

Abstract
Bacteria harbor an immense reservoir of potentially new and therapeutic small molecules in the form of ‘silent’ biosynthetic gene clusters (BGCs). These BGCs can be identified bioinformatically but are sparingly-expressed under normal laboratory growth conditions, or not at all, and therefore do not produce significant levels of the corresponding small molecule product. Several methods have been developed for activating silent BGCs. A major limitation for nearly all methods is that they require genetic procedures and/or do not report on the bioactivity of the cryptic metabolite. We herein report ‘Bioactivty-HiTES’, an approach that links the bioactivity of cryptic metabolites to their induction, while at the same time obviating the need for genetic manipulations. Using this method, we detected induction of cryptic antibiotics in three actinomycete strains tested. Follow-up studies in one case allowed us to structurally elucidate two cryptic metabolites, elicited by the beta-blocker atenolol in Streptomyces hiroshimensis, with selective growth-inhibitory activity against Gram-negative bacteria, notably Escherichia coli and Acinetobacter baumannii. Atenolol turned out to be a global elicitor of secondary metabolism, and characterization of additional cryptic metabolites led to the discovery of a novel naphthoquinone epoxide. Bioactivity-HiTES is a general, widely-applicable procedure that will be useful in identifying cryptic bioactive metabolites in the future.
Introduction
Bacteria produce a diverse array of secondary metabolites, or natural products, the discovery of which has been an important driving force for various disciplines in chemistry, biology, and pharmacology.1 Natural products account for more than 50% of the drugs approved by the FDA in the United States in the past ~35 years and >70% of the current clinical antibiotics.2–4 These data provide a strong impetus for the continued discovery and development of new natural products.
By the early 2000s, the secondary metabolic potential of well-known bacterial genera appeared to have been exhausted. Rediscovery of old molecules was a major bottleneck, leading to a sharp decline in natural product research in the pharmaceutical industry.5–7 However, innovations that led to next generation sequencing and the ensuing accumulation of thousands of complete bacterial genomes, along with advances in bioinformatic identification of biosynthetic genes, revealed that the biosynthetic potential of bacteria, even well-studied ones, had been vastly underestimated.8–14 We now know that bacteria harbor many secondary metabolite biosynthetic gene clusters (BGCs) that do not give rise to appreciable levels of a small molecule product under normal laboratory conditions. These BGCs are referred to as ‘silent’ or ‘cryptic’ and the current consensus is that they outnumber constitutively active ones by a factor of 5–10. Silent BGCs, therefore, represent a massive reservoir of potentially new small molecules, the underlying chemistry and biology of which remain to be explored.
With the realization of the abundance of silent BGCs, feverish activity has followed to devise methods for harvesting cryptic metabolites.15–19 We have contributed to the available approaches with our development of HiTES (high-throughput elicitor screening).20,21 In this approach, a reporter gene is placed inside the gene cluster of interest and small molecule libraries are then screened against the reporter strain to find suitable transcriptional elicitors or inducers. With the inducer identified, the small molecule product can be characterized along with the regulation of the silent BGC, an area that remains largely in its infancy.22 The disadvantage of HiTES and those of most other available techniques is that genetic manipulations and/or molecular biology approaches are required. These slow down the pace of discovery, especially in the talented but genetically-intractable rare actinomycetes, which are prolific producers of secondary metabolites and harbor stores of silent BGCs.12 A second drawback of most available approaches, with some notable exceptions,23 is that they do not link bioactivity to a given cryptic metabolite or BGC. Therefore, activity assays must be conducted upon discovery of a cryptic metabolite. Given that natural products have a virtually unlimited potential for biological function, both in the environment and in medicine, a method that directly connects bioactivity to a cryptic metabolite prior to its tedious isolation and structural elucidation would be highly beneficial.
We herein introduce ‘Bioactivity-HiTES’, an approach that addresses both limitations mentioned above and obviates the need for genetic manipulations by identifying cryptic metabolites with the desired bioactivity: a wild-type microorganism is subjected to elicitor screening in a high-throughput fashion. The resulting ‘induced metabolomes’ are screened for a chosen bioactivity. Activity-guided fractionation in combination with the elicitor then allow for isolation and characterization of the cryptic metabolite. We demonstrate the feasibility of this approach by screening for cryptic antibiotics and report two pyrimidine natural products that are toxic to and mildly selective for Gram-negative bacteria. Follow-up studies allowed us to identify the cognate BGC along with other novel cryptic metabolites that are pleiotropically induced by the same elicitor, notably a novel pyranonaphthoquinone epoxide that we call hiroshidine.
Results
Rationale for Bioactivity-HiTES.
HiTES consists of two steps, the activation of silent BGCs by elicitor screening followed by a genetic read-out, which so far has relied on transcriptional or translational reporter assays. Most recently, we have applied an imaging mass spectrometry approach as the detection step to remove the reliance on genetic procedures.24 While cryptic bioactive metabolites can be, and have been, discovered with these approaches, neither reports directly on the bioactivity of the desired metabolite. To address this limitation, we considered a workflow, in which a bacterium would be subjected to elicitor screening and the resulting ~500–1000 induced metabolomes would be screened for a desired biological activity (Fig. 1). Wells that contained the active component(s) would be pursued further to identify the cryptic metabolite of interest. Naturally, the approach can only be applied to bacteria that do not constitutively produce the chosen bioactivity. Because of the well-documented need for new antibiotics and the continuing development of resistance to extant ones, we focused on cryptic metabolites that exhibit inhibitory activity against Gram-positive (B. subtilis) and Gram-negative (E. coli) bacteria.
Figure 1.

Summary of the Bioactivity-HiTES workflow. A wt microorganism is subjected to elicitor screening. Each well is then screened for a desired biological activity, such as antibiosis. The desired elicitor-cryptic metabolite pair is subsequently characterized.
Development of Bioactivity-HiTES.
We began by screening several actinomycete bacteria in our strain library, starting with Streptomyces lavendulae, Amycolatopsis kerathiniphila, and Streptomyces hiroshimensis, the producers, among others, of streptothricin, keratinimicin, and prodigiosin, respectively. We verified that neither produced an antibiotic under our experimental conditions in R4 growth medium. We then subjected each strain to elicitor screening from a single 96-well plate of a larger, commercial natural products library. After growth of each strain under 80–96 different conditions, the supernatants were used directly in disc diffusion assays against B. subtilis. The resulting agar plates were assessed visually, and the frequency and size of growth-inhibition zones were tabulated. Somewhat surprisingly, even with this limited screen, we observed a number of conditions that gave rise to a zone of inhibition (Fig. 2, Figs. S1–S3). Some of these were likely the result of residual antibiotics from the natural products library. But in most case, the elicitor was not known to exhibit antibiotic properties. For example, in S. lavendulae, 7,2′-dimethoxyflavone, isobutylmethylxanthine, and methoxyvone appeared to induce synthesis of anti-B. subtilis metabolite(s) (Fig. S1). For A. keratiniphila, baccatin III and isoscopoletin gave rise to zones of inhibition that were otherwise not observed (Fig. S2). Lastly, with S. hiroshimensis, several clinical drugs, including the antimalarial primaquine and the anesthetic procaine appeared to elicit anti-B. subtilis metabolite(s) (Fig. S3). The stimulatory activities of these metabolites had previously not been reported.
Figure 2.

Identification of antibiotic-eliciting small molecules from three actinomycetes. Summary of results from agar plate assays in which S. lavendulae, A. keratiniphila, and S. hiroshimensis were challenged with 80–96 elicitors and the resulting metabolomes were screened for antibiotic activity against B. subtilis. The plates are shown in Figs. S1–S3. The size of the zones of inhibition observed correspond to: none (n), weak (w, 5–6 mm), medium (m, 6–7 mm), and strong (s, >7 mm).
We also carried out assays against E. coli. In this case, we conducted broader elicitation screens using a ~500-member natural product library as the source of potential inducers. To increase the throughput of the assays, we transitioned to liquid bioassays. Rather than manually spotting each disc, we robotically transferred the supernatant from the elicitor plate to 96-well plates inoculated with the E. coli test strain. After a defined period, growth inhibition was assessed by monitoring the optical density at 600 nm (OD600 nm) using a plate reader. To account for growth variation from one plate to another, the percent growth for each well was plotted, that is, the OD600 nm in the presence of an elicitor relative to the average OD600 nm for all the wells in a given plate. The results for the entire library are shown for S. lavendulae (Fig. 3A), A. keratiniphila (Fig. 3B), and S. hiroshimensis (Fig. 3C). Most wells gave a value close to 100 ± 30%, suggesting minimal effect on growth. Some induced metabolomes enhanced E. coli growth, while others inhibited it. With S. lavendulae, metabolomes induced by the heart block agent isoproterenol and the bile acid chenodiol seemed to support E. coli growth best, while those induced by inositol and allantoin inhibited it (blue and red spheres, Fig. 3A). Similarly, aspartame- and tandutinib-induced metabolomes of A. keratiniphila resulted in enhanced E. coli growth while those induced by the anti-HIV medication AZT and the anti-psychotic sulpiride seemed to produce growth-inhibitory compounds (blue and red spheres, Fig. 3B). Lastly, with S. hiroshimensis, the metabolomes that most enhanced E. coli growth were elicited by aceclidine (Fig. 3D, 1), an acetylcholine agonist used to treat glaucoma, glucosamine (2), and dexchlorpheniramine (3), a clinical antihistamine. The metabolomes that resulted in the lowest percent-growth, and inhibited E. coli growth best, were induced by atenolol (Fig. 3D, 4), a beta-blocker clinically used to treat hypertension, the Alzheimer drug memantine (5), and the ergot alkaloid methylergonovine (6). These results provide ample opportunities for follow-up studies to investigate the effects of these unsuspected modulators of actinomycete physiology.
Figure 3.
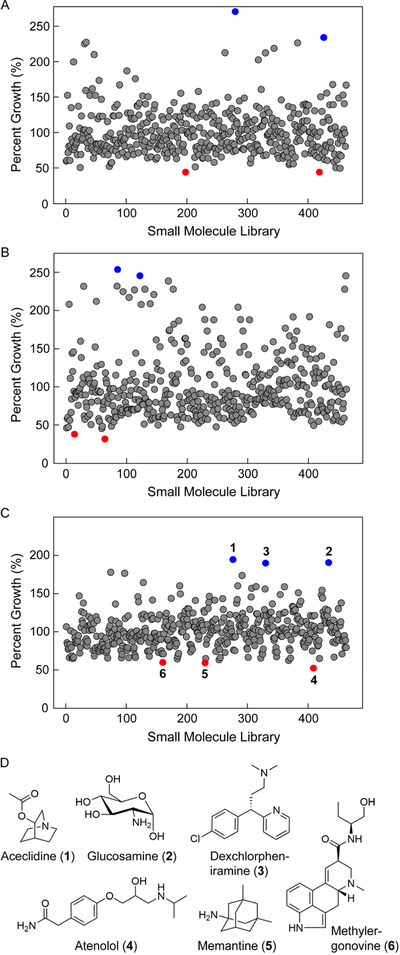
Cryptic metabolites with inhibitory bioactivity against E. coli. (A–C) Bioactivity-HiTES of S. lavendulae (A), A. keratiniphila (B), and S. hiroshimensis (C) against E. coli. Each bacterium was challenged with a ~500-member natural product library. After a suitable growth period, the supernatants from each of the ~500 wells were transferred to 96-well plates inoculated with E. coli, the plates were cultured for a defined period, and OD600 nm was subsequently recorded. For each well, percent growth, that is, OD600 nm for a given well relative to the average OD600 nm for the entire plate, was calculated and plotted. This normalization eliminated differences in growth kinetics that can arise from plate-to-plate variation. With S. lavendulae and A. keratiniphila, the two wells that stimulated (blue dots) or inhibited (red dots) E. coli growth most are highlighted. With S. hiroshimensis the three wells that resulted in the optimal stimulation (1, 2, 3, blue circles) or inhibition (4, 5, 6, red circles) of E. coli growth are marked. (D) Structures of elicitors 1–6 from Bioactivity-HiTES with S. hiroshimensis.
Discovery of New, Cryptic Antibiotics.
The observation of numerous conditions that seem to elicit biomolecules with growth-inhibitory activities suggest that cryptic antibiotics can be identified using Bioactivity-HiTES. The results provided several options for follow-up studies. Because of the dearth of treatments against Gram-negative bacteria, we next sought to verify production of cryptic metabolites that inhibit E. coli growth. To do so, we focused on S. hiroshimensis, because relatively few studies have investigated its natural product output. The atenolol-induced metabolome was most effective at inhibiting E. coli and further experiments were carried out with this elicitor. We validated the effects of atenolol using disc diffusion assays and HPLC-MS analysis. S. hiroshimensis culture supernatants induced by atenolol resulted in a clear zone of inhibition on E. coli agar plates and an orange hue, while uninduced cultures exhibited neither (Fig. 4A). Moreover, differential HPLC-MS profiling revealed numerous new peaks induced by the beta-blocker, indicative of global induction of secondary metabolism (Fig. 4B). Much like other elicitors that we have explored in the past, atenolol too appears to have a pleiotropic effect on secondary metabolism in S. hiroshimensis.
Figure 4.

Identification of cryptic metabolites induced by atenolol. (A) Agar disc diffusion assays of uninduced (‘−’) and atenolol-induced (‘+’) S. hiroshimensis cultures against E. coli, verifying the induction of compounds with growth-inhibitory activity against E. coli. (B) Comparative HPLC-MS profile of S. hiroshimensis cultures grown in the absence (black trace) or presence (red trace) of 33 μM atenolol. Identified peaks are numbered with the structures shown in panel C. Note that compounds 7/8 and 10/11 had similar retention times. Several newly-induced metabolites remain unidentified (question mark). (C) Taylorflavins, pyridindolol, two isocoumarins and hiroshidine were identified from atenolol-induced S. hiroshimensis cultures. Relevant NMR data for taylorflavin A and hiroshidine are shown. Qinimycin is the closest structural homolog of hiroshidine.
We set out to first determine the cryptic anti-E. coli antibiotic that was induced by atenolol and elucidate the structures of the additional cryptic metabolites. Bioactivity assays of the induced peaks showed that the antibiotic activity was associated with compounds 7 and 8 (Fig. 4B). Large-scale production cultures in the presence of atenolol followed by activity-guided fractionation allowed us to purify two related metabolites. HR-MS suggested that both compounds were nitrogen-rich with predicted molecular formulas of C7H7N5O3 and C8H9N5O3 (Table S1). UV-vis (λmax = ~240 nm and 420 nm) and NMR analysis were in line with a 7-azapteridine scaffolds with three ketone carbon shifts at 149.8, 153.2, and 158.4 ppm in the first analog as well as two N-methyl groups with 1H shifts at 3.22 and 3.67 ppm (Fig. S4). Detailed analysis of 1D/2D NMR data ultimately revealed that the two compounds consisted of a new toxoflavin-type analog (7) and 2-methylfervenulone (8), a metabolite that was previously isolated from an unidentified actinomycete (Table S2, Fig. S4–S5).25,26 Interestingly, compound 8 and a series of other pyrimidine natural products were synthesized by Taylor and colleagues, a research program that culminated in the drug Alimta.27,28 We have therefore named these pyrimidine compounds taylorflavin A (Fig. 4C, 7) and B (8), in honor of Ted Taylor.
In-house bioactivity assays showed taylorflavin B to be a more potent inhibitor of E. coli growth. We submitted this compound for broad bioactivity assays against a battery of pathogenic bacteria; a more limited set was tested with taylorflavin A. Consistent with our screen, the results yielded minimal inhibitory concentrations (MICs) of 4 μg/mL against Neisseria ghonorrheae and 16 μg/mL against E. coli, Acinetobacter baumannii, and Vibrio cholerae for taylorflavin B (Table 1). Conversely, the antibiotic did not inhibit the growth of a collection of Gram-positive bacteria. Likewise, taylorflavin A displayed MICs of 25 μg/mL and 37.5 μg/mL against P. aeruginosa and E. coli, respectively. Significantly weaker activity was observed against B. subtilis and S. aureus (Table 1) In contrast to most antibiotics, the taylorflavins exhibited Gram-negative-selective antibacterial activity. These results validate the use of Bioactivity-HiTES for the discovery of cryptic antibiotics.
Table 1.
MIC values (in μg/mL) of taylorflavins against select bacteria.a
| Bacterium | Taylorflavin A | Taylorflavin B |
|---|---|---|
| Gram-Negative | ||
| E. coli | 37.5 | 16 |
| A. baumannii | n.d. | 16 |
| V. cholerae | n.d. | 16 |
| N. gonorrhoeae | n.d. | 4 |
| P. aeruginosa | 25 | >64 |
| K. pneumoniae | n.d. | >64 |
| Gram-Positive | ||
| S. aureus | >64 | 64 |
| S. aureus MRSA | n.d. | 64 |
| S. pneumoniae PSPP | n.d. | 32 |
| S. pyogenes | n.d. | 32 |
| S. agalactiae | n.d. | 32 |
| E. faecalis VSE | n.d. | >64 |
| E. faecalis VRE | n.d. | 64 |
| B. subtilis | >64 | 64 |
| C. difficile | n.d. | >64 |
| B. fragilis | n.d. | >64 |
See SI for details. Significant bioactivities (MIC ≤ 25 μg/mL) are shown in bold.
Pleiotropic Stimulation of Secondary Metabolism.
Differential metabolic profiling shows that atenolol is not a specific inducer of the toxoflavin-like pteridines, but rather a global elicitor of secondary metabolism (Fig. 4B). As few metabolites have been reported from S. hiroshimensis, we characterized the additional compounds observed in the presence of atenolol. 1D/2D NMR and/or HR-MS identified one of these, produced under normal conditions but further induced by atenolol, as pyridindolol (Fig. 4C, 9), a secondary metabolite previously isolated Streptomyces alboverticillatus with inhibitory activity against β-galactosidase (Table S1, Fig. S6).29 Two further metabolites, whose production was also enhanced by atenolol, were determined to be 6,7,8-trimethoxy-3-methylisocoumarin (10) and 6,8-dihydroxy-3-methylisocoumarin (11), previously isolated from Streptomyces sp. ANK302 with zoosporicidal, but lacking antibacterial, bioactivity (Figs. S6–S7, Table S3).30 Lastly, we focused on peak 12 (Fig. 4C). Initial analysis by HR-MS and comparison with a database of known natural products indicated that it was likely new. Structural elucidation by 1D/2D NMR revealed a naphthoquinone-type scaffold that was fused to a substituted tetrahydropyran group. 13C and HMBC were consistent with the presence of an epoxide group bridging the pyrano and naphthoquinone moieties, with characteristic 13C shifts at 61.9 and 66.8 ppm (Fig. S8–S9). The naphthoquinone was modified with a spin system containing a contiguous, substituted –CH-CH2-CH2-CH-CH-CH3– chain as revealed by COSY spectra. HSQC and HMBC analysis indicated it consisted of an N-methyl-1-hydroxyethylsubstitued pyrrolidine, thus completing the two-dimensional structure of the compound, to which we have assigned the trivial name hiroshidine (Fig. 4C, 12, Table S3).
Hiroshidine’s structure is closest to qinimycin A (13), recently identified by the van Wezel group from Streptomyces sp. MBT76.31 It contains 9 stereocenters. Preliminary attempts to crystallize hiroshidine failed. Based on NMR NOESY constraints, we propose the relative stereochemistry shown for some of the stereocenters (Fig. 4C). Further work is required to experimentally assign the absolute stereochemistry at these positions as well as that of the epoxide group in hiroshidine.
Biosynthetic Gene Clusters for Taylorflavins and Hiroshidine.
Given the unusual structural features of taylorflavins and hiroshidine, we investigated the BGCs for the two groups of metabolites. We suspected that taylorflavins may follow a similar biosynthetic pathway as toxoflavin, which has been shown be composed of GTP and Gly.32–34 Indeed, growth of S. hiroshimensis in the presence of atenolol and 2-13C-Gly revealed taylorflavins that were now 1 Da heavier, consistent with incorporation of Gly (Table S1). This result suggested that we could identify the taylorflavin cluster by scouring the genome of S. hiroshimensis for genes encoded in the toxoflavin cluster (tox).
To find a tox-like cluster, the genome sequence of S. hiroshimensis was determined with 70-fold coverage using Illumina sequencing technology, resulting in a single chromosome at 8.4 Mbp. Analysis by antiSMASH revealed a total of 47 BGCs, suggesting that S. hiroshimensis harbors a prolific potential for secondary metabolite synthesis. The BGC of toxoflavin has been characterized in Burkholderia glumae and Pseudomonas protegens and the reactions of some of the enzymes have been examined in vitro.32–34 The biosynthetic pathway begins with the action of a GTP cyclohydrolase encoded by toxB. We searched the genome of S. hiroshimensis using toxB from Pseudomonas protegens Pf-5 as query and obtained a single hit. Several genes involved in toxoflavin biosynthesis could be identified, including toxA (SAM-dependent methyltransferase), toxC (WD40-repeat protein with yet unknown function), and toxE (a ribD homolog) (Fig. 5A, Table S4). Unique to this cluster were a second putative SAM-dependent methyltransferase (tflA), a Ser/Thr kinase (tflB), an ORF lacking similarity to known proteins (tflC), an oxidoreductase (tflD), possibly involved in introduction of the additional carbonyl functionality, and a transcriptional regulator (tflE).
Figure 5.

Biosynthetic gene clusters for taylorflavins and hiroshidine. (A) The tfl/tox BGC consists of genes tflABCDE as well as toxABCDE, which are shared with the tox gene cluster. (B) The hrs gene cluster is homologous to qin and primarily encodes sugar and polyketide biosynthetic enzymes. (C) Extracted ion chromatogram for taylorflavin B (m/z 224.1) in wt (red trace) and toxB–tflA::apr (black trace) S. hiroshimensis cultures. (D) Extracted ion chromatogram for hiroshidine (m/z 478.2) in wt (red trace) and hrsC–hrsJ::apr (black trace) S. hiroshimensis cultures.
To find the hiroshidine BGC, we used a type II polyketide synthase (PKS) from the qinimycin gene cluster (qin) in Streptomyces sp. MBT76 as query.31 Again, we obtained a single hit inside a ~40 kb gene cluster, containing many of the same genes as the qin BGC. This cluster, which we annotate as hrs, is significantly smaller than qin, but encodes type II PKSs as well as a number of genes involved in sugar biosynthesis and other modifications (Fig. 4B, Table S5). A biosynthetic pathway for qinimycin has been proposed but awaits experimental verification.31
We used a genetic approach to verify that the two gene clusters tfl/tox and hrs code for taylorflavins and hiroshidine. The tfl/tox locus was inactivated by replacing the six genes between toxB–tflA with an apramycin resistance gene (apr) using insertional mutagenesis, thus giving rise to a toxB–tflA::apr mutant strain (See SI Methods, Table S6). Likewise, hrsC–hrsJ::apr was generated to investigate the role of the hrs BGC in hiroshidine production. The wt and both mutants were subsequently cultured in the presence of atenolol and the supernatants examined by HPLC-MS. In the wt, we could clearly see production of both metabolites. In the toxB–tflA::apr strain, production of taylorflavins was abolished (Fig. 4C), though hiroshidine was observed under these conditions (Fig. S10). Similarly, hiroshidine was not observed in the hrsC–hrsJ::apr strain, though it was induced in both the wt and toxB–tflA::apr strains in response to atenolol (Fig. 4D, Fig. S10). These results are consistent with a role for the tfl/tox and hrs clusters in biosynthesizing taylorflavins and hiroshidine, respectively. With the BGCs identified, future studies will be able to assess the biosynthesis of both compounds, especially the interesting structural features, such as the epoxide or pyrrolidine functionalities in hiroshidine as well as the N-N bond in the 7-azapteridine scaffold and the oxidation and methylation patterns in taylorflavins.
Discussion
Bioactivity-guided fractionation has long been used to find natural products with the desired properties. Traditional methods, however, have focused on the bioactivity of the ‘constitutive’ metabolome, that is, natural products that are synthesized under standard growth conditions.35 We now know that constitutively-generated natural products are significantly outranked in abundance by the ‘inducible’ metabolome, that is, metabolites induced by generally unknown cues or signals. By combining HiTES with bioactivity assays, our approach allows access to this inducible metabolome resulting from activation of silent BGCs.
We validate Bioactivity-HiTES by reporting cryptic antibiotics from S. hiroshimensis. Two of these, taylorflavins A and B, belong to the toxoflavin family of molecules and exhibit the desired bioactivity we screened for, growth-inhibition of E. coli. Indeed, taylorflavins were more potent against Gram-negative than Gram-positive bacteria. Because of the selective outer membrane and the hydrolytic enzymes in the periplasmic space, Gram-negative bacteria tend to be less susceptible to antibacterial action by small molecules.36 Several structural elements of the taylorflavins are likely responsible for their unusual selective phenotype: the taylorflavins are small and are therefore not discriminated against by porins in the outer membrane of E. coli. They do not contain typical structural features that are degraded by well-known hydrolytic enzymes in the periplasm. And the likely target of taylorflavins, the electron transport chain (where toxoflavins act),37 resides in the inner bacterial membrane, which may be more easily accessed in Gram-negative than in Gram-positive bacteria, due to the extensive peptidoglycan layer in the latter. These features could provide guidelines for designing or finding other potential Gram-negative-selective antibacterial compounds.
Bioactivity-HiTES adds to the available approaches that have similarly leveraged cryptic metabolites in bioactivity assays. Notably, Bachmann and coworkers have used a small number of challenge conditions along with creative eukaryotic reporter assays to examine cryptic metabolites that modify human cell line behavior.23 Co-culture screens have been used by several groups to look for induction of cryptic metabolites with a selected biological activity using agar overlay assays.38,39 OSMAC-type methods have also been coupled to bioactivity assays to find new, induced metabolites.40 The differentiating feature of Bioactivity-HiTES is the throughput, as hundreds to thousands of conditions can be examined for induction of cryptic metabolites in a rapid manner. Moreover, the elicitor is linked directly to production of a cryptic, bioactive metabolite, thus enabling downstream mechanistic investigations that can address the basis of the stimulatory activity.
Additional applications of Bioactivity-HiTES are likely to lead to other cryptic metabolites with desired biological properties. The mechanism underlying HiTES is also an emerging area that will gain traction. Our previous applications of HiTES have repeatedly revealed antibiotics as inducers of silent BGCs, highlighting their stimulatory effects at sub-inhibitory concentrations in contrast to inhibitory ones at higher doses.41 Atenolol, an unsuspected anti-hypertension medicine, is unlike these previous elicitors in that it does not exhibit growth-inhibitory activity against S. hiroshimensis. It not only induced production of the taylorflavins, but generally enhanced the secondary metabolome of S. hiroshimensis, which in turn led to the characterization of pyridindolol, two isocoumarins, and hiroshidine along with its BGC. These results indicate that future studies addressing the mode of induction by atenolol will deliver further insights into the regulatory elements that exert cell-wide control over secondary metabolism as well as the hormetic functions of antibiotics in S. hiroshimensis and beyond.42
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (DP2-AI-124786 to M.R.S.), the Searle Scholars Program, and the Princeton University IP Accelerator Fund for support of this work.
Footnotes
The authors declare no competing financial interests.
References
- 1.Clardy J, and Walsh C (2004) Lessons from natural molecules. Nature 432, 829–837. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, and Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 3.Cragg GM, and Newman DJ (2013) Natural products: a continuing source of novel drug leads. Biochim. Biophys. Acta 1830, 3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan C (2004) Antibiotics at the crossroads. Nature 431, 899–902. [DOI] [PubMed] [Google Scholar]
- 5.Bérdy J (2005) Bioactive microbial metabolites. J. Antibiot 58, 1–26. [DOI] [PubMed] [Google Scholar]
- 6.Koehn FE, and Carter GT (2005) The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov 4, 206–220. [DOI] [PubMed] [Google Scholar]
- 7.Projan SJ (2003) Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol 6, 427–430. [DOI] [PubMed] [Google Scholar]
- 8.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S Huang CH, Kieser T, Larke L, Murphy L, Oliver K O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, and Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, and Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol 21, 526–531. [DOI] [PubMed] [Google Scholar]
- 10.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, and Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol 25, 447–453. [DOI] [PubMed] [Google Scholar]
- 11.Nett M, Ikeda H, and Moore BS (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep 26, 1362–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baltz RH (2017) Gifted microbes for genome mining and natural product discovery. J. Ind. Microbiol. Biotechnol 44, 573–588. [DOI] [PubMed] [Google Scholar]
- 13.Katz L, and Baltz RH (2016) Natural product discovery: past, present, and future. J. Ind. Microbiol. Biotechnol 43, 155–176. [DOI] [PubMed] [Google Scholar]
- 14.Challis GL (2008) Genome mining for novel natural product discovery. J. Med. Chem 51, 2618–2628. [DOI] [PubMed] [Google Scholar]
- 15.Rutledge PJ, and Challis GL (2015) Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol 13, 509–523. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Sandiford SK, and van Wezel GP (2014) Triggers and cues that activate antibiotic production by actinomycetes. J. Ind. Microbiol. Biotechnol 41, 371–386. [DOI] [PubMed] [Google Scholar]
- 17.Ochi K, and Hosaka T (2013) New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol 97, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao D, Okada BK, Wu Y, Xu F, and Seyedsayamdost MR (2018) Recent advances in activating silent biosynthetic gene clusters in bacteria. Curr. Opin. Microbiol 45, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Tian W, Wang S, Yan X, Jia X, Pierens GK, Chen W, Ma H, Deng Z, and Qu X (2017) Activation of natural products biosynthetic pathways via a protein modification level regulation. ACS Chem. Biol 12, 1732–1736. [DOI] [PubMed] [Google Scholar]
- 20.Seyedsayamdost MR (2014) High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc. Natl. Acad. Sci. USA 111, 7266–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu F, Nazari B, Moon K, Bushin LB, and Seyedsayamdost MR (2017) Discovery of a cryptic antifungal compound from Streptomyces albus J1074 using high-throughput elicitor screens. J. Am. Chem. Soc 139, 9203–9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada BK, and Seyedsayamdost MR (2017) Antibiotic dialogues: induction of silent biosynthetic gene clusters by exogenous small molecules. FEMS Microbiol. Rev 41, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Earl DC, Ferrel PB Jr., Leelatian N, Froese JT, Reisman BJ, Irish JM, and Bachmann BO (2018) Discovery of human cell selective effector molecules using single cell multiplexed activity metabolomics. Nat. Commun 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu F, Wu Y, Zhang C, Davis KM, Moon K, Bushin LB, and Seyedsayamdost MR (2019) A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat. Chem. Biol 15, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Damme PA, Johannes AG, Cox HC, and Berends W (1960) On toxoflavin, the yellow poison of Pseudomonas cocovenenans. Recl. Trav. Chim. Pays-Bas 79, 255–267. [Google Scholar]
- 26.Miller TW, Chaiet L, Arison B, Walker RW, Trenner NR, and Wolf FJ (1963) Isolation and characterization of antibiotic MSD-92, a pyrimidotriazine antibiotic. Antimicrob. Agents Chemother. 161, 58–62. [PubMed] [Google Scholar]
- 27.Taylor EC, and Sowinski F (1969) Structure and total synthesis of the pyrimido(5,4-e)-as-triazine antibiotic, 2-methylfervenulone. J. Am. Chem. Soc 91, 2143–2144. [DOI] [PubMed] [Google Scholar]
- 28.Taylor EC (1993) Design and synthesis of inhibitors of folate-dependent enzymes as antitumor agents. Adv. Exp. Med. Biol 338, 387–407. [DOI] [PubMed] [Google Scholar]
- 29.Aoyagi T, Kumagai M, Hazato T, Hamada M, and Takeuchi T (1975) Pyridindolol, a new beta-galactosidase inhibitor produced by actinomycetes. J. Antibiot 28, 555–557. [DOI] [PubMed] [Google Scholar]
- 30.Zinad DS, Shaaban KA, Abdalla MA, Islam MT, Schüffler A, and Laatsch H (2011) Bioactive isocoumarins from a terrestrial Streptomyces sp. ANK302. Nat. Prod. Commun 6, 45–48. [PubMed] [Google Scholar]
- 31.Wu C, Du C, Ichinose K, Choi YH, and van Wezel GP (2017) Discovery of C-glycosylpyranonaphthoquinones in Streptomyces sp. MBT76 by a combined NMR-based metabolomics and bioinformatics workflow. J. Nat. Prod 80, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenwick MK, Philmus B, Begley TP, and Ealick SE (2011) Toxoflavin lyase requires a novel 1-His-2-carboxylate facial triad. Biochemistry 50, 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philmus B, Shaffer BT, Kidarsa TA, Yan Q, Raaijmakers JM, Begley TP, and Loper JE (2015) Investigations into the biosynthesis, regulation, and self-resistance of toxoflavin in Pseudomonas protegens Pf-5. Chembiochem 16, 1782–1790. [DOI] [PubMed] [Google Scholar]
- 34.Fenwick MK, Philmus B, Begley TP, and Ealick SE (2016) Burkholderia glumae toxA is a dual-specificity methyltransferase that catalyzes the last two steps of toxoflavin biosynthesis. Biochemistry 55, 2748–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachmann BO, Van Lanen SG, and Baltz RH (2014) Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J. Ind. Microbiol. Biotechnol 41, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silhavy TJ, Kahne D, and Walker S (2010) The bacterial cell envelope. Cold Spring Harb. Perspect. Biol 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latuasan HE, and Berends W (1961) On the origin of the toxicity of toxoflavin. Biochim. Biophys. Acta 52, 502–508. [DOI] [PubMed] [Google Scholar]
- 38.Petit RK (2009) Mixed fermentation for natural product drug discovery. Appl. Microbiol. Biotechnol 83, 19–25. [DOI] [PubMed] [Google Scholar]
- 39.Seyedsayamdost MR, Traxler MF, Clardy J, and Kolter R (2012) Old meets new: using interspecies interactions to detect secondary metabolite production in actinomycetes. Methods Enzymol. 517, 89–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bode HB, Bethe B, Höfs R, and Zeeck A (2002) Big effects from small changes: possible ways to explore nature’s chemical diversity. Chembiochem 3, 619–627. [DOI] [PubMed] [Google Scholar]
- 41.Rosen PC, and Seyedsayamdost MR (2017) Though much is taken, much abides: finding new antibiotics using old ones. Biochemistry 56, 4925–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davies J, Spiegelman GB, and Yim G (2006) The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol 9, 445–453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.