterozygous carriers of the mutation.
Figure 4.
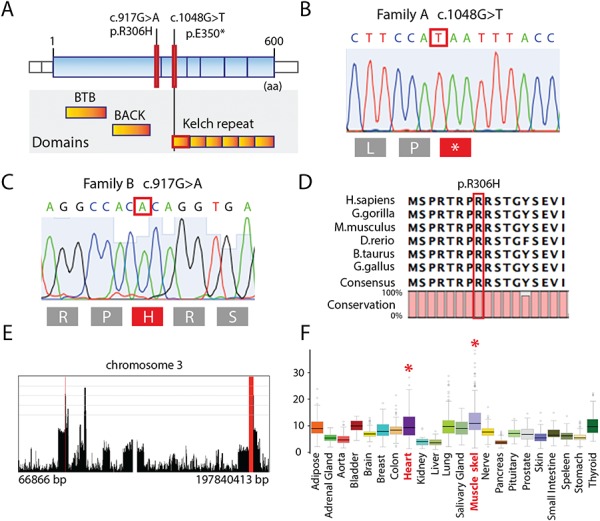
Molecular genetics analysis. (A) Illustration showing the different domains in the KLHL24 protein; mutations are indicated by red bars. (B) Chromatogram demonstrating the homozygous mutation c.1048G>T (p.Glu350*) in family A. (C) Chromatogram demonstrating the homozygous mutation c.917G>A (p.Arg306His) in family B. (D) Illustration showing the evolutionary conservation of the amino acids. The mutated residue (p.Arg306His) is indicated by the red bar. (E) Homozygosity mapping results from Family B showing homozygous regions in a view of chromosome 3 that reveals the longest run of homozygosity containing the candidate variant and spans the coordinates chr3:182,207,825-185,614,988 (rs9877496 to rs73175592) that is approximately 3.4 Mb in length. (F) Gene expression for KLHL24 in the GTEx Portal Database with the highest expression in skeletal muscle, followed by lung and the left ventricle of the heart; data source: GTEx Analysis Release V6p (dbGaP Accession phs000424.v6.p1).
In family B, linkage mapping of the five affected individuals revealed one large (with a homozygosity interval greater than 1 Mb) region of homozygosity on chromosome 3 with an estimated logarithm of odds (LOD) score of 3.6, assuming an autosomal recessive mode of inheritance (Supplementary Material, Fig. S3A). The ~3.4 Mb homozygous region on chromosome 3 (hg19, chr3:182,207,825-185,614,988) is defined by rs9877496 to rs73175592 (Fig. 4E, Supplementary Material, Table S3 and Supplementary Material, Fig. S3A–B). Copy number variant (CNV) analysis of genome-wide single nucleotide polymorphism (SNP)-array genotyping data did not highlight any potentially pathogenic shared CNVs in the affected individuals. Exome sequencing data from individuals V:2 and V:4 identified only one homozygous likely disease-causing variant in this region. The missense mutation at position c.917G>A in the KLHL24 gene changes the amino acid arginine to histidine at position 306 (Fig. 4C). The Arg306 residue is highly conserved among species (Fig. 4D) as well as among different KLHL proteins (Supplementary Material, Fig. S4A). Sanger sequencing confirmed the mutation, and genetic screening demonstrated the same homozygous mutation in the two additional affected individuals in Family B (V:5, V:7). The parents of the homozygous affected individuals in Family B were heterozygous carriers of the mutation (IV:5, IV:6). The affected offspring of an additional branch of Family B (IV:9) was also found to be homozygous for the p.Arg306His mutation and the parents (IV:10 and IV:11) heterozygous, confirming the autosomal recessive (AR) pattern of inheritance.
Neither of the identified KLHL24 mutations was represented in the Greater Middle Eastern Variome including populations from Iran and Iraq. The p.Arg306His mutation was also not found in 500 ethnically matched in-house exomes.
In the Genotype-Tissue Expression (GTEx) Portal Database (http://www.gtexportal.org), KLHL24 shows the highest expression in skeletal muscle, followed by lung and the left ventricle of the heart (Fig. 4F), supporting a role for this protein in muscle (3).
Desmin is upregulated in KLHL24-associated cardiomyopathy
Dominant translation initiation codon mutations in KLHL24 were recently demonstrated to be associated with a type of epidermolysis bullosa in two independent studies (4,5). Both studies indicated that intermediate filament proteins might be substrates for the E3 ubiquitin ligase KLHL24. However, no substrate for KLHL24 has yet been identified in skeletal and heart muscle, despite its expression in these tissues. Based on the finding of accumulation of intermediate filaments in the heart and skeletal muscle by electron microscopy and accumulation of desmin as revealed by immunohistochemistry, we performed western blot analysis of desmin, which was markedly upregulated (Fig. 5).
Figure 5.
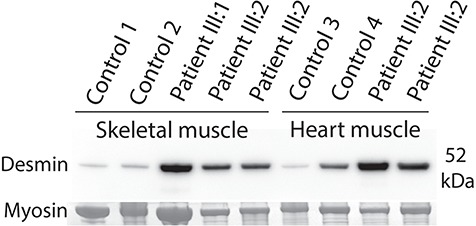
Western blot analysis of desmin in protein extracted from skeletal muscle biopsies and heart muscle specimens showed upregulation of desmin compared to control sample both in the skeletal muscle and the heart muscle. The band corresponding to myosin heavy chain was used as loading control. Each lane represents one unique specimen. Control 3 is a normal heart, whereas control 4 is a heart explant of a patient with cardiomyopathy with an expected moderate upregulation of desmin (27).
Inactivation of klhl24a in zebrafish results in cardiac failure
To assess the function of KLHL24 in heart development, we used zebrafish as a vertebrate model system. Zebrafish have two KLHL24 homologues, Klhl24a and Klhl24b, with 78% and 85% identity to the human protein, respectively (Supplementary Material, Fig. S6). We determined the spatiotemporal mRNA expression of klhl24a and klhl24b with whole-mount in situ hybridization. The klhl24a mRNA was detected at early time points and was, by 22 h post fertilization (hpf), expressed in the cardiac cone, especially in the central region harbouring ventricular myocytes (Fig. 6A). At 72 hpf, when the heart was an S-shaped loop, klhl24a transcripts were detected in the ventricle and at a lower level in the atrium (Fig. 6C).
Figure 6.
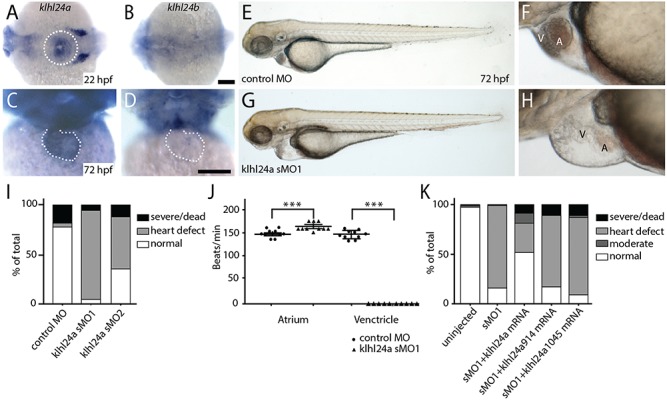
Expression and functional analysis of klhl24 in zebrafish. (A–D) Whole-mount in situ hybridization of klhl24a and klhl24b at 22 hpf (A,B; dorsal view, head left) and 72 hpf (C,D; front view, dorsal up). Expression of klhl24a mRNA in the cardiac cone at 22 hpf (A, dotted circle) and heart (C, dotted line) at 72 hpf. (E–H) Morphology of embryos injected with control antisense morpholino (E,F) or klhl24a sMO1 (G, H) at 72 hpf with close-up on the heart region (F, H). Scale bar, 100 μm. V; ventricle, A; atrium. (I) Phenotypic distribution of embryos injected with control or klhl24a morpholinos. Embryos were categorized as normal (normal appearance), heart defect (otherwise normal), moderate (non-cardiac related abnormalities) or severe/dead (severely altered morphology or dead). (J) Contractions of atrium or ventricle as beats per minute analysed with the non-parametric Mann–Whitney t-test, with standard error of the mean (SEM). ***P < 0.001. (K) Phenotypic distribution in percentage of un-injected embryos or injected with sMO1, sMO1 + 12.5 pg klhl24a mRNA, sMO1 + 12.5 pg klhl24a 914 mRNA or sMO1 + 12.5 pg klhl24a1045.
Contrary, klhl24b expression was not detected in the developing heart although observed in other tissues (Fig. 6B–D). The heart specific localization of klhl24a, therefore, led us to focus on the involvement of klhl24a in cardiac development.
To address the role of klhl24a, we used an antisense morpholino oligonucleotide (MO) technique to knockdown the protein. The general development of morpholino-injected embryos was normal; however, defects in heart function started to become detectable after 48 hpf. Heart defects initially manifested as pericardial oedema, changed heart rate and reduced blood circulation (Fig. 6J, Supplementary Material, Videos 1 and 2), later resulted in ventricular failure and blocked blood circulation in 90% of the klhl24a morphants (n = 179; Fig. 6G–I) as compared to 4% in embryos injected with control morpholino (n = 119; Fig. 6E, F and I). Injection of a second morpholino (sMO2), targeting the boundary between exon and intron 4, resulted in similar phenotypes as with sMO1 (Fig. 6I). A change in klhl24a mRNA splicing, resulting in a premature stop codon in exon 4, was confirmed with reverse transcription polymerase chain reaction (RT-PCR) using primers specific for surrounding exons (Fig. 7A). The identity of the atrium and ventricle was maintained in klhl24a sMO1-injected embryos as shown by the cardiac myosin light chain 2 gene (cmlc2) expression at 48 hpf (Fig. 7B), but the morphology of the ventricle was changed as compared to control MO-injected embryos (Fig. 7B).
Figure 7.
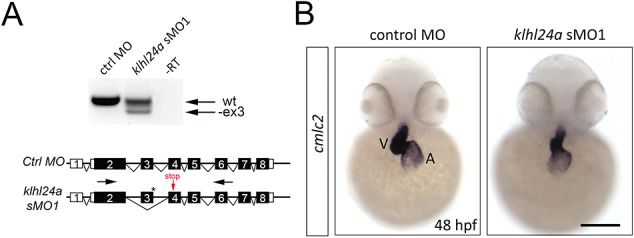
RT-PCR analysis of splicing of klhl24a mRNA using primers (black arrows) located in exon 2 and 6 surrounding the binding site of the sMO1 (asterisk). Control and klhl24a sMO1-injected embryos were analysed at 48 hpf. No reverse transcriptase (RT) served as negative control (−RT). One PCR fragment of sMO1-injected embryos is shorter than that of control-injected embryos. Sanger sequencing of the smaller PCR product showed that exon 3 is skipped in sMO1-injected embryos, resulting in a premature stop in exon 4 (red arrow). (B) Expression of cmlc2 in heart of control and klhl24a sMO1 morpholino-injected embryos at 48 hpf. Ventricle (V), Atrium (A). Scale bar, 100 μm.
Immunoblotting using an antibody against desmin (D8281 Sigma-Aldrich, Saint Louis, Missouri, USA 1:100) could not detect any obvious change in the protein expression of desmin in klh24a sMO1 embryos at 52 hpf relative to control MO-injected embryos. The short time span of the fish experiments and differences in cell metabolism between fish and human may explain this difference.
To confirm the specificity of the morpholino, we addressed if full-length klhl24a mRNA could rescue the heart defect. Co-injections of mRNA and sMO1 resulted in an increased number of embryos with normal heart formation (51.5%, n = 178) as compared to embryos injected with klhl24a sMO1 alone (16%, n = 108). We then addressed the effect of the human KLHL24 mutations on protein function in zebrafish and made site-specific mutagenesis at the corresponding conserved amino acids in the zebrafish klhl24a gene. The lack of one amino acid in the N-terminal of zebrafish Klhl24a compared to human KLHL24 (Supplementary Material, Fig. S6) makes the homologous mutation shifted three nucleotides and was thus made at 914G>A (R305H) and at 1045G>T (E349*). Co-injection of sMO1 and klhl24a 914 mRNA (n = 123) or klhl24a 1045 mRNA (n = 107) gave rise to heart defects in 71.5% and 77.5% of all embryos, respectively, and were thus not able to rescue the knockdown of the endogenous klhl24a (Fig. 6K).
Together, these results show that klhl24a has a role during heart development, especially in the formation of a functional ventricle, and support that both mutations found in human KLHL24 with HCM are loss-of-function mutations.
Discussion
We describe a new cardiomyopathy that mimics HCM and is associated with biallelic mutations in KLHL24. By morphological studies on heart and skeletal muscle, molecular genetic investigations and experimental studies on zebrafish, we provide evidence that the disease is caused by inactivation of KLHL protein 24.
In the GTEx Portal Database, KLHL24, which encodes a member of the KLHL protein family, shows the highest expression in skeletal muscle, followed by lung and the heart, supporting a role for this protein in muscle (3). The importance of KLHL24 is further supported by a high degree of conservation throughout the protein in vertebrates. The KLHL24 gene has a Residual Variation Intolerance Score of −0.78 and a percentile of 13.22, meaning it is among 13.22% of human genes most intolerant to functional genetic variants. Furthermore, missense and loss of function variants in this gene are extremely rare. By exome sequencing, supported by genome-wide linkage analysis, we identified homozygous and most likely pathogenic mutations in KLHL24 in affected individuals of both families.
To gain additional support for a causal association between the KLHL24 mutations and cardiac function, we investigated the tissue-specific expression and effect of downregulation of klhl24a in the zebrafish. The strong klhl24a expression in early ventricular myocytes and later in the established heart ventricle suggests a pivotal role for Klhl24a during cardiogenesis. Further, the lack of function of zebrafish klhl24a mRNA carrying the human mutations (917 or 1048) strongly suggests that both mutations result in a loss-of-function protein. In humans, the pathogenesis of HCM includes various mechanisms including structural abnormalities and deficiencies in the contractile machinery. Zebrafish has gained increasing attention as a vertebrate model system in investigating the molecular basis of heart development and disease. However, heart disease in the zebrafish frequently appears different from that in humans in spite of the same genetic or cellular deficit. Previously reported studies on downregulations or mutations in orthologues to TNNC1 (6), MYBPC (7) MYH7 (8) and TNNT2 (9), all known to cause HCM in humans, demonstrated decreased ventricle size and heart failure in zebrafish similar to that reported by us in this study. We therefore conclude that our result strengthens a role for KLHL24 in HCM since knockdown of Klhl24a displays phenotypes similar to those observed with loss of function studies of other HCM genes in zebrafish.
The KLHL proteins are involved in a variety of cellular processes such as cytoskeletal organization, regulation of cell morphology, cell migration, protein degradation and gene expression (10–13). Many KLHL proteins have been identified as adaptors for the recruitment of substrates to Cul3-based E3 ubiquitin ligases (14–16). Furthermore, there are many examples of KLHL proteins associated with disorders of the sarcomere (17). The involvement of KLHL proteins in muscle structural protein turnover thus appears to be an important result and a research field to be further explored.
Since KLHL24 is highly expressed in striated muscle cells, it may be important for the processing of intermediate filaments specific for muscle, such as desmin. The accumulation of desmin observed in both heart and skeletal muscle may be an effect of insufficient degradation due to a lack of functional KLHL24. It is well established that desmin is important for both structure and function of muscle cells, and dominant mutations in desmin cause a severe form of cardiomyopathy with desmin accumulation (18–21). Therefore, the identified upregulation of desmin in our patients may be part of the pathogenesis. The unique pathological alterations with accumulation of desmin, glycogen and tubular structures in skeletal muscle that were present in individuals from both families further support shared disease aetiology. While no skin abnormalities were noted in our families, patients with epidermolysis bullosa caused by a dominant initiation codon mutation in KLHL24 were also described to develop a dilated cardiomyopathy, a finding that further supports the concept that mutations in KLHL24 are associated with cardiomyopathy (4,22,23).
We identified accumulation of polyglucosan in the heart. Cardiomyopathies with polyglucosan accumulation are restricted to a few diseases, which are generally disorders of glycogen metabolism (24). However, no pathogenic variants were identified in genes known to be associated with polyglucosan storage diseases. The pathogenesis of the polyglucosan storage in our patients remains unknown but may serve as a diagnostic marker.
In conclusion, we describe a new cardiomyopathy that mimics HCM and is associated with mutations in KLHL24. It is histologically characterized by polyglucosan accumulation in some cardiomyocytes and with accumulation of glycogen, desmin and tubular structures in the cardiomyocytes and in skeletal muscle fibres. The skeletal muscle biopsies showed unique pathological alterations not previously described. Since the jagged structure of the periphery of the muscle fibres gave them a cogwheel appearance, we suggest that this pathological change is referred to as ‘cogwheel’ fibres that may be used as a diagnostic marker. Several individuals suffered fatal sudden cardiac arrest. Experience from additional cases may clarify if arrhythmias are a common complication in KLHL24-associated cardiomyopathy and increase the need for early diagnosis.
Materials and Methods
Morphological investigations of myocardium and skeletal muscle
Endomyocardial biopsy was performed in individual III:1 and III:2 in Family A. The specimens were fixed in paraformaldehyde for paraffin embedding or glutaraldehyde for electron microscopy. In individual III:2, an additional myocardial specimen was fresh frozen. After cardiac transplantation in individual III:2, the cardiac explant was fixed in paraformaldehyde, and specimens were embedded in paraffin for histological examination. Routine staining methods were applied including hematoxylin–eosin, van Gieson and Periodic acid and Schiff (PAS) staining for glycogen before and after digestion with alpha-amylase. Specimens fixed in glutaraldehyde were postfixed in osmium tetroxide and embedded in resin. Ultrathin sections were contrasted with uranyl acetate and lead citrate and examined by electron microscopy.
Skeletal muscle biopsy was performed in individual III:1 and III:2 in Family A and V:5 in family B. Specimens were snap frozen, and sections for histochemical investigations were prepared in a cryostat. A battery of histochemical staining was applied and included hematoxylin–eosin (general morphology), Gömöri trichrome (general morphology), myofibrillar adenosine triphosphatase (ATPase) (different muscle fibre types), NADH-TR (sarcoplasmic reticulum and mitochondria), SDH (mitochondria), cytochrome c oxidase (mitochondria), PAS (glycogen) and oilred O (fat) (25). Immunohistochemical staining included sarcolemmal proteins such as dystrophin, different sarcoglycans, alpha-dystroglycan and spectrin. Immunohistochemical staining of desmin as a muscle specific intermediate filament was performed, and lysosomal-associated membrane protein-2 was included as a marker for lysosomes (25).
Molecular genetic analysis
We performed exome sequencing on genomic DNA from individuals III:1 and III:2 in family A and individuals V:2 and V:4 in family B. Filtering was performed for high quality variants that were classified as deleterious (missense, nonsense, indel and splice-site variants +/− 5 bp around exon boundaries) and rare (<0.5% minor allele frequency in the ExAC Browser, gnomAD or 1000 Genomes). Assuming a recessive mode of inheritance for the clinical phenotype in the studied families and considering the consanguineous marriage, we compared the exomes of the two affected siblings in each family in a search for homozygous variants in genes associated with inherited cardiac conditions (gene panel of 88 genes) or polyglucosan storage disease (9 genes) (Supplementary methods). After excluding these candidate genes, rare variants shared between the two affected sibs in each family were selected and with the assumption of recessive inheritance. Variants of interest were further evaluated by the following prediction tools: PhyloP, SIFT, PolyPhen-2 and MutationTaster (Supplementary Material, Table S1–S2).
In family B, homozygosity mapping was performed under the assumption that the causative variant would be homozygous and identical by descent in the affected children. Genomic DNA samples from the five affected individuals (V:2, V:4, V:5, V:7, V:9) were subjected to genotyping using the Infinium Global Screening Array-24 v1.0 BeadChip (Illumina, San Diego, CA). This array contains 642 824 markers selected from over 26 global populations and has a mean marker density of one marker per ~4.5 kb. Arrays were performed in accordance with the manufacturer’s protocols. Genotyping data were analysed using Homozygosity Mapper to identify common homozygous intervals among the affected individuals (PMID: 19465395). Runs of homozygosity with a maximum threshold of 0.99 were included in the analysis. These regions were further cross-referenced to support results.
Protein expression by immunoblotting
Western blot analyses were performed on protein extract from fresh frozen skeletal muscle and cardiac muscle biopsy specimens from patient III:1 and III:2 from family A. The protein extractions were performed by denaturing the samples using Laemmli sample buffer with 5% β-mercaptoethanol, incubating 4 min at 95°C and a final centrifugation for 10 min. The supernatants including protein were loaded and separated on 4–12% Bis-Tris gel (Novex; Life Technologies, Grand Island, NY) followed by electroblotting. The membranes were incubated with primary antihuman-desmin antibody (Dako, M0760; clone D33); 1:250. Western Breeze Chromogenic kit (Life Technologies) was used for antibody detection.
Cloning and mutagenesis of zebrafish klhl24a
The spatiotemporal expression of klhl24a and klhl24b was analysed with whole-mount in situ hybridization using transcribed antisense mRNA probes on embryos treated with 0.003% 1-phenyl 2-thiourea. Full-length klhl24a and klhl24b were amplified from total RNA at 2 days post fertilization (dpf) using gene-specific primers, cloned into the pCS2+ vector and sequenced to confirm maintained reading frame. Site-specific mutagenesis of zebrafish klhl24a was performed, following the instructions in the QuikChange II site-directed mutagenesis protocol (Agilent Technologies, Santa Clara, CA) (Supplementary Material, Table S5).
Functional analysis of KLHL24 in zebrafish
Zebrafish of AB background were maintained in a 14 h:10 h light:dark cycle at 28.5°C, at the facility of the Institute of Neuroscience and Physiology, University of Gothenburg.
To determine the function of klhl24a in heart development, we used an antisense MO technique to knockdown the protein (klhl24a sMO1, 3 ng and klhl24a sMO2, 6 ng) and a standard control MO at an equal amount. Morpholino-modified splice-targeting antisense oligonucleotides (MOs) were injected at the one- to two-cell stage, and the specificity of the MOs analysed with RT-PCR on total RNA extracted from 2 dpf with TRI Reagent (Sigma-Aldrich, Saint Louis, Missouri, USA 93289) according to the manufacturer’s protocol (26). Injection of a sMO2, targeting the splice site of exon 4 was also performed, as assessed by RT-PCR using primers specific for surrounding exons (Supplementary Material, Table S5).
Rescue experiments used in vitro transcribed full-length mRNA from klhl24a-pCS2+ plasmid with mMessenge mMachine SP6 kit (Ambion, Carlsbad, CA, USA, AM1340). Co-injections were performed at one- to two cell stage with 2 ng sMO1 and 12.5 pg klhl24a mRNA or containing the 914 or 1045 mutations.
Study approval and consent to participate. The regional ethical review board in Gothenburg, Sweden approved the present study. The study complies with the Declaration of Helsinki and informed consent has been obtained from the patients.
Supplementary Material
Acknowledgements
We are grateful to the patients and their families who contributed to this study. We’d also like to thank Professor Thomas Haaf for his support on the mapping work that was carried out.
Conflict of Interest statement. None declared.
Funding
Research Fund for Neuromuscular Disorders in West Sweden; Knut and Alice Wallenberg Foundation; Swedish Research Council (2012-2014 to A.O., 2013-2546 to H.Z.; Swedish Heart-Lung Foundation (20180236 to A.O.).
References
- 1. Sen-Chowdhry S., Jacoby D., Moon J.C. and McKenna W.J. (2016) Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat. Rev. Cardiol., 13, 651–675. [DOI] [PubMed] [Google Scholar]
- 2. Alfares A.A., Kelly M.A., McDermott G., Funke B.H., Lebo M.S., Baxter S.B., Shen J., McLaughlin H.M., Clark E.H., Babb L.J. et al. (2015) Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med., 17, 880–888. [DOI] [PubMed] [Google Scholar]
- 3. Consortium G.T. (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He Y., Maier K., Leppert J., Hausser I., Schwieger-Briel A., Weibel L., Theiler M., Kiritsi D., Busch H., Boerries M. et al. (2016) Monoallelic mutations in the translation initiation codon of KLHL24 cause skin fragility. Am. J. Hum. Genet., 99, 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin Z., Li S., Feng C., Yang S., Wang H., Ma D., Zhang J., Gou M., Bu D., Zhang T. et al. (2016) Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat. Genet., 48, 1508–1516. [DOI] [PubMed] [Google Scholar]
- 6. Sogah V.M., Serluca F.C., Fishman M.C., Yelon D.L., Macrae C.A. and Mably J.D. (2010) Distinct troponin C isoform requirements in cardiac and skeletal muscle. Dev. Dyn., 239, 3115–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y.H., Pai C.W., Huang S.W., Chang S.N., Lin L.Y., Chiang F.T., Lin J.L., Hwang J.J. and Tsai C.T. (2013) Inactivation of Myosin binding protein C homolog in zebrafish as a model for human cardiac hypertrophy and diastolic dysfunction. J. Am. Heart. Assoc., 2, e000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bainbridge M.N., Davis E.E., Choi W.Y., Dickson A., Martinez H.R., Wang M., Dinh H., Muzny D.M., Pignatelli R., Katsanis N. et al. (2015) Loss of function mutations in NNT are associated with left ventricular noncompaction. Circ Cardiovasc. Genet., 8, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sehnert A.J., Huq A., Weinstein B.M., Walker C., Fishman M. and Stainier D.Y.R. (2002) Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Gen., 31, 106–110. [DOI] [PubMed] [Google Scholar]
- 10. Dhanoa B.S., Cogliati T., Satish A.G., Bruford E.A. and Friedman J.S. (2013) Update on the Kelch-like (KLHL) gene family. Hum. Genomics, 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prag S. and Adams J.C. (2003) Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics, 4, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stogios P.J., Downs G.S., Jauhal J.J., Nandra S.K. and Prive G.G. (2005) Sequence and structural analysis of BTB domain proteins. Genome Biol., 6, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams J., Kelso R. and Cooley L. (2000) The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol., 10, 17–24. [DOI] [PubMed] [Google Scholar]
- 14. Genschik P., Sumara I. and Lechner E. (2013) The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J., 32, 2307–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sambuughin N., Swietnicki W., Techtmann S., Matrosova V., Wallace T., Goldfarb L. and Maynard E. (2012) KBTBD13 interacts with Cullin 3 to form a functional ubiquitin ligase. Biochem. Biophys. Res. Commun., 421, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu L., Wei Y., Reboul J., Vaglio P., Shin T.H., Vidal M., Elledge S.J. and Harper J.W. (2003) BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature, 425, 316–321. [DOI] [PubMed] [Google Scholar]
- 17. Papizan J.B., Garry G.A., Brezprozvannaya S., McAnally J.R., Bassel-Duby R., Liu N. and Olson E.N. (2017) Deficiency in Kelch protein Klhl31 causes congenital myopathy in mice. J. Clin. Invest., 127, 3730–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hnia K., Ramspacher C., Vermot J. and Laporte J. (2015) Desmin in muscle and associated diseases: beyond the structural function. Cell Tissue Res., 360, 591–608. [DOI] [PubMed] [Google Scholar]
- 19. Dalakas M.C., Park K.Y., Semino-Mora C., Lee H.S., Sivakumar K. and Goldfarb L.G. (2000) Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med., 342, 770–780. [DOI] [PubMed] [Google Scholar]
- 20. Lorenzon A., Beffagna G., Bauce B., De Bortoli M., Li Mura I.E., Calore M., Dazzo E., Basso C., Nava A., Thiene G. et al. (2013) Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol., 111, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hedberg C., Melberg A., Kuhl A., Jenne D. and Oldfors A. (2012) Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur. J. Hum. Genet., 20, 984–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yenamandra V.K., van der Akker P.C., Lemmink H.H., Jan S.Z., Diercks G.F.H., Vermeer M., van der Berg M.P., van der Meer P., Pasmooij A.M.G., Sinke R.J. et al. (2018) Cardiomyopathy in epidermolysis bullosa simplex patients with mutations in the KLHL24 gene. Br. J. Dermatol., 179, 1181–1183. [DOI] [PubMed] [Google Scholar]
- 23. Schwieger-Briel A., Fuentes I., Castiglia D., Barbato A., Greutmann M., Leppert J., Duchatelet S., Hovnanian A., Burattini S., Yubero M.J. et al. (2019) Epidermolysis bullosa simplex with KLHL24 mutations is associated with dilated cardiomyopathy. J. Invest. Dermatol., 139, 244–249. [DOI] [PubMed] [Google Scholar]
- 24. Hedberg-Oldfors C. and Oldfors A. (2015) Polyglucosan storage myopathies. Mol. Aspects Med., 46, 85–100. [DOI] [PubMed] [Google Scholar]
- 25. Dubowitz V., Sewry C.A. and Oldfors A. (2013) Muscle Biopsy: A Practical Approach, 4th edn. Elsevier, Philadelphia. [Google Scholar]
- 26. Abramsson A., Kettunen P., Banote R.K., Lott E., Li M., Arner A. and Zetterberg H. (2013) The zebrafish amyloid precursor protein-b is required for motor neuron guidance and synapse formation. Dev. Biol., 381, 377–388. [DOI] [PubMed] [Google Scholar]
- 27. Heling A., Zimmermann R., Kostin S., Maeno Y., Hein S., Devaux B., Bauer E., Klovekorn W.P., Schlepper M., Schaper W. et al. (2000) Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res., 86, 846–853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.