

Abstract
Objective
To identify the genetic cause of autosomal dominant spinocerebellar ataxia and retinitis pigmentosa in a large extended pedigree.
Methods
Clinical studies were done at 4 referral centers. Ten individuals in the same extended family participated in at least a portion of the study. Records were obtained from an 11th, deceased, individual. Neurologic and dermatological examinations were performed. Ophthalmologic evaluation including funduscopic examination and in some cases ocular coherence tomography were used to identify the presence of retinal disease. Whole exome sequencing (WES), in conjunction with Sanger sequencing and segregation analysis, was used to identify potential genetic mutation.
Results
Affected individuals reported slowly progressive cerebellar ataxia with age at onset between 38 and 57. Imaging demonstrated cerebellar atrophy (3/3). WES identified a novel heterozygous mutation in the elongation of very long chain fatty acids 4 (ELOVL4) gene (c.512T>C, p.Ile171Thr) that segregated with ataxia in 7 members tested. Four of 8 members who underwent ophthalmologic evaluation were found to have retinitis pigmentosa. No skin findings were identified or reported. Ocular movement abnormalities and pyramidal tract signs were also present with incomplete penetrance.
Conclusions
We report a family with both spinocerebellar ataxia and retinal dystrophy associated with an ELOVL4 mutation. In addition, to supporting prior reports that ELOVL4 mutations can cause spinocerebellar ataxia, our findings further broaden the spectrum of clinical presentations associated with spinocerebellar ataxia 34.
The spinocerebellar ataxias (SCAs) are a group of autosomal dominant disorders characterized by progressive cerebellar ataxia. Some subtypes of SCA are associated with other neurologic dysfunction including ocular movement abnormalities, peripheral neuropathy, pyramidal or extrapyramidal signs, and in some cases non-neurological signs. To date, over 40 causative genes have been described.1
Mutations in the elongation of very long chain fatty acids 4 (ELOVL4) gene have been associated with spinocerebellar ataxia SCA34 (OMIM 133190) in 3 families and 2 other individuals. One French Canadian family and 2 individuals were reported to have spinocerebellar ataxia with erythrokeratodermia variabilis (EKV) while 2 Japanese families have a pure SCA with no skin findings.2–5 Although ELOVL4 mutations are also known to cause autosomal dominant Stargardt-like macular dystrophy, none of the previous reports identified ophthalmologic abnormalities.
We report an American family with a late onset SCA in which many affected individuals also have retinitis pigmentosa on ophthalmologic evaluation. Whole exome sequencing (WES) with confirmative Sanger sequencing identified a novel mutation in the ELOVL4 gene (c512.T>C, pIle171Thr) that segregated with the disease in this family.
Methods
Ten individuals with ataxia from the same extended family located in 8 different cities participated in the elements of the study based on logistical availability. The pedigree can be found in figure 1. DNA was collected from 7 affected and 2 unaffected individuals. Clinical studies were done at 4 referral centers. Participants were referred for neurologic, dermatologic, and ophthalmic evaluations. Participants who were unable to reach a referral center were interviewed remotely, and clinical records were obtained when possible. Six individuals participated in neurologic and dermatologic examination. On neurologic evaluation, both Scale for the Assessment and Rating of Ataxia and Inventory of Non-Ataxia Signs scales were recorded. Seven individuals participated in ophthalmic examination. Clinical records of one deceased individual (IV-1) were also obtained.
Figure 1. Pedigree of an American Family with a Novel Mutation in ELOVL4.

Pedigree of a large extended family including all members of our study. Individuals who participated in genetic testing for p.I171T variant denoted in + for presence and − for absence.
Genetic analysis was done independently at 3 Clinical Laboratory Improvement Amendments-certified centers. Two individuals (V-1 and V-I12) were evaluated for ataxia via targeted analysis of WES. Details of sequencing and bioinformatics used are previously published. See supplemental materials NGS Pipeline and Bioinformatics (links.lww.com/NXG/A182) for details. In conjunction, Sanger sequencing and segregation analysis were used to confirm and support the genetic mutation. Another individual (IV-1), now deceased, underwent allele-specific prescreen followed by targeted analysis of WES at a third center for familial retinitis pigmentosa.
Standard protocol approvals, registrations, and patient consents
Elements of this study involving human subjects were approved by the institutional review board of participating institutions (University of Iowa ID# 200202022 and University of Chicago 14707A-CR004). Other studies were standard of care. All participants provided informed written consents prior to initiating research and data collection.
Data availability
The authors confirm that the deidentified data supporting the findings of this study are available within the article and supplementary materials (links.lww.com/NXG/A181).
Results
A summary of clinical findings can be found in the table. The age at ataxia onset was in early 40s for most individuals, though this ranged from 38 to 57. All 10 affected individuals reported slowly progressive gait instability, 6 of 10 complained of dysarthria, and 8 of 10 noted handwriting changes. Three individuals have progressed to using a wheelchair (average 18.3 years after onset). One of 10 complained of diplopia, 7 of 10 wear corrective lenses. Two of 10 complained of nighttime vision loss corresponding to onset of ataxia. All persons denied bowel/bladder issues as well as history of dermatologic disease. Disease progression was extremely slow. Of the 10 persons interviewed, one required a cane and 2 required wheelchairs at the time of interview.
Table.
Comparison of clinical characteristics among patients with spinocerebellar ataxia 34

Genetic testing
Data from WES on 2 individuals with ataxia (V1 and VI12) were analyzed independently by different labs and resulted in identification of a variant in ELOVL4 (c.512T>C, pIle171Thr). The variant was deemed likely pathogenic based on American College of Medical Genetics Standards and Guidelines. DNA from 5 more affected and 2 unaffected members of this family was analyzed for the presence of this mutation by Sanger sequencing. All affected family members carried the mutation, while the 2 unaffected members lacked the mutation. A third lab independently identified the same mutation in ELOVL4 in a now deceased individual (IV1) during evaluation for retinal pigmentary changes; this individual was reported to have ataxia. A representation of the novel variant can be found in figure 2; its relation to previously described variants is detailed in figure 3.
Figure 2. Novel Mutation in ELOVL4.

(A) Whole-exome sequencing identified a novel heterozygous mutation in the elongation of very long chain fatty acids 4 (ELOVL4) gene (c.512T>C, p.Ile171Thr) that segregated with symptoms in 9 individuals tested. (B) The nucleic acid change is highly conserved across species and is predicted to be likely pathogenic.
Figure 3. Location of Novel ELOVL4 Mutation on Topographic Prediction of Protein.

Illustration of topographic prediction of the ELOVL4 protein based on the Uniprot database and location of our mutation relative to previously reported disease-causing mutations.
Neurologic evaluation
On neurologic examination, affected individuals had gait ataxia (6/6), limb ataxia (5 of 6), dysarthria (3 of 6) along with ocular movement abnormalities (5 of 6), and pyramidal tract signs (6 of 6).
MRI of the brain was done in 3 affected individuals, all showing cerebellar and pontine atrophy that correlated with their disease duration. Representative images can be found in figure 4.
Figure 4. Brain MRIs of an American Family with Novel ELOVL4 Mutation.

MRI demonstrates cerebellar and pontine atrophy in 2 of 3 participants who were imaged. The third younger individual had mild changes.
Dermatological workup
None of the 10 affected individuals had a history of skin findings that could be attributed to a form of EKV. A complete skin examination done by a dermatologist found no evidence of EKV or unusual skin lesions in 4 of 4 participants.
Ophthalmologic evaluation
Clinical images can be found in figure 5. Three of 7 affected individuals had peripheral bone-spicule-like pigmentary changes on funduscopic examination consistent with retinitis pigmentosa. Records obtained from an eighth, now deceased, individual (IV1) also showed peripheral bone-spicule-like pigmentary changes consistent with retinitis pigmentosa. Electroretinogram results on this individual showed rod-cone photoceptor degeneration (figure 6).
Figure 5. Fundus Photographs of an American Family with Novel ELOVL4 Mutation.

Color fundus photographs demonstrating ophthalmic examination findings. Four patients were found to have ophthalmic findings consistent with retinitis pigmentosa. Color fundus photographs of patient IV1 (A, B) demonstrate bone spicule-like pigmentary changes. Montage color photographs of patient V4 demonstrate mild optic disc pallor, arteriolar attenuation, and peripheral retinal pigment epithelial changes (C = right eye, D = left).
Figure 6. Electroretinogram Data for Individual IV1.
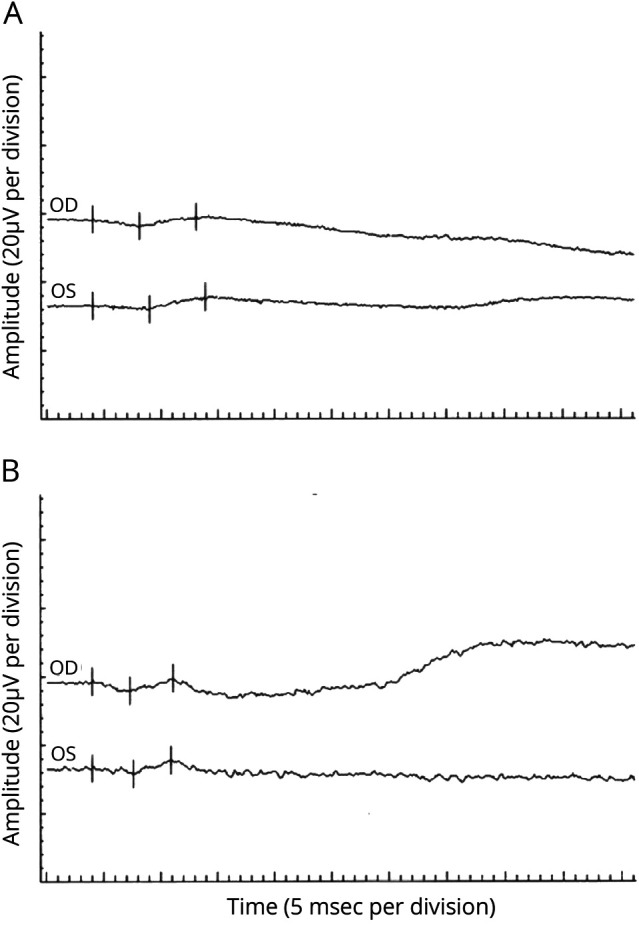
(A) Scotopic electroretinogram: After 30 minutes of dark adaptation, a bright flash stimulus resulted in b-waves of 37.8 µV OD (implicit time 45.5 msec) and 50 µV OS (implicit time 49.5 msec). Normal for age is greater than 229.2 µV with an implicit time of less than 53.4 msec. (B) Photopic lectroretinogram: Under light-adapted conditions, a bright flash stimulus resulted in b-waves of 19.8 µV OD (implicit time 35.5 msec) and 21 µV OS (implicit time 34.5 msec). Normal for age is greater than 27.2 µV with an implicit time of less than 32.4 msec. The rod-selective responses were nonrecordable. The scotopic bright flash responses were markedly reduced in amplitude (A). The photopic responses were reduced in amplitude and delayed (B).
Discussion
We identified a novel missense variant in ELOVL4 in a large American family with SCA. This variant is in a highly conserved amino acid residue, is predicted to be likely pathogenic by American College of Medical Genetics criteria and is nearby a previously reported mutation (P168F) in a large French Canadian family with SCA34. In addition, affected persons have relatively high incidence of pyramidal tract signs and oculomotor signs more consistent with those reported in the 2 Japanese families. Unlike the Japanese families, we did not see the loss of transverse pontine fibers on neuroimaging for members of our cohort. Several members of our family reported subjectively anosmia around the onset of gait ataxia. The clinical significance of this is unclear.
The relevant new finding in our family is the presence of retinitis pigmentosa. Although RP is not a typical presentation for Stargardt type 3, previous work on the more common autosomal recessive form of Stargardt disease, caused by mutations in the ABCA4 gene, has shown RP and macular dystrophy to be part of the same phenotypic spectrum due to buildup of a toxic product in the retinal pigmented epithelium layer.6 The presence of inclusions in the retina of an ELOVL4 transgenic mouse model could also point to a toxic gain of function mechanism.7
While ELOVL4 mutations have been associated with autosomal dominant Stargardt-like macular dystrophy (OMIM 600110) in the past, ophthalmologic pathology in the previously reported SCA34 families has not been identified. The mechanism by which ELOVL4 mutations lead to both Stargardt disease or SCA34 is currently unknown. ELOVL4 encodes a protein that contributes to elongation of fatty acids longer than 26 carbons and is expressed in the retina, brain, testis, skin, and thymus.8 We did not measure very long chain fatty acids (VLCFA) due to technical constraints as standard assays cannot resolve the necessary carbon length. Further study is necessary to determine whether the pathogenesis of ELOVL4 related disorders can be directly linked to VLCFA production. Based on the lack of overlapping ophthalmologic and neurologic phenotypes in affected patients with ELOVL4 mutations, the mechanism for the 2 phenotypes is thought to be preferential disruption of polyunsaturated (PUFA) vs saturated (SFA) very long chain fatty acid biosynthesis, important in retina and brain/skin respectively.9 This model predicts patients carrying the Thr171Ile mutation would have decreased PUFA and SFA synthesis while persons carrying previous mutations in SCA families would only have decreased SFA synthesis. Since our mutation is in close proximity to prior reported mutations in the 4th transmembrane domain of the ELOVL4 protein, which were not associated with retinal phenotypes, it is unclear why it would produce a different functional effect. Recent work found ELOVL4 synthesis of VLCFAs play an important role in tight junction stability in the blood-retinal barrier in mice.10 This could suggest blood-brain and blood-retinal barrier instability as a potential mechanism of disease in late-onset SCA. Speculation on potential mechanisms of cerebellar and retinal pathogenesis is limited by the lack of a robust biomarker for the synthesis of either VLC-PUFA or VLC-SFA in human subjects as well as limited understanding of the biological function of ELOVL4 and its very long chain FA products on health and disease.
ELOVL4 mutations have previously been associated with inherited retinopathy and SCA independently of each other. We report an American family with concurrent spinocerebellar ataxia and retinal dystrophy due to an ELOVL4 mutation. In addition, to supporting prior reports that ELOVL4 mutations can cause an autosomal dominant spinocerebellar ataxia, our findings further broaden the spectrum of clinical presentations associated with SCA34.
Glossary
- EKV
erythrokeratodermia variabilis
- ELOVL4
elongation of very long chain fatty acids 4
- PUFA
poly-unsaturated
- SCA
spinocerebellar ataxias
- SFA
saturated
- WES
whole exome sequencing
Appendix. Authors

Study funding
No targeted funding reported.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Autosomal dominant hereditary ataxia—NORD (National Organization for Rare Disorders). NORD (National Organization for Rare Disorders) (blog). Available at: rarediseases.org/rare-diseases/autosomal-dominant-hereditary-ataxia/. Accessed June 26, 2018. [Google Scholar]
- 2.Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and Erythrokeratodermia. JAMA Neurol 2014;71:470–475. 10.1001/jamaneurol.2013.6337. [DOI] [PubMed] [Google Scholar]
- 3.Ozaki K, Doi H, Mitsui J, et al. A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 2015;72:797–805. 10.1001/jamaneurol.2015.0610. [DOI] [PubMed] [Google Scholar]
- 4.Bourassa CV, Raskin S, Serafini S, et al. A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol 2015;72:942–943. 10.1001/jamaneurol.2015.0888. [DOI] [PubMed] [Google Scholar]
- 5.Bourque PR, Warman-Chardon J, Lelli DA, et al. Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet 2018;4:e263 doi: 10.1212/NXG.0000000000000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheffield VC, Stone EM. Genomics and the eye. N Engl J Med 2011;364:1932–1942. 10.1056/NEJMra1012354. [DOI] [PubMed] [Google Scholar]
- 7.Karan G, Lillo C, Yang Z, et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci U S A 2005;102:4164–4169. 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ELOVL4—elongation of very long chain fatty acids protein 4—Homo sapiens (human)—ELOVL4 gene & protein. Available at: uniprot.org/uniprot/Q9GZR5#sequences. Accessed March 5, 2018.
- 9.Agbaga MP. Different mutations in ELOVL4 affect very long chain fatty acid biosynthesis to cause variable neurological disorders in humans. Adv Exp Med Biol 2016;854:129–135. 10.1007/978-3-319-17121-0_18. [DOI] [PubMed] [Google Scholar]
- 10.Kady NM, Liu X, Lydic TA, et al. ELOVL4-Mediated production of very long chain ceramides stabilizes tight junctions and prevents diabetes-induced retinal vascular permeability. Diabetes 2018;67:769–781. 10.2337/db17-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the deidentified data supporting the findings of this study are available within the article and supplementary materials (links.lww.com/NXG/A181).