

Abstract
The regenerative healing response of injured skeletal muscle is dependent upon an appropriately timed switch from a local type-I to a type-II immune response. Biologic scaffolds derived from extracellular matrix (ECM) have been shown to facilitate a macrophage phenotype transition that leads to downstream site-appropriate functional tissue deposition and myogenesis. However, the mechanisms by which ECM directs the switching of immune cell phenotype are only partially understood. Herein, we provide the first evidence that matrix bound nanovesicles (MBV) embedded within ECM-scaffolds are a rich and stable source of interleukin-33 (IL-33), an alarmin/cytokine with emerging reparative properties. We show that IL-33 encapsulated within MBV bypass the classical IL33/ST2 receptor signaling pathway to direct macrophage differentiation into the reparative, pro-remodeling M2 phenotype, which in turn facilitates myogenesis of skeletal muscle progenitor cells. Our results suggest the potential of IL-33+ MBV as a clinical therapy to augment the restorative efficacy of existing ECM-based and non-ECM based approaches.
Keywords: Extracellular Matrix, Matrix bound nanovesicles, Interleukin-33, Macrophage polarization
Graphical Abstract
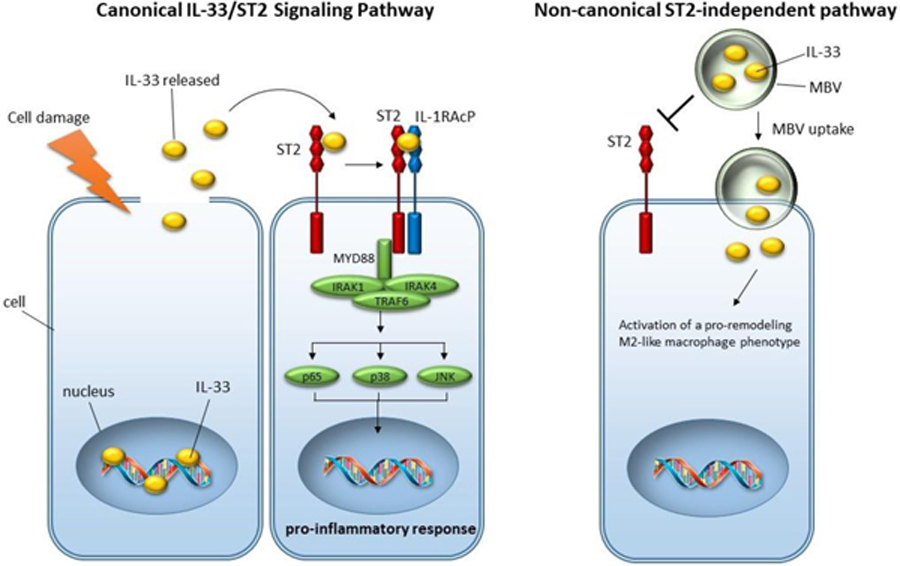
1. Introduction
Skeletal muscle has a remarkable capacity for repair in response to mild trauma(1). However, skeletal muscle damage associated with volumetric muscle loss (VML) overwhelms the regenerative process, ultimately resulting in scar tissue deposition, loss of function, and aesthetic deformities. There are very limited therapeutic options for massive loss of skeletal muscle tissue subsequent to trauma, surgical excision of neoplasms, and related conditions. Autologous muscle grafts or muscle transposition represent optional salvage procedures for restoration of muscle tissue, however these approaches are limited by the availability of donor tissue(2, 3). Alternatively, cell-based therapies have been explored, but legitimate issues remain associated with ES/iPS cell therapies such as immunogenicity, the desirable requirement for source, and a favorable environment to maintain cell viability(4). Moreover, development of a therapy that avoids the collection, isolation and/or ex-vivo expansion and purification of autologous stem cells with subsequent re-introduction to the patient would almost certainly reduce the regulatory hurdles for clinical translation, reduce the cost of treatment, and avoid the risks associated with cell-based approaches. There is an unmet need for therapeutic strategies that can enhance the innate regenerative ability of skeletal muscle following VML. Acellular biologic scaffolds composed of mammalian (typically porcine) extracellular matrix (ECM) have been investigated in preclinical in-vivo models of VML(5) and in a recent cohort study involving the use of ECM bioscaffolds in 13 patients with VML(6, 7). Outcomes have shown partial restoration of both structure and function, and support the translational aspects of an ECM-bioscaffold based approach. These ECM-based materials are most commonly xenogeneic in origin, and are prepared by the decellularization of a source tissue such as dermis, urinary bladder or small intestinal submucosa (SIS), among others(8). Use of these naturally occurring biomaterials is typically associated with at least partial restoration of functional, site-appropriate tissue; a process referred to as “constructive remodeling”( 9). Arguably, the major determinant of downstream functional remodeling outcome is the early innate immune response to ECM bioscaffolds(10, 11). ECM bioscaffolds, or more accurately, the degradation products of ECM bioscaffolds, have been shown to direct tissue repair by promoting a transition from a pro-inflammatory M1-like macrophage and Th1 T cell phenotype to a pro-remodeling M2-like macrophage and T helper Type 2 (Th2) cell response(12–15). Numerous studies have shown that an appropriately timed transition in macrophage activation state is required for promotion of tissue remodeling and wound healing processes rather than scar tissue formation in numerous anatomic sites including skeletal muscle(16, 17), and cardiovascular systems(18, 19). Importantly, this transition is not immunosuppression, but rather a constructive form of immunomodulation that promotes a phenotypic change in local macrophage phenotype(20, 21).
Emerging evidence shows that degradation of the scaffold material and subsequent release of matrix-bound-nanovesicles (MBV), that harbor bioactive components which have been only partially defined, are critical for the activation of a reparative and anti-inflammatory M2 macrophage phenotype(22, 23). MBV are nanometer-sized, membranous vesicles that are embedded within the collagen network of the ECM and protect biologically active signaling molecules (microRNAs and proteins) from degradation and denaturation(22). We have previously shown that ECM bioscaffolds and their resident MBV can activate macrophages toward a M2-like, pro-remodeling phenotype(22, 23). However, the molecular components of MBV and the mechanisms by which these nanovesicles direct the switching of immune cell phenotype are poorly understood.
Herein, it is shown that MBV are a rich source of extra-nuclear interleukin-33 (IL-33). IL-33 is an IL-1 family member that is typically found in the nucleus of stromal cells and generally regarded as an alarmin, or a self-derived molecule that is released after tissue damage to activate immune cells via the IL-33 receptor, ST2(24). Emerging evidence, however, indicates that IL-33 may function as a promoter of skeletal muscle repair by stimulating ST2+ regulatory T cells (Treg)(25). Intracellular IL-33 protein has been suggested to modulate gene expression through interactions with chromatin or signaling molecules via the IL-33 N-terminus(24, 26). As of yet, however, intracellular IL-33 activities have not been shown to control any immune cell functions. Herein, evidence is provided that IL-33, stably stored within the ECM and protected from proteolytic cleavage by incorporation into the lumen of MBV, is a potent mediator of M2 macrophage activation through an uncharacterized, non-canonical ST2-independent pathway. Specifically, MBV isolated from il33+/+ mouse tissue ECM, but not MBV from il33−/−, direct st2−/−macrophage activation toward the reparative, pro-remodeling M2 activation state. This capacity of IL33+ MBV is distinct from the well characterized IL-4/IL-13-mediated M2 macrophage differentiation pathway, as IL33+ MBV generate M2-like macrophages independent of Stat6 phosphorylation. Moreover, the secreted products from IL33+ MBV-treated macrophages promote myogenesis of skeletal muscle progenitor cells. In total, our results suggest that MBV can mediate receptor-free delivery of cytokine cargo to immune cells to orchestrate cell function.
2. Materials and Methods
2.1. Animals
C57BL/6 (B6) and Bm12 mice were purchased from Jackson Laboratories. The il33−/− mice were a gift from S. Nakae (University of Tokyo, Tokyo, Japan)(27). St2−/− mice were originally generated on a BALB/c background as described(28) and obtained from Dr. Anne Sperling (University of Chicago) after they were backcrossed 7 times onto the C57BL/6 background. These mice were then backcrossed 3 additional times onto the C57BL/6 background here at the University of Pittsburgh before use in our experiments. Animals were housed in a specific pathogen-free facility maintained by the University of Pittsburgh. The studies conformed to the principles set forth by the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of animals in biomedical research.
2.2. Decellularization of mouse intestines
Mouse small intestine was decellularized as previously described(29) with minor modifications. Fresh small intestines were obtained from adult wt B6 mice or adult IL-33−/− B6 mice. Small intestines were washed in phosphate buffered saline (PBS) to completely remove all the intestinal contents, and cut into 1.5 cm-length fragments for immediate decellularization. Samples were immersed in 5M NaCl for 72 hr under continuous soft agitation. The decellularization solution was replaced every 24 hr. Decellularized intestines were then washed three times in distilled water. The native and decellularized tissue was prepared for histologic analysis to determine decellularization efficacy by fixing in 10% neutral buffered formalin, or lyophilized and powdered using a Wiley Mill with a #40 mesh screen.
2.3. Determining decellularization efficacy
Native mouse small intestine tissue and decellularized mouse intestines were fixed in 10% neutral buffered formalin for 24 h. The fixed samples were then paraffin embedded, and 5 µm sections were cut onto slides. Slides were stained with hematoxylin and eosin (H&E) or 4’,6-diamidino-2-phenylindole (DAPI) to visualize the presence of nuclear material. Residual DNA content of the ECM was quantified by powdering samples with a Wiley Mill using a 40-mesh from separate preparations (n=4) of lyophilized native mouse intestine tissue or decellularized mouse intestine. Samples (50 mg) were digested in 0.1 mg/mL proteinase K digestion buffer at 50°C for 24 h. DNA was extracted twice in phenol/chloroform/isoamyl alcohol and centrifuged at 10,000g (10 min at 48C). The aqueous phase, containing the DNA was then mixed with 3M sodium acetate and 100% ethanol, centrifuged at 10,000 × g (10 min at 4C), pouring off the supernatant, adding 70% ethanol, repeating centrifugation, removing supernatant, and drying the remaining DNA pellet. When dry, the pellet was resuspended in TE buffer (10 mM Tris, pH 7.5/1 mM EDTA), and the DNA concentration was quantified utilizing a PicoGreen Assay (Invitrogen) following manufacturer’s instructions.
2.4. Preparation of Dermal ECM
Dermal ECM was prepared as previously described(30). Briefly, full-thickness skin was harvested from market-weight (~110 kg) pigs (Tissue Source Inc.), and the subcutaneous fat and epidermis were removed by mechanical delamination. This tissue was then treated with 0.25% trypsin (Thermo Fisher Scientific) for 6 hours, 70% ethanol for 10 hours, 3% H2O2 for 15 min, 1% Triton X-100 (Sigma-Aldrich) in 0.26% EDTA/0.69% tris for 6 hours with a solution change for an additional 16 hours, and 0.1% peracetic acid/4% ethanol (Rochester Midland) for 2 hours. Water washes were performed between each chemical change with alternating water and phosphate-buffered saline (PBS) washes following the final step. All chemical exposures were conducted under agitation on an orbital shaker at 300 rpm. Dermal ECM was then lyophilized and milled into particulate using a Wiley Mill with a #40 mesh screen.
2.5. Preparation of urinary bladder matrix (UBM)
UBM was prepared as previously described(31). Porcine urinary bladders from market-weight animals were acquired from Tissue Source, LLC. Briefly, the tunica serosa, tunica muscularis externa, tunica submucosa, and tunica muscularis mucosa were mechanically removed. The luminal urothelial cells of the tunica mucosa were dissociated from the basement membrane by washing with deionized water. The remaining tissue consisted of basement membrane and subjacent lamina propria of the tunica mucosa and was decellularized by agitation in 0.1% peracetic acid with 4% ethanol for 2 hours at 300 rpm. The tissue was then extensively rinsed with PBS and sterile water. The UBM was then lyophilized and milled into particulate using a Wiley Mill with a #60 mesh screen.
2.6. Preparation of small intestinal submucosa (SIS)
SIS was prepared as previously described(32). Briefly, jejunum was harvested from 6-month-old market-weight (~110 to ~120 kg) pigs and split longitudinally. The superficial layers of the tunica mucosa were mechanically removed. Likewise, the tunica serosa and tunica muscularis externa were mechanically removed, leaving the tunica submucosa and basilar portions of the tunica mucosa. Decellularization and disinfection of the tissue were completed by agitation in 0.1% peracetic acid with 4% ethanol for 2 hours at 300 rpm. The tissue was then extensively rinsed with PBS and sterile water. The SIS was then lyophilized and milled into particulate using a Wiley Mill with a #60 mesh screen.
2.7. Preparation of cardiac ECM
Cardiac ECM was prepared as previously described(33). Briefly, porcine hearts were obtained immediately following euthanasia and frozen at −80°C for at least 16 h and thawed. The aorta was cannulated and alternately perfused with type 1 reagent grade (type 1) water and 2× PBS at 1 liter/min for 15 min each. Serial perfusion of 0.02% trypsin/0.05% EDTA/0.05% NaN3 at 37°C, 3% Triton X-100/0.05% EDTA/0.05% NaN 3, and 4% deoxycholic acid was conducted (each for 2 h at approximately 1.2 liters/min). Finally, the heart was perfused with 0.1% peracetic acid/4% EtOH at 1.7 liters/min for 1 h. After each chemical solution, type 1 water and 2× PBS were flushed through the heart to aid in cell lysis and the removal of cellular debris and chemical residues. The cardiac ECM was then lyophilized and milled into particulate using a Wiley Mill with a #60 mesh screen.
2.8. Isolation of matrix bound nanovesicles (MBV)
MBV were isolated as previously described(22) with minor modifications. Briefly, 100mg of powdered ECM was enzymatically digested with 100 ng/ml Liberase DL (Roche) in buffer (50mM Tris pH 7.5, 5mM CaCl2, 150mM NaCl) for 12 hr at room temperature on an orbital rocker. The digested ECM was then subjected to centrifugation at 10,000 × g for 30 min to remove ECM debris, and the supernatant passed through a 0.22 µm filter (Millipore). The clarified supernatant containing the liberated MBV was then centrifuged at 100,000 × g (Beckman Coulter Optima L-90K Ultracentrifuge) at 4°C for 70 min to pellet the MBV. The MBV pellet was then washed and resuspended in 1X PBS, and stored at −20°C until further use.
2.9. Transmission Electron Microscopy (TEM)
TEM imaging was conducted on MBV loaded on carbon-coated grids and fixed in 4% paraformaldehyde as previously described(22). Grids were imaged at 80 kV with a JEOL 1210 TEM with a high-resolution Advanced Microscopy Techniques digital camera. Size of MBV was determined from representative images using JEOL TEM software.
2.10. Cytokine antibody array
Cytokines stored within MBV were analyzed using the Mouse XL Cytokine Array Kit (R&D Systems; Minneapolis, MN, USA) according the manufacturer’s instructions. Extracts were prepared from MBV isolated from decellularized wt B6 mouse intestine (n=3) or decellularized il33−/− B6 mouse intestine (n=3) by incubation with 1% Triton X-100. Extracts were diluted and incubated overnight with the array membrane. The array was rinsed to remove unbound protein, incubated with an antibody cocktail, and developed using streptavidin–horseradish peroxidase and chemiluminescent detection reagents. Mean spot pixel density was quantified using Image J software.
2.11. Size Exclusion Chromatography (SEC)
Fractionation of MBV by SEC was performed as previously described(34). Briefly, 15 ml of Sepharose CL-2B resin (Sigma Aldrich) was stacked in a 1cm × 20cm glass column and equilibrated with 1X PBS. 1ml of intact MBV, or 1ml of MBV lysed with 1% Triton X-100 were loaded onto the column and fraction collection (0.35 ml per fraction, and a total of 30 fractions collected) started immediately under gravity flow using PBS as the elution buffer. Eluted fractions were continuously monitored by UV 280nm using the Biologic LP system (BioRad).
2.12. Biotinylation and streptavidin pull down of MBV proteins
Biotinylation of MBV surface proteins was performed as previously described(35) with minor modifications. One hundred micrograms of intact MBV were incubated in the absence or presence of 10mM Sulfo-NHS-Biotin at 4°C for 2 hr. After incubation, 100mM glycine was added to quench the reaction, and excess biotin reagent and byproducts were removed using a 10 kDa MWCO filtration column. MBV were then lysed in 1% Triton X-100, and concentrated to a final volume of 50µl. Biotinylation of MBV extracts was performed by first lysing 100 micrograms of MBV in 1% Triton X-100. After lysis, buffer exchange was performed to replace the 1% Triton X-100 solution with 1X PBS. The MBV extract was then incubated in the absence or presence of 10mM Sulfo-NHS-Biotin at 4°C for 2 hr. After incubation, 100mM glycine was added to quench the reaction, and excess biotin was removed using a 10 kDa MWCO filtration column. Biotinylated MBV extracts were concentrated to a final volume of 50µl. MBV ±biotin or MBV extract ±biotin were then diluted to 200µl in 1X PBS and incubated with 50µl prewashed streptavidin-sepharose resin (Sigma Aldrich). After incubation on an orbital rocker for 2 hr at room temperature, the streptavidin-sepharose resin was pelleted by centrifugation at 10,000 × g for 5 min. The supernatant representing the unbound fraction was transferred to a fresh tube, and the resin was washed 5 times in 300mM NaCl. Bound proteins were eluted from the resin by incubating with 200µl elution buffer (2% SDS, 6M Urea) for 15 minutes at room temperature and then 15 minutes at 96°C. Equal volumes of unbound protein fraction, and streptavidin pull-down fractions were analyzed by immunoblot for IL-33.
2.13. Immunoblot assays
MBV protein concentration was determined using the bicinchoninic acid assay quantification kit (Pierce Chemical). 20µg MBV were resuspended in Laemmli buffer (R&D Systems) containing 5% β-mercaptoethanol (Sigma-Aldrich), resolved on a 4 to 20% gradient SDS-PAGE (Bio-Rad), and then transferred onto a PVDF membrane. Porcine derived MBV samples were analyzed using a porcine-specific IL-33 antibody (LSBio; cat# C285540). Murine-derived MBV samples were analyzed using a mouse-specific IL-33 antibody (R&D systems; cat# AF3626). Macrophages were lysed in RIPA buffer, and 60µg of cell lysate was resuspended in Laemmli buffer (R&D Systems) containing 5% β-mercaptoethanol (Sigma-Aldrich) and resolved on a 4 to 20% gradient SDS-PAGE (Bio-Rad), and then transferred onto a PVDF membrane. Membranes were incubated with primary antibodies against p38, phospho-p38, p65, phospho-p65, Arginase-1, Stat6, phospho-Stat6 (all from Cell Signaling Technology), or GAPDH (Abcam).
2.14. Proteinase K protection assay
Proteinase K protection assay was performed as previously described(36). Briefly, MBV were incubated in either PBS or increasing concentrations of Proteinase K in PBS, with or without the presence of 1% Triton X‐100, in a final volume of 20 µl per sample for 1 hr at 37°C. The assay was stopped by addition of 20 µl 2× Laemmli Buffer with 10 mM DTT. After a 5 min incubation at 96°C, the samples were resolved by SDS-PAGE and analyzed by immunoblot for IL-33.
2.15. Isolation and activation of macrophages
Murine bone marrow-derived macrophages (BMDM) were isolated and characterized as previously described(22). Briefly, bone marrow was harvested from 6- to 8-week-old B6 mice, B6 st2−/− mice, or Balb/c st2−/− mice. Harvested cells from the bone marrow were washed and plated at 2 × 106 cells/mL and were allowed to differentiate into macrophages for 7 days in the presence of macrophage colony-stimulating factor (MCSF) with complete medium changes every 48 h. Macrophages were then activated for 24 h with one of the following: 1) 20 ng/mL Interferon-γ (IFNγ) and 100 ng/mL lipopolysaccharide (LPS) (Affymetrix eBioscience, Santa Clara, CA; Sigma Aldrich) to promote an MIFNγ+LPS phenotype (M1-like); 2) 20 ng/mL interleukin (IL)-4 (Invitrogen) to promote an MIL-4 phenotype (M2-like); 3) 20 ng/ml IL-33 (Peprotech), or 4) 25 µg/mL of wt mouse MBV, IL-33−/− mouse MBV, or porcine SIS-MBV. After the incubation period at 37°C, cells were washed with sterile PBS and lysates prepared using RIPA buffer for immunoblot analysis, or cells fixed with 2% paraformaldehyde (PFA) for immunolabeling.
2.16. Macrophage immunolabeling
To prevent nonspecific binding, the cells were incubated in a blocking solution composed of PBS, 0.1% Triton-X, 0.1% Tween-20, 4% goat serum, and 2% bovine serum albumin for 1 h at room temperature. The blocking buffer was then removed and cells were incubated in a solution of one of the following primary antibodies: 1) monoclonal anti-F4/80 (Abcam, Cambridge, MA) at 1:200 dilution as a pan-macrophage marker; 2) polyclonal anti-inducible nitric oxide synthase (iNOS) (Abcam, Cambridge, MA) at 1:100 dilution as an M1-like marker, and 3) anti-Arginase1 (Abcam, Cambridge, MA) at 1:200 dilution, as an M2-like marker. The cells were incubated at 4°C for 16 h, the primary antibody was removed, and the cells washed with PBS. A solution of fluorophore-conjugated secondary antibody (Alexa donkey anti-rabbit 488 or donkey anti-rat 488; Invitrogen, Carlsbad, CA) was added to the appropriate well for 1 h at room temperature. The antibody was then removed, the cells washed with PBS, and the nuclei were counterstained using DAPI. Cytokine-activated macrophages were used to establish standardized exposure times (positive control), which were held constant throughout groups thereafter. CellProfiler (Broad Institute, Cambridge, MA) was used to quantify images. Data were analyzed for statistical significance using a one-way analysis of variance with Tukey’s post-hoc test for multiple comparisons. Data are reported as mean ± standard deviation with a minimum of N = 3. p-values of <0.05 were considered to be statistically significant.
2.17. Myogenesis assay
Bone marrow-derived macrophages (BMDM) from Balb/c st2−/− mice were used to generate macrophage supernatents. Murine C2C12 myoblasts were cultured in DMEM containing 10% FBS, 100 µg/ml streptomyocin and 100 U/ml penicillin. Cells were maintained at 37 °C, 5% CO2. High serum media (10% fetal bovine serum) maintains cell proliferation within the cell cycle and inhibits differentiation. Conversely, low serum media (1% fetal bovine serum, 1% horse serum) induces cell-cycle exit and myotube formation providing a positive control. These will be referred to as proliferation media and differentiation media, respectively. Myogenic differentiation potential was determined by examining myotube formation. Myoblasts were cultured in proliferation media until they reached approximately 80% confluence. Media was then changed to treatment media consisting of a 1:1 solution of macrophage supernatants and 20% FBS media (final concentration equivalent to proliferation media), or controls of proliferation media or differentiation media. Following 2 and 5 days, cells were fixed for immunolabeling with 2% paraformaldehyde. Fixed cells were blocked according to the previous described protocol for 1 h at room temperature and incubated in anti-sarcomeric myosin primary antibody (Developmental Studies Hybridoma Bank) at a dilution of 1:500 for 16 h at 4 °C. Following primary incubation, cells were washed with PBS and incubated in Alexa Fluor donkey anti-mouse 488 secondary antibody at a dilution of 1:200 for 1 h at room temperature and counterstained with DAPI. Images of three 20× fields were taken for each well (n = 3 biological replicates) using a Zeiss Axiovert microscope, and the number of myotubes was quantified and averaged. The data were then analyzed by one-way ANOVA followed by Tukey’s Multiple Comparison’s Test, with a p < 0.05 considered significant.
3. Results
3.1. Matrix-bound nanovesicles isolated from ECM bioscaffolds contain full length IL-33
The isolation of MBV from ECM bioscaffolds and characterization of the miRNA cargo has been previously described(22, 37). To identify protein signaling molecules associated with MBV, a preliminary cytokine, chemokine, and growth factor screen was performed with MBV isolated from porcine small intestinal submucosa (SIS) ECM bioscaffolds using the Mouse XL Cytokine Array Kit from R&D system. Results showed robust expression of IL-33 associated with porcine SIS MBV (fig. S1A). To confirm and extend this observation, MBV were isolated from decellularized wild type (wt) mouse small intestine or decellularized il33−/− mouse small intestine. Using well established decellularization criteria(38, 39), we showed that no residual nuclei were visible in H&E and DAPI images of decellularized small intestine ECM compared to native tissue (Fig. 1A), and quantification of double stranded DNA (dsDNA) showed that decellularized intestine retained <50ng dsDNA per mg dry ECM (Fig. 1B). Following verification of efficient decellularization, MBV were isolated from decellularized ECM by enzymatic digestion and imaged by transmission electron microscopy (Fig. 1C). The cytokine cargo of MBV isolated from decellularized wt mouse intestine or decellularized il33−/− mouse intestine was analyzed using the mouse cytokine array (fig. S1B). Quantitation of proteins with the highest expression levels in MBV showed that IL-33 was highly expressed in MBV isolated from wt mice (IL33+ MBV) compared to MBV isolated form il33−/− mice (IL33- MBV), with minimal differences in the expression of the other proteins present in isolated MBV (Fig. 1D). These results were validated by immunoblot analysis which showed that IL-33 associated with MBV was the full-length (32 kDa) form of the protein (Fig. 1E) and not smaller described cleavage products(40, 41). The presence of full-length IL-33 expression in MBV was subsequently observed in ECM surgical meshes commonly used in clinical applications, which included laboratory-produced and commercially available equivalents of urinary bladder matrix (UBM) and ACell® MatriStem™; small intestinal submucosa (SIS) and Cook Biotech® BioDesign™; dermis and BD® XenMatrix™; and cardiac ECM ( Fig. 1F). Results showed that laboratory-produced scaffolds had similar IL-33 expression levels relative to their respective commercially available counterparts, indicating that these results were not an artifact of laboratory manufacturing protocols.
Fig. 1: MBV isolated from ECM bioscaffolds contain full-length (32 kDa) IL-33.
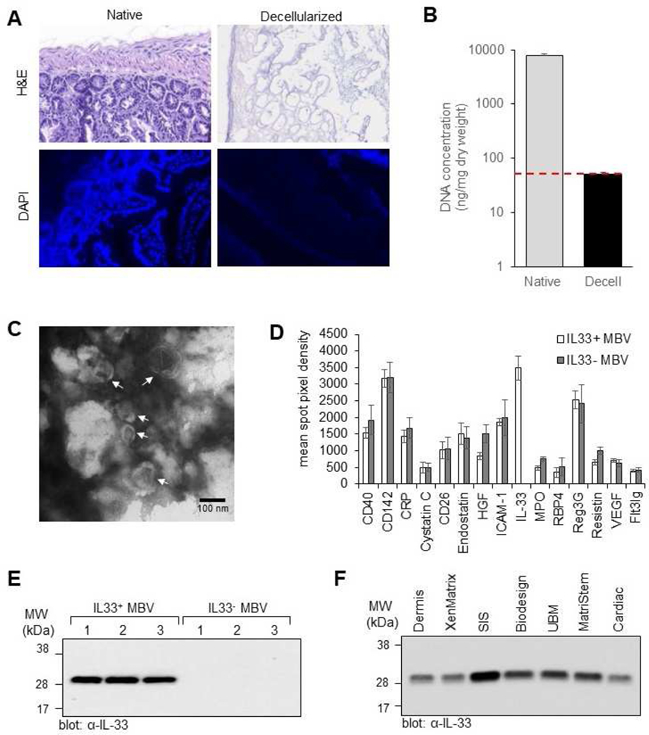
(A, B) Decellularization efficacy. The presence of nuclei in native mouse small intestine tissue and decellularized mouse intestine tissue was assessed by hemotoxylin and eosin (H&E) staining and 4’,6-diamidino-2-phenylindole (DAPI) staining (A). Concentration of double stranded DNA was quantified using the PicoGreen assay; y-axis is log scale; n = 4; p <0.005. Red line denotes established DNA criteria against which decellularization efficacy was verified. Native dsDNA concentration = 7747.34 ± 748; Decellularized dsDNA concentration = 49.87 ± 4.84 (B). (C) Transmission electron microscopy imaging of MBV isolated from decellularized mouse intestine. (D) Graphic representation of the quantitation of 15 cytokines with the highest expression levels in MBV isolated from decellularized wt and il33−/− mouse intestines. Shown are mean ± s.d, n=3 biological replicates. (E) Immunoblot analysis of IL-33 expression levels in MBV isolated from three decellularized wt mouse intestines (IL33+ MBV) or three decellularized il33−/− mouse intestines (IL33- MBV). (F) Immunoblot analysis of IL-33 expression levels in MBV isolated from laboratory produced porcine urinary bladder matrix (UBM), small intestinal submucosa (SIS), dermis, and cardiac ECM; and three commercially available ECM bioscaffold equivalents: ACell® MatriStem™ (porcine UBM), BD® XenMatrix™ (porcine dermis), and Cook Biotech® Biodesign™ (porcine SIS).
3.2. IL-33 is stored within the lumen of MBV and protected from proteolytic degradation
To verify that detected IL-33 was not a contaminant of the MBV isolation process, MBV were further purified by size exclusion chromatography (SEC) using a Sepharose CL-2B resin with continuous monitoring of eluted fractions by UV absorbance at 280nm (Fig. 2A). The results show that MBV eluted primarily in the heavy fractions as assessed by transmission electron microscopy of pooled fractions 6–8 (Fig. 2B). In addition, immunoblot analysis confirmed the presence of IL-33 in the heavy MBV fractions (Fig. 2C, top panel). In a separate experiment, MBV were first lysed with 1% Triton X-100 and the extracts then analyzed by SEC. Results show that the molecular components from lysed MBV eluted primarily in the lighter fractions as determined by the shift in the UV chromatogram (Fig. 2A), and immunoblot analysis (Fig. 2C, bottom panel). These results confirmed that IL-33 was associated with the MBV, and not a soluble contaminant of the MBV isolates. We next determined if IL-33 was present on the surface membrane of MBV or stored within the lumen. MBV pooled from fractions 6–8 were biotinylated with NHS-LC-Biotin. The sulfonate group prevents the biotin from permeating the lipid membrane, thereby labeling only the outer surface proteins(35). After biotinylation, MBV were lysed and subjected to a streptavidin pull down assay to fractionate the surface proteins from the unbound luminal components. Immunoblot analysis showed that IL-33 was present only in the unbound fraction and was not pulled down by the streptavidin (SA) beads (Fig. 2D). In a separate experiment, MBV were first lysed with 1% Triton X-100 and then subjected to biotinylation. This allowed for biotinylation of both the surface and luminal components of MBV. Immunoblot analysis showed that following streptavidin pull down, IL-33 was associated with the SA beads (Fig. 2D). Cumulatively, these data suggested that IL-33 was stored within the lumen of MBV. To confirm these results, we performed a proteinase K protection assay. MBV from pooled fractions 6–8 were incubated with increasing concentrations of proteinase K for 30 min at 37°C in the absence or presence of 1% Triton X-100. As shown by immunoblot analysis (Fig. 2E), in the absence of Triton X-100, IL-33 was not degraded by Proteinase K. Permeabilization of the MBV membrane by Triton X-100, however, makes IL-33 accessible and susceptible to proteinase K, resulting in its degradation (Fig. 2E). These results confirmed that MBV-associated IL-33 was present in the lumen of the vesicle membrane where it was protected from proteolytic degradation.
Fig. 2: Full-length IL-33 stored in the ECM is protected from proteolytic degradation by incorporation into the lumen of MBV.
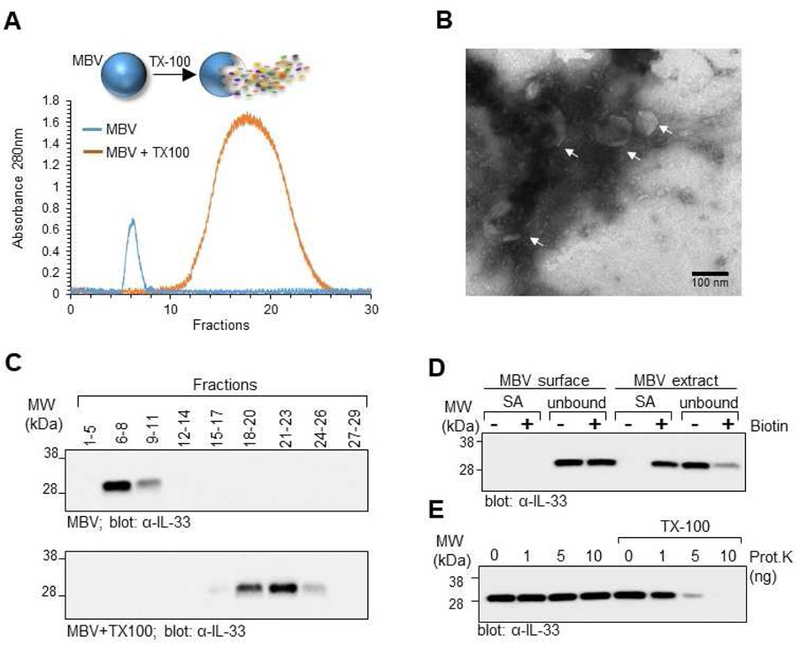
(A) MBV isolated from decellularized wt mouse intestines were fractionated by size exclusion chromatography (SEC) with continuous monitoring of eluted fractions by UV absorbance at 280nm. In a separate experiment, MBV were first lysed with Triton X-100 and then fractioned by SEC. Overlay of the two UV chromatograms shows that intact MBV eluted in the heavier fractions, whereas the molecular components of lysed MBV eluted primarily in the lighter fractions. (B) Pooled fractions 6–8 of the intact MBV sample were imaged by transmission electron microscopy to confirm the presence of MBV in the pooled fraction. (C) Eluted fractions from the intact MBV sample (top panel) or lysed MBV (bottom panel) were pooled as indicated and analyzed by immunoblot for IL-33. (D) Pooled fractions 6–8 of intact MBV were either directly biotinylated to label the MBV surface proteins, or first lysed with Triton X-100 and the MBV extract biotinylated to label the luminal and surface proteins. Proteins isolated after streptavidin pull down (SA) and the unbound fraction representing proteins that did not bind to the streptavidin beads (unbound) were analyzed by immunoblot for the presence of IL-33. (E) Proteinase K protection assay. Pooled fractions 6–8 of intact MBV were treated with indicated amounts of Proteinase K in the absence or presence of Triton X-100 and analyzed by immunoblot for IL-33.
3.3. IL33+ MBV activate a pro-remodeling macrophage phenotype via a non-canonical ST2-independent pathway
Given the location of IL-33 within the lumen of MBV, we hypothesized that encapsulation of IL-33 prevents binding to its cognate ST2 receptor, suggesting the presence of an ST2-independent transduction mechanism. To investigate this scenario, bone marrow-derived macrophages (BMDM) isolated from wt (Fig. 3A) or st2−/− (Fig. 3B) mice were stimulated with interferon-γ (IFN-γ) and lipopolysaccharide (LPS) to induce an M1-like macrophage phenotype, interleukin-4 (IL-4) to induce an M2-like phenotype, recombinant IL-33, MBV isolated from decellularized wt (IL33+ MBV) or il33−/− (IL33- MBV) mouse intestine, or MBV isolated from porcine small intestinal submucosa (SIS MBV). Results showed that macrophages expressed Arginase 1 (Arg-1) in response to SIS MBV and IL33+ MBV, similar to the expression pattern of the IL-4–stimulated (M2) cells. In contrast, IL33- MBV induced the expression of iNOS but not Arg-1 (Fig. 3A, C, D). A similar effect was observed with macrophages isolated from st2−/− mice (Fig. 3B). Specifically, IL33+ MBV, but not IL33- MBV, directed st2−/− macrophage activation into the reparative, pro-remodeling M2-like phenotype (Fig. 3B, C, D).
Fig. 3: MBV containing luminal IL-33 activate a pro-remodeling macrophage phenotype (F4/80+iNOS−Arg+) via a non-canonical ST2-independent pathway.
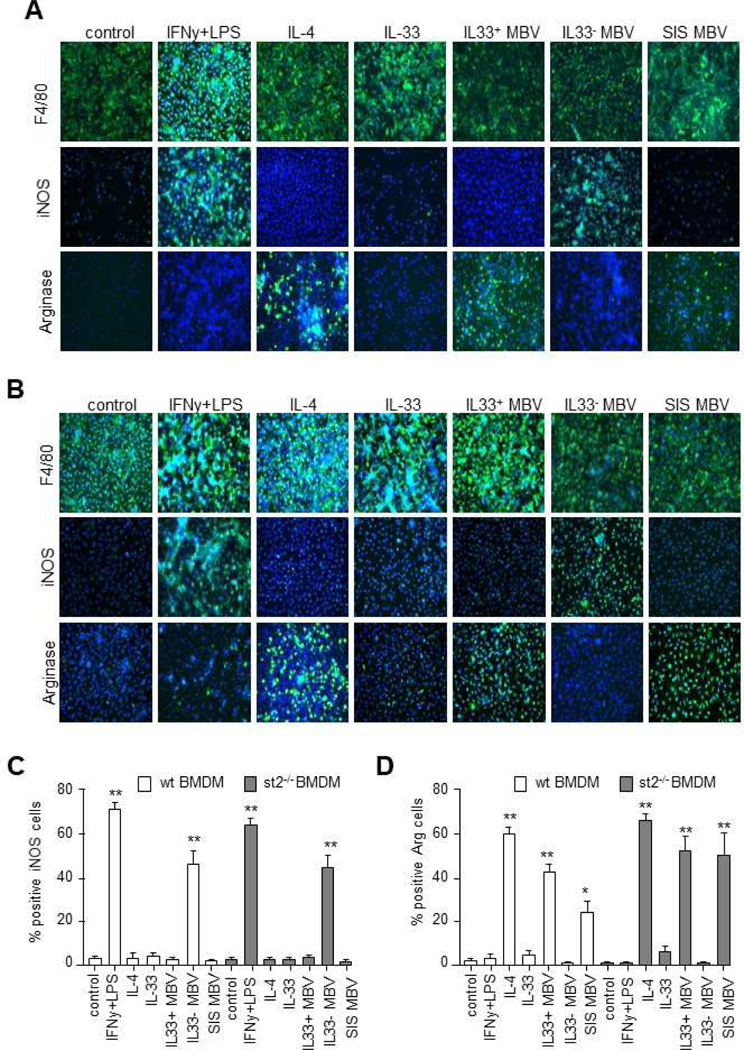
(A, B) Bone Marrow-Derived Macrophages (BMDM) harvested from wt (A) or st2−/− (B) mice were untreated (control) or treated with the following test articles for 24 hours: IFNɣ+LPS, IL-4, IL-33, MBV isolated from decellularized wt mouse intestine (IL33+ MBV), MBV isolated from decellularized il33−/− mouse intestine (IL33- MBV), or MBV isolated from porcine small intestinal submucosa (SIS MBV). Cells were immunolabeled with F4/80 (macrophage marker), iNos (M1 marker), or Arg1 (M2 marker). (C) Quantification of iNOS immunolabeling (** indicates p < 0.01; compared to negative control, error bars represent SEM, n=3 biological replicates analyzed in triplicate). (D) Quantification of arginase immunolabeling (**indicates p < 0.01; * indicates p < 0.05 compared to negative control, error bars represent SEM, n=3 biological replicates analyzed in triplicate).
Results from the immunolabeling assay were subsequently confirmed by western blot analysis which showed that stimulation of macrophages with IL33+ MBV, but not IL33- MBV, could induce the upregulation of Arg-1 expression (Fig. 4A). In addition, this capacity of IL33+ MBV to induce Arg-1 expression was shown to be distinct from the well characterized IL-4/IL-13-mediated M2 macrophage differentiation pathway, as IL33+ MBV activate M2 macrophages independently of STAT6 phosphorylation (Fig. 4B). As an important control, we show that recombinant IL-33, but not IL33+ MBV, was able to activate the ST2-dependent signaling pathway as evidenced by p38 and p65 phosphorylation in wt macrophages but not in the st2−/−macrophages (Fig. 4B). These data demonstrate that MBV-associated IL-33 modulates macrophage activation through an uncharacterized, non-canonical ST2-independent pathway.
Fig. 4: MBV upregulate Arginase expression independent of Stat6 phosphorylation.
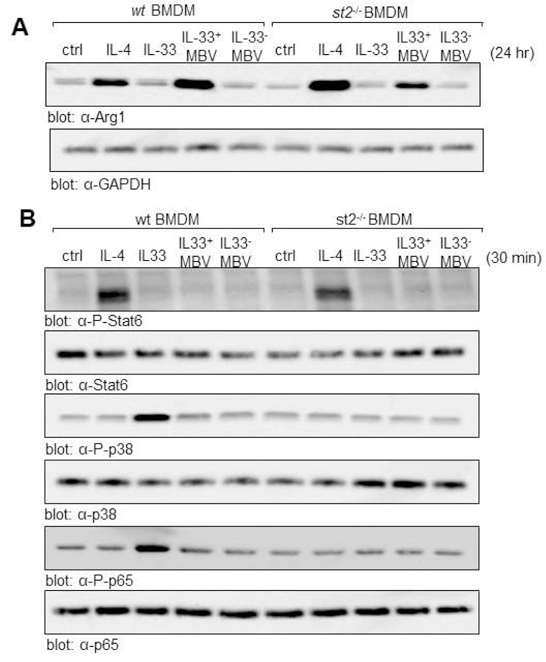
(A, B) BMDM harvested from wt or st2−/− mice were untreated (ctrl), or stimulated for 24 hr (A) or 30 min (B) with IL4, recombinant IL-33, IL33+ MBV, or IL33- MBV). Cell lysates were analyzed by immunoblot for Arginase-1 expression (A) and phosphorylation of Stat6 (B). Activation of the canonical IL-33 signaling cascade was analyzed by immunoblot for p38 and p65 phosphorylation (B).
3.4. Evaluation of myogenesis of skeletal muscle progenitor cells following exposure to macrophage secreted products
It has been shown that the secretome associated with alternatively activated M2 macrophages is myogenic for skeletal muscle myoblasts(42, 43). Previously, we have shown that media conditioned by ECM-treated macrophages promoted myotube formation and sarcomeric myosin expression of C2C12 myoblasts(15). The present study shows similar results in that media conditioned by macrophages stimulated with IL33+ MBV, but not IL33- MBV, promoted myotube formation of C2C12 myoblasts similar to the biologic activity of IL-4-induced M2-like macrophages (Fig. 5A, B, C)
Fig. 5: Secreted products of IL33+ MBV-treated macrophages are myogenic for progenitor cells.
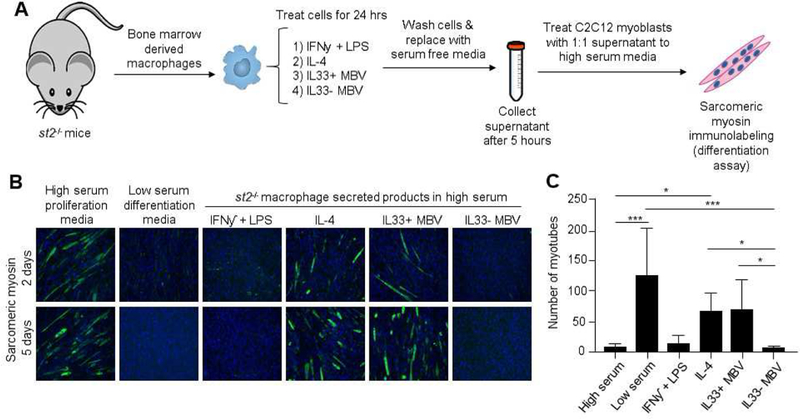
(A) Experimental design. C2C12 myoblasts were cultured to confluence and treated with high serum proliferation media, low serum differentiation media, or proliferation media supplemented with media conditioned by st2−/−macrophages stimulated with the indicated test articles (B) Cells were allowed to differentiate and were immunolabeled for sarcomeric myosin. (C) Quantification of myotube formation at day 5 (**indicates p < 0.001; * indicates p < 0.05, error bars represent SEM, n=3 biological replicates analyzed in triplicate).
4. Discussion
In the present study, we provide the first evidence that IL-33, stably stored within the ECM and protected from proteolytic cleavage by incorporation into MBV, is a potent mediator of M2-like macrophage activation through an uncharacterized, non-canonical ST2-independent pathway. Specifically, we show that IL33+ MBV, but not IL33- MBV, direct st2−/− macrophage activation towards a reparative, pro-remodeling M2-like phenotype. IL33- MBV instead activated a pro-inflammatory M1-like phenotype. The ability of IL33+ MBV, but not IL33- MBV to upregulate Arginase1 expression, is ST2 and Stat6-independent. In total, our studies establish that IL-33 encapsulated within lipid-membrane nanovesicles mediates the differentiation of myeloid cells away from pro-inflammatory subsets independent of the classical ST2 receptor. Of potential clinical significance, IL-33 was also shown to be present in MBV isolated from commercially manufactured ECM bioscaffolds including those used in the 13 patient cohort study of VML(6, 7). Biologic scaffold materials composed of ECM and configured as surgical meshes, powders, and hydrogels have been successfully used in a variety of tissue engineering/regenerative medicine and general surgery applications both in preclinical studies and in clinical applications(8). If not chemically crosslinked, such scaffolds are readily degradable and associated with favorable tissue remodeling properties including angiogenesis, stem cell recruitment, and modulation of macrophage phenotype toward an anti-inflammatory effector cell type(8). However, the biologic mechanisms by which these events and functional tissue restoration are mediated are largely unknown. Separate from the mechanical and structural functions of ECM-scaffolds, the molecular components of ECM, including growth factors, cytokines, and matricryptic peptides, have been extensively investigated for their ability to confer bioactivity(44). However, there is legitimate controversy concerning not only the relevant importance of these biochemical and structural components, but also the potential influence that the decellularization, disinfection, and sterilization methods used during the manufacturing process have on their biological activity. We have recently shown that MBV are a potent bioactive component of ECM scaffolds, and that these MBV shield their cargo from degradation and denaturation during the ECM-scaffold manufacturing process(22). Given their location within the ECM of soft tissue, and their incorporation into the collagen network of the matrix itself, MBV can be separated from the matrix only after enzymatic digestion of the ECM-scaffold material, similar to the functional activity attributed to the degradation of implanted biologic scaffolds used in clinical applications. These findings suggest that MBV are only available for cellular uptake during matrix remodeling events, such as those that occur during normal physiologic processes like wound healing and mechanical stress, or during degradation initiated by the host response to implanted biologic scaffolds. The early macrophage response to biomaterials has been shown to be a critical and predictive determinant of downstream outcomes(11, 45). When properly prepared (i.e. thoroughly decellularized and not chemically crosslinked), bioscaffolds composed of mammalian ECM have been shown to promote a transition in macrophage behavior from a pro-inflammatory to a regulatory/anti-inflammatory phenotype, which in turn has been associated with constructive and functional tissue repair(8). We have previously shown that MBV are critical for the activation of macrophages towards a M2-like, pro-remodeling phenotype(22, 23). Although the mechanism(s) by which MBV promote this phenotypic transition are poorly understood, results from the present study suggest that MBV-associated IL-33 plays a critical role in this phenotypic transition. Furthermore, results from the present study show that the secretome associated with IL33+ MBV-stimulated macrophages is myogenic for skeletal muscle myoblasts. However, future verification of these finding using appropriate in-vivo animal models will be required to determine the effect of modulating IL-33 in ECM scaffolds upon functional remodeling after volumetric muscle loss.
Conceptually, the discovery that MBV embedded within ECM-scaffolds are a rich source of extra-nuclear IL-33, is novel in itself and has not been previously reported. Full length IL-33 is generated as an approximately 32 kDa protein with an IL-1 family C-terminal cytokine domain and an N-terminal domain consisting of a non-classical nuclear localization sequence and a DNA binding domain, as well as a chromatin-binding domain(46, 47). Given this N-terminus and the lack of a secretion signaling sequence, current dogma is that IL-33 is found only in the nucleus and released upon necrotic cell death(48). Thus, IL-33 is defined as an alarmin, or an endogenous molecule that alerts the immune system to cell injury and tissue damage. IL-33 released from necrotic cells has been shown to activate the canonical ST2 signaling pathway in a variety of ST2+ immune cells to drive broad immune responses that include Type 1, Type 2, and regulatory immune responses(24, 49). Full length IL-33 is fully capable of activating ST2-dependent signaling, but can be converted by inflammatory proteases to an 18 kDa cytokine that is a more potent activator of ST2 signaling(50, 51). The ST2-dependent functions of IL-33 are negatively regulated by the apoptotic caspases-3 and −7, which cleave IL-33 in the cytokine domain, and a soluble and antagonistic form of ST2(24). More recently, it was revealed that soluble IL-33 released into the extracellular environment is rapidly oxidized to inactivate the cytokine domain and limit its range and duration of action(52). Several studies have shown that cytokines that lack a signal peptide sequence can be packaged and secreted within extracellular vesicles, including the IL-1 family members IL-1β, IL-1α, and IL-18(53–56). IL-33 has been reported to be present in exosomes released from Epstein-Barr Virus (EBV)-infected cells as a strategy to reduce innate immunity activation(57). Furthermore, it was previously demonstrated that IL-33 can efflux from the nucleus where it is packaged within membrane vesicles in the cytosol(58). In the present study, we provide the first evidence that IL-33 can also be stably compartmentalized within the extracellular matrix in a wide variety of tissues through incorporation into the lumen of MBV thereby preventing IL-33 degradation and inactivation.
The findings of the present study show that MBV-mediated delivery of IL-33 to immune cells can upregulate Arginase-1 expression independent of Stat6 phosphorylation. Previous reports have shown that epigenetic regulation of histone modification, including acetylation/deacetylation and chromatin remodeling are mechanistically important for activation of the M2 macrophage phenotype(59, 60). More specifically, Arg-1 expression in macrophages has been shown to be upregulated in a Stat6-independent manner via the AP-1 binding site in the Arg-1 promoter(61). Our results showing that IL-33 stored within the lumen of MBV is the full length (32kDa) form of the cytokine, which likely retains its N-terminal chromatin binding motif, offers some insight into the molecular mechanism by which MBV-delivered IL-33 regulates gene expression. Although it has been reported that IL-33 binds chromatin and may act as a transcriptional repressor(62), to date, no bona fide IL-33 regulated genes have been identified. Our data suggest that IL-33 delivered to cells via MBV may translocate to the nucleus and bind directly to genomic DNA to modulate gene expression. Conversely, we show that IL-33 deficient MBV support M1-like macrophage activation, which suggests that the absence of IL-33 within MBV may either drive uncharacterized gene expression or that IL-33 may repress genes leading to M1 macrophage activation after MBV uptake. Future studies aimed at a rigorous molecular characterization of IL-33 regulated genes will be required to fully explore the biology of not only MBV, but also IL-33, and will yield important information on their potential use in not only regenerative medicine applications, but also in the mitigation of adverse tissue responses; for example, to promote allograft survival following heart transplant. Given our finding that MBV can mediate receptor-free delivery of protein cargo to immune cells and orchestrate their function, it is easy to appreciate how the present study may support the development of tailored vesicles (e.g., MBV, exosomes, or engineered liposomes) as next generation immunomodulatory therapeutics. Separately, the use of ECM-based therapies can now be examined by new directions and will help guide the design of next generation ECM-based materials.
Supplementary Material
Highlights.
Matrix bound nanovesicles (MBV) isolated from ECM bioscaffolds are a rich and stable source of interleukin-33 (IL-33).
IL-33 is stored within the lumen of MBV and protected from proteolytic degradation.
IL-33+ MBV bypass the classical IL-33 receptor signaling pathway after myeloid cell uptake of MBV to direct macrophage differentiation into the reparative M2 subset via a previously undescribed non-canonical ST2-independent mechanism.
IL33+ MBV-induced macrophage secretome promotes myoblast differentiation.
Acknowledgements:
We thank Anna Lucas for her technical assistance and animal husbandry support. This work was supported by the following grants: NIH R01AR073527 to S.F.B and H.R.T; and R01HL122489 to H.R.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Statement:
The authors declare that they have no competing interests.
References:
- 1.Tidball JG, Mechanisms of muscle injury, repair, and regeneration. Compr Physiol 1, 2029–2062 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Goldman SM, Corona BT, Co-delivery of micronized urinary bladder matrix damps regenerative capacity of minced muscle grafts in the treatment of volumetric muscle loss injuries. PloS one 12, e0186593 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corona BT, Rivera JC, Wenke JC, Greising SM, Tacrolimus as an adjunct to autologous minced muscle grafts for the repair of a volumetric muscle loss injury. Journal of experimental orthopaedics 4, 36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herberts CA, Kwa MS, Hermsen HP, Risk factors in the development of stem cell therapy. Journal of translational medicine 9, 29 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarrafian TL, Bodine SC, Murphy B, Grayson JK, Stover SM, Extracellular matrix scaffolds for treatment of large volume muscle injuries: A review. Veterinary Surgery 47, 524–535 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Sicari BM et al. , An acellular biologic scaffold promotes skeletal muscle formation in mice and humans with volumetric muscle loss. Sci Transl Med 6, 234ra258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dziki J et al. , An acellular biologic scaffold treatment for volumetric muscle loss: results of a 13-patient cohort study. NPJ Regenerative medicine 1, 16008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussey GS, Dziki JL, Badylak SF, Extracellular matrix-based materials for regenerative medicine. Nature Reviews Materials, (2018).
- 9.Badylak SF, The extracellular matrix as a biologic scaffold material. Biomaterials 28, 3587–3593 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Julier Z, Park AJ, Briquez PS, Martino MM, Promoting tissue regeneration by modulating the immune system. Acta biomaterialia 53, 13–28 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Brown BN et al. , Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater 8, 978–987 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadtler K et al. , Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 352, 366–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadtler K et al. , Proteomic composition and immunomodulatory properties of urinary bladder matrix scaffolds in homeostasis and injury. Semin Immunol, (2017). [DOI] [PMC free article] [PubMed]
- 14.Huleihel L et al. , in Seminars in immunology (Elsevier, 2017), vol. 29, pp. 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sicari BM et al. , The promotion of a constructive macrophage phenotype by solubilized extracellular matrix. Biomaterials 35, 8605–8612 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Tidball JG, Wehling-Henricks M, Shifts in macrophage cytokine production drive muscle fibrosis. Nature medicine 21, 665 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Arnold L et al. , Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. Journal of Experimental Medicine 204, 1057–1069 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Troidl C et al. , Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. Journal of cellular and molecular medicine 13, 3485–3496 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nahrendorf M et al. , The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. Journal of Experimental Medicine 204, 3037–3047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keane TJ et al. , Restoring Mucosal Barrier Function and Modifying Macrophage Phenotype with an Extracellular Matrix Hydrogel: Potential Therapy for Ulcerative Colitis. J Crohns Colitis 11, 360–368 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Martinez FO, Gordon S, The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 6, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huleihel L et al. , Matrix-bound nanovesicles within ECM bioscaffolds. Sci Adv 2, e1600502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huleihel L et al. , Matrix bound nanovesicles recapitulate extracellular matrix effects on macrophage phenotype. Tissue engineering. Part A, (2017). [DOI] [PMC free article] [PubMed]
- 24.Liew FY, Girard J-P, Turnquist HR, Interleukin-33 in health and disease. Nature Reviews Immunology 16, 676–689 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Kuswanto W et al. , Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrels B et al. , IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal 10, eaan8355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oboki K et al. , IL-33 is a crucial amplifier of innate rather than acquired immunity. Proceedings of the National Academy of Sciences 107, 18581–18586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN, T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. Journal of Experimental Medicine 191, 1069–1076 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira AC et al. , Evaluation of small intestine grafts decellularization methods for corneal tissue engineering. PloS one 8, e66538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reing JE et al. , The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials 31, 8626–8633 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mase VJ et al. , Clinical application of an acellular biologic scaffold for surgical repair of a large, traumatic quadriceps femoris muscle defect. Orthopedics 33, (2010). [DOI] [PubMed] [Google Scholar]
- 32.Badylak SF, Lantz GC, Coffey A, Geddes LA, Small intestinal submucosa as a large diameter vascular graft in the dog. J Surg Res 47, 74–80 (1989). [DOI] [PubMed] [Google Scholar]
- 33.Wainwright JM et al. , Preparation of cardiac extracellular matrix from an intact porcine heart. Tissue Engineering Part C: Methods 16, 525–532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Böing AN et al. , Single-step isolation of extracellular vesicles by size-exclusion chromatography. Journal of extracellular vesicles 3, 23430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diaz G, Wolfe LM, Kruh-Garcia NA, Dobos KM, Changes in the membrane-associated proteins of exosomes released from human macrophages after Mycobacterium tuberculosis infection. Scientific reports 6, 37975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jong OG, Balkom BW, Gremmels H, Verhaar MC, Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. Journal of cellular and molecular medicine 20, 342–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huleihel L et al. , Matrix-Bound Nanovesicles Recapitulate Extracellular Matrix Effects on Macrophage Phenotype. Tissue Eng Part A 23, 1283–1294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crapo PM, Gilbert TW, Badylak SF, An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Londono R, Badylak SF, Biologic scaffolds for regenerative medicine: mechanisms of in vivo remodeling. Annals of biomedical engineering 43, 577–592 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Lefrançais E et al. , IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proceedings of the National Academy of Sciences 109, 1673–1678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cayrol C et al. , Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nature immunology 19, 375 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Ruffell D et al. , A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proceedings of the National Academy of Sciences 106, 17475–17480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG, IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. The Journal of Immunology, 1103180 (2012). [DOI] [PMC free article] [PubMed]
- 44.Hussey GS, Keane TJ, Badylak SF, The extracellular matrix of the gastrointestinal tract: a regenerative medicine platform. Nature Reviews Gastroenterology and Hepatology 14, 540 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF, Macrophage polarization: an opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials 33, 3792–3802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lingel A et al. , Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors— insight into heterotrimeric IL-1 signaling complexes. Structure 17, 1398–1410 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baekkevold ES et al. , Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. The American journal of pathology 163, 69–79 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pichery M et al. , Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33–LacZ gene trap reporter strain. The Journal of Immunology 188, 3488–3495 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Kakkar R, Lee RT, The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nature reviews. Drug discovery 7, 827–840 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefrancais E et al. , Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proceedings of the National Academy of Sciences of the United States of America 111, 15502–15507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lefrancais E et al. , IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proceedings of the National Academy of Sciences of the United States of America 109, 1673–1678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen ES et al. , Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nature communications 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacKenzie A et al. , Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15, 825–835 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Berda-Haddad Y et al. , Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proceedings of the National Academy of Sciences of the United States of America 108, 20684–20689 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gulinelli S et al. , IL-18 associates to microvesicles shed from human macrophages by a LPS/TLR-4 independent mechanism in response to P2X receptor stimulation. European journal of immunology 42, 3334–3345 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa H, Thomas HJ, Schooley K, Born TL, Native IL-32 is released from intestinal epithelial cells via a non-classical secretory pathway as a membrane-associated protein. Cytokine 53, 74–83 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Ansari MA et al. , Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol 87, 8606–8623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kakkar R, Hei H, Dobner S, Lee RT, Interleukin 33 as a mechanically responsive cytokine secreted by living cells. Journal of Biological Chemistry 287, 6941–6948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishii M et al. , Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapellos TS, Iqbal AJ, Epigenetic Control of Macrophage Polarisation and Soluble Mediator Gene Expression during Inflammation. Mediators Inflamm 2016, 6591703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharda DR et al. , Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol 187, 2181–2192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carriere V et al. , IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proceedings of the National Academy of Sciences 104, 282–287 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.