

Abstract
Objective
Obesity and type-2 diabetes (T2D) are metabolic diseases that represent a critical health problem worldwide. Metabolic disease is differentially associated with fat distribution, while visceral white adipose tissue (VAT) is particularly prone to obesity-associated inflammation. Next to their canonical function of immune suppression, regulatory T cells (Tregs) are key in controlling adipose tissue homeostasis. Towards understanding the molecular underpinnings of metabolic disease, we focus on how environmental-metabolic stimuli impinge on the functional interplay between Tregs and adipose tissue. Here, cold exposure or beta3-adrenergic signaling are a promising tool to increase energy expenditure by activating brown adipose tissue, as well as by reducing local inflammation within fat depots by supporting immunosuppressive Tregs. However, in humans, the underlying mechanisms that enable the environmental-immune crosstalk in the periphery and in the respective tissue remain currently unknown.
Methods
We used combinatorial approaches of next generation humanized mouse models and in vitro and in vivo experiments together with beta3-adrenergic stimulation to dissect the underlying mechanisms of human Treg induction exposed to environmental stimuli such as cold. To test the translational relevance of our findings, we analyzed samples from the FREECE study in which human subjects were exposed to individualized cooling protocols. Samples were analyzed ex vivo and after in vitro Treg induction using qRT-PCR, immunofluorescence, as well as with multicolor flow cytometry and cell sorting.
Results
In vivo application of the beta3-adrenergic receptor agonist mirabegron in humanized mice induced thermogenesis and improved the Treg induction capacity of naïve T cells isolated from these animals. Using samples from the human FREECE study, we demonstrate that a short-term cold stimulus supports human Treg induction in vitro and in vivo. Mechanistically, we identify BORCS6 encoding the Ragulator-interacting protein C17orf59 to be significantly induced in human CD4+ T cells upon short-term cold exposure. Strong mTOR signaling is known to limit successful Treg induction and thus likely by interfering with mTOR activation at lysosomal surfaces, C17orf59 improves the Treg induction capacity of human naïve T cells upon cold exposure.
Conclusions
These novel insights into the molecular underpinnings of human Treg induction suggest an important role of Tregs in linking environmental stimuli with adipose tissue function and metabolic diseases. Moreover, these discoveries shed new light on potential approaches towards tailored anti-inflammatory concepts that support human adipose tissue homeostasis by enabling Tregs.
Keywords: Regulatory T cell, Human adipose tissue, Beta3-adrenergic stimulation, Mirabegron, Immunometabolism, Humanized mice
Highlights
-
•
Beta3-adrenergic stimulation enhances human Tregs in humanized mice.
-
•
Short-term cold stimulation increases human Treg induction in vitro and in vivo.
-
•
Short-term cold exposure elevates human Treg signatures genes.
-
•
Short-term cold induces BORCS6 encoding C17orf59 in human CD4+T cells.
-
•
C17orf59 limits mTOR signaling and thereby supports human Treg induction.
Abbreviations
- ADRB3
beta3-adrenergic receptor
- AT
adipose tissue
- BAT
brown adipose tissue
- BMI
body mass index
- BORCS6
BLOC-1 related complex subunit 6
- Cidea
cell death-inducing DFFA-like effector a
- CTLA-4
cytotoxic T lymphocyte associated protein 4
- FOXP3
forkhead box protein 3
- FREECE
FTO-Genotype on Resting Energy Expenditure after defined Cold Exposure
- i.p.
intraperitoneal
- IPEX
immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome
- LAG3
lymphocyte activating gene 3
- mTOR
mechanistic target of rapamycin
- NSG
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ
- NST
non-shivering thermogenesis
- PBMC
peripheral blood mononuclear cell
- PI3K
phosphoinositide 3 kinase
- PPARγ
Peroxisome proliferator-activated receptor gamma
- SAT
subcutaneous adipose tissue
- T2D
Type 2 diabetes
- TCR
T cell receptor
- UCP1
Uncoupling protein 1
- Treg
regulatory T cell
- VAT
omental visceral adipose tissue
1. Introduction
Obesity and Type 2 diabetes (T2D) are metabolic diseases that represent critical health threats to modern societies. Metabolic disease is differentially associated with local fat depots, with visceral white adipose tissue (VAT) being especially prone to obesity-associated inflammation [1].
In addition to their critical role of immune suppression, regulatory T cells (Tregs) are key in controlling tissue homeostasis and function including local fat depots. Tregs are characterized by the expression of CD4, CD25, and the transcription factor FOXP3, which functions as the master regulator for their function and development [2], [3]. Mutations in the Foxp3 gene have deleterious consequences, leading to autoimmune and severe inflammatory phenotypes in both mice (scurfy mice) and humans (IPEX – immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome), highlighting the crucial role of Foxp3 for Treg function [4], [5], [6]. In humans, activated T cells can express low amounts of FOXP3 [7], [8], while immunosuppressive Tregs are characterized by high expression of FOXP3 together with low expression of CD127 [9], [10]. Here, FOXP3 confers suppressive capacity by ensuring the expression of immunosuppressive molecules such as CTLA-4, LAG3, and IL-10 [11].
Tregs can be induced in the peripheral immune system, a process referred to as Treg conversion or induction [12]. Efficient Treg induction from naïve CD4+ T cells can be achieved using antigenic stimulation under so-called subimmunogenic conditions avoiding the activation of antigen-presenting cells and T cells [13], [14]. In contrast, immunogenic stimulation and strong costimulatory signals activate the phosphoinositide 3 kinase (PI3K)/Akt/mechanistic target of the rapamycin (mTOR) pathway, thereby interfering with Treg induction [12], [13], [15], [16], [17], [18], [19].
In order to maintain local homeostasis, Tregs take residence in tissues such as VAT, followed by tissue-specific adaptations of their functions, e.g. by expression of the transcription factor Peroxisome proliferator-activated receptor gamma (PPARγ) [20]. Adipocytes are surrounded by several immune cell types. Immune cell type, number, and function dramatically change in VAT following overnutrition. Fat-residing Tregs control local inflammatory processes. Specifically, upon hypercaloric challenge Treg frequencies in VAT are severely reduced accompanied by increased inflammation [21].
Importantly, Tregs possess the ability to respond to environmental and metabolic signals. It is becoming increasingly evident that the disruption of these interactions between immune and environmental-metabolic responses critically contributes to the emergence of metabolic diseases.
Towards understanding the molecular underpinnings of these metabolic diseases, we have recently provided an example of how environmental-metabolic stimuli impinge on the functional interplay between Tregs and adipose tissue [22]. Specifically, and in line with a critical role of cold exposure in interfering with metabolic impairments in obesity, we recently showed that short-term cold exposure or beta3-adrenergic stimulation induce murine Tregs in vitro as well as in vivo [22].
In line with these observations, another study had suggested an impact of cold exposure on brown adipose tissue (BAT) Tregs [23]. Mechanistically, using CD4+ T cell proteomes, we uncovered Treg induction and higher protein expression of Foxp3 regulatory networks following cold exposure or beta3-adrenergic stimulation [22]. Using loss- and gain-of-function studies we highlighted that a T cell-specific Stat6/Pten axis adjusts Foxp3+Treg induction and adipose tissue function according to the degree of sympathetic tone and environmental temperature [22].
In contrast to white adipose tissue, the main function of which is the storage of lipids, BAT critically contributes to non-shivering thermogenesis (NST), burns energy, and dissipates heat. This is facilitated by the expression of uncoupling protein 1 (UCP1) that uncouples mitochondrial respiration from ATP synthesis [1]. Upon exposure to cold or beta3-adrenergic stimulation, white adipocytes can undergo a process referred to as browning that induces the expression of UCP1 in white adipocytes [24].
In humans, recent studies have uncovered cold-inducible BAT in adults [25], [26]. Stimulating BAT energy expenditure through activation of the beta3-adrenergic receptor (ADRB3), which is expressed on human adipocytes and other tissues including human peripheral blood mononuclear cells [26], [27] was also achieved by the ADRB3 agonist mirabegron [28], [29]. Moreover, cold exposure was shown to provide beneficial effects on whole-body and skeletal muscle insulin sensitivity in patients with obesity and T2D [30]. Accordingly, mirabegron was reported to stimulate human BAT thermogenesis and may therefore be a promising future treatment option for metabolic disease [31].
Despite these mechanistic insights in Treg adipose crosstalk in the murine setting together with research efforts focusing on metabolic readouts upon cold exposure in humans, the impact of human Tregs in guiding the functional immunometabolic interplay in response to cold exposure in vivo remains currently unknown.
To fill this knowledge gap, here we studied human Treg induction following beta3-adrenergic stimulation in next generation humanized mice as well as upon short-term cold exposure in humans. Specifically, we report that a short-term cold stimulus improves human Treg induction in vitro and in vivo. Mechanistically, we find that BLOC-1 related complex subunit 6 (BORCS6) encoding the Ragulator-interacting protein C17orf59 is significantly enhanced in human CD4+ T cells upon short-term cold exposure. C17orf59 was demonstrated to interfere with mTORC1 activity [32], thereby likely supporting enhanced Treg induction.
2. Material and methods
2.1. Mice
Humanized mice lack murine MHC class II and transgenically express human HLA-DQ8 or HLA-DR4 (#026561 NOD.Cg-PrkdcscidH2-Ab1tm1DoiIl2rgtm1WjlTg (HLA-DQA1,HLA-DQB1)1Dv/SzJ and #017637 NOD.Cg-PrkdcscidIl2rgtm1WjlH2-Ab1tm1DoiTg (HLA-DRB1)31Dmz/SzJ, Jackson Laboratories, (herein abbreviated as NSG)) were reconstituted with 107 PBMCs from healthy subjects. Mice were maintained group-housed on a 12 h/12 h light dark cycle at 25 °C under specific pathogen free (SPF) conditions and had ad libitum access to food (Altromin, #1314, Lage, Germany) and water. Mice were randomized to test groups. For in vivo beta3-adrenergic stimulation, mice were injected i.p. with 1 mg/kg mirabegron (Selleckchem, CAS 223673-61-8) on three consecutive days two weeks after reconstitution. 0.9% NaCl was used as vehicle control. No animals were excluded due to illness or outlier results; therefore, no exclusion determination was required. The investigators were not blinded to group allocation or to the assessment of experimental end points. All animal care was executed according to guidelines established by the Institutional Animal Committees. Ethical approval for all mouse experimentations has been received by corresponding local animal welfare authorities (approval number #ROB-55.2-2532.Vet_02-17-130, District Government of Upper Bavaria, Germany).
2.2. Human subjects and samples
Venous blood was collected from healthy adults (n = 2, females, age 25) who consented to the Munich Bioresource project (approval number #5049/11, Technische Universität München, Munich, Germany) in heparinized blood collection tubes (BD Vacutainer Blood Collection Tubes, Becton Dickinson).
For RT-qPCR analyses of human paired samples of subcutaneous (SAT) and omental AT (VAT), samples were obtained from lean individuals (n = 6, mean BMI: 23.2 kg/m2). Phenotypic characterization of the study participants and AT sample collection during open or laparoscopic abdominal surgery was performed as previously described [33]. The study was approved by the Ethics Committee of the University of Leipzig (approval number #159-12-21052012 and #017-12-23012012) and performed in accordance to the declaration of Helsinki.
For in vivo cold exposure in the FREECE study, subjects were recruited at the Institute for Nutritional Medicine, Technical University of Munich, Munich, Germany. The study protocol was approved by the ethical committee of the Faculty of Medicine of the Technical University of Munich (approval number 236/16S and 113/19S), and registered at the German Clinical Trials Register (ID: DRKS00010489). This ongoing study examines the effects of short-term mild cold exposure on thermogenesis and metabolism of healthy individuals. Inclusion criteria were healthy non-smoking adults. Exclusion criteria were severe diseases (such as diagnosed hypertension or established diabetes), pregnancy or lactation, weight gain or loss of more than 3% of body weight within 3 months and more than 10 h rigorous exercise per week. The study was conducted at both locations of the Institute for Nutritional Medicine in Munich and in Freising, Germany.
For needle aspiration fat biopsy, participants were invited one week prior to the examinations. Subjects were informed in detail about the procedure and written informed consent was obtained. Blood was drawn to test for the prothrombin time, and the abdomen was examined for sufficient SAT by the study physicians. Subjects were studied in the morning after a 10-hour overnight fast, and subjects were not allowed to exercise 24 h prior to the study nor to drink any caffeinated beverages or alcohol. Upon arrival, height was measured using a stadiometer (Seca, Hamburg, Germany) to the nearest 0.1 cm. Body weight and body composition were measured using the TANITA Body Composition Analyser Type BC-418 MA (Tanita Europe GmbH, Sindelfingen, Germany). Measurements were performed barefoot, in underwear, and after voiding the urinary bladder. Nine wireless iButtons were attached to the skin to measure skin temperature. Core temperature was monitored by a zero-heat flux thermometer, which was attached to the forehead.
Resting energy expenditure was determined via indirect calorimetry for 30 min under thermoneutral conditions using a ventilated hood system (COSMED Quark RMR, Fridolfing, Germany). The conversion of the measurements of VO2 (consumption of oxygen (ml/min)) and VCO2 (production of CO2 (ml/min)) to energy expenditure [kcal/day] was done by application of the shortened Weir equation [34].
For the individualized cooling protocol, subjects lay between two water-perfused mattresses in a supine position. Water temperature was gradually decreased until subjects reported shivering. Subsequently, water temperature was increased for 2 °C to prevent muscle shivering. Subjects remained for 2 h between the mattresses at this temperature and another 30 min for the second indirect calorimetry (in total 150 min).
Blood was drawn and fat biopsy samples were taken directly after each indirect calorimetry. To obtain 1–2 g of SAT, the paraumbilical area was treated with lidocaine and tissue was obtained by needle biopsy. 0.3–0.8 g AT were used for flow cytometric analyses.
Where indicated, subjects were grouped according to their BMI in lean (BMI <25 kg/m2), overweight (25 kg/m2 ≤ BMI < 30 kg/m2) and obese (BMI ≥ 30 kg/m2).
Immediately after each indirect calorimetry, blood was taken (EDTA KE monovettes, Nümbrecht, Sarstedt) and centrifuged at 2,500 g for 10 min at room temperature. An enzyme-linked immunosorbent assay was used to determine plasma leptin levels (Duoset ELISA human, R&D, Wiesbaden, Germany).
For human studies, written informed consent was obtained from all subjects and experiments were carried out in accordance with the Declaration of Helsinki.
For subject biometrics, please see Table 1.
Table 1.
Subject biometrics analyzed from the FREECE study.
| FREECE | subjects | range |
|---|---|---|
| n (males/females) | 29 (11/18) | |
| age males (mean ± SD) | 29.4 ± 8.9 | (22–51) |
| age females (mean ± SD) | 30.1 ± 8.3 | (20–52) |
| BMI males (mean ± SD) | 30.2 ± 6.8 | (21.5–40.6) |
| BMI females (mean ± SD) | 32.3 ± 7.2 | (19.7–47.6) |
2.3. Isolation of CD4+ T cells from murine lymphoid organs
Lymph nodes and spleens were mashed through 70 μm cell strainers in HBSS+ (supplemented with 5% FCS and 10 mM HEPES) and further processed for flow cytometry.
2.4. Human T cell isolation from blood
Venous blood was collected in heparinized tubes. Peripheral blood mononuclear cells (PBMCs) were isolated by density centrifugation over Ficoll-Paque PLUS (GE Healthcare). Human CD4+ T cells were isolated from fresh PBMCs via MACS enrichment with CD4+ microbeads following the manufacturer's protocol. For cytospins, untouched EasySep human CD4+ T cell enrichment kit (Stem Cell) was used according to the manufacturer's instructions.
2.5. Isolation of CD4+ T cells from human SAT
SAT biopsies were collected by needle aspiration, washed intensively with 0.9% NaCl to remove blood contaminants, stored in PBS supplemented with 0.5% BSA and digested with collagenase II solution (4 mg/ml collagenase II, Sigma Aldrich, EC#3.4.24.3 and 10 mM CaCl2) for 5–9 min at 37 °C in a water bath, slightly shaking. Cell suspensions were passed through a 200 μm nylon mesh and centrifuged (380×g, 5 min, 4 °C) to separate the stromal vascular fraction from adipocytes. Pelleted cells were re-suspended in HBSS+ and stained for flow cytometric analysis.
2.6. Cell staining for flow cytometry
Following antibodies were used for flow cytometry (reactivity, fluorochrome, clone, manufacturer): CD11B, Pacific Blue, ICRF44, BioLegend; CD127, PE-Cy7, A019D5, BioLegend; CD14, Pacific Blue, HCD14, BioLegend; CD19, Pacific Blue, HIB19, BioLegend; CD25, APC, 2A3, BD Biosciences; CD3, PerCP-Cy5.5, HIT3a, BioLegend; CD3, Alexa Fluor 700, HI30, BioLegend; CD4, V500, RPA-T4, BD Biosciences; CD62L, PE, DREG-56, BioLegend; CD45RA, FITC, HI100, BioLegend; CD45RO, APC-H7, UCHL1, BD Biosciences; CD8A, Pacific Blue, RPA-T8, BioLegend; FOXP3, Alexa Fluor 700, PCH101, eBioscience; FOXP3, PE, 236A/E7, eBioscience; KI67, APC, 16A8, BioLegend; KI67, Brilliant Violet 605, 16A8, BioLegend. Unspecific binding of antibodies was prevented by incubation of cell suspensions with Fc-Block (Human TruStain FcX, BioLegend, 1:20) for 5 min at RT, followed by FACS staining for 20 min at RT in the dark. Cells were passed through a 40 μm cell strainer (NeoLab) to remove large debris.
To detect intracellular protein expression, T cells were fixed and permeabilized using the Foxp3 Staining Buffer Set (eBioscience) after surface staining as recommended by the manufacturer.
Cells were acquired on BD FACSAriaIII flow cytometer using FACSDiva software V6.1.3 or V8.0.1 with optimal compensation and gain settings based on unstained and single-color stained samples. Doublets were excluded based on SSC-A vs. SSC-W and FSC-A vs. FSC-W plots. Live cell populations were gated on the basis of cell side and forward scatter and the exclusion of cells positive for Sytox Blue (Life Technologies) or Fixable Viability Dye eFluor450 (eBioscience). Samples were analyzed using FlowJo software V7.6.1 or V10.4.2 (TreeStar Inc., OR).
2.7. In vitro Treg induction assays
96 well U-bottom plates (Greiner bio-one) were pre-coated with 5 μg/ml anti-CD3e (UCHT1, BioLegend) and 15 μg/ml anti-CD28 (CD28.2, BioLegend) in 0.1 M sodium bicarbonate buffer pH = 8.2 for 1 h at 37 °C, followed by 4 °C. Cells were sorted with a FACSAriaIII (BD) cell sorter for purity. Human naïve live CD8a−CD11b−CD14−CD19-CD3+CD4+CD45RA+CD45RO−CD127+CD25- T cells were cultured in x-Vivo15 Medium supplemented with 2 mM glutamine, penicillin (50 U/ml), streptomycin (50 μg/ml), and 5% (vol/vol) heat-inactivated human AB serum (Invitrogen) in the presence of 100 U/ml human recombinant IL-2 (Peprotech) in an humidified incubator at 37 °C with 5% CO2. TCR stimulation was limited to 18 h by transferring cells into uncoated wells. Cells were cultured for additional 36 h without further TCR stimulation.
2.8. Quantitative analysis of mRNA abundance
Total RNA was extracted from 300,000 human MACS-purified CD4+ T cell populations using QIAzol Lysis Reagent/miRNeasy Micro Kit according to the manufacturer's instructions. 200 ng total RNA were converted to first strand cDNA using iScript™Advanced cDNA Synthesis Kit (Bio-Rad) in a peqStar2X thermal cycler (Peqlab). Quantitative real-time PCR (RT-qPCR) analyses were performed using SsoFast Evagreen Supermix (Bio-Rad) and gene-specific primer sets on a CFX96 Touch real time system (Bio-Rad). HISTONE RNA levels were used for normalization of target gene expression levels. The following primers were used for RT-qPCR analyses (mRNA target, forward primer (5′-3′), reverse primer (5′-3′), manufacturer): HISTONE, ACTGGCTACAAAAGCCG, ACTTGCCTCCTGCAAAGCAC, Sigma Aldrich; Hs_CD4, TTGCTTCTGGTGCTGCAACT, CAGCGCGATCATTCAGCTTG, Sigma Aldrich; Hs_FOXP3, order nr Hs_Foxp3_1_SG, Qiagen; Hs_IKZF2, GTGACGTCTGTGGCATGGT, ACCCACAGAATGGGTCCTGA, Sigma Aldrich; Mm_Cidea, TCAGACCTTAAGGGACAACACGCA, TTCTTTGGTTGCTTGCAGACTGGG, Sigma Aldrich; Mm_Ucp1, GGCCTCTACGACTCAGTCCA, TAAGCCGGCTGAGATCTTGT, Sigma Aldrich.
2.9. Immunofluorescence
CD4+ T cells, enriched with the Stem Cell Kit, were fixed with 4.5% Histofix (Carl Roth) for 10 min at room temperature, washed with HBSS and used for cytospins. Stainings were carried out using rabbit-anti-human antibodies for C17orf59/BORCS6 (MyBiosource) and mouse-anti-huCD3 (BioLegend), and as secondary antibodies goat-anti-rabbit AlexaFluor594 and goat-anti-mouse AlexaFluor488 (both Thermofisher). Negative control slides were incubated with secondary antibodies only. Nuclei were stained with Hoechst-33342 before final analysis by confocal Leitz microscopy (TCS-SP5II).
2.10. Quantification and statistical analysis
Data are presented as box-and-whisker plots with min and max values for data distribution or as percentage where appropriate or as summary plots for paired analyses. Student's paired t-test was used for treatment analyses and Student's unpaired t-test to compare independent groups. For mirabegron titration, one-way ANOVA with Dunnett's multiple comparison test was used and mirabegron treated groups vs. vehicle compared. Correlations were analyzed with either Pearson or nonparametric Spearman as indicated in the respective figures and/or figure legends. For all tests, a two-tailed p value of <0.05 was considered to be significant. Statistical significance is shown as * = p < 0.05; ** = p < 0.01; *** = p < 0.001. Analyses were performed using Prism v6.0.1 or v7.04 (GraphPad).
3. Results
3.1. Beta3-adrenergic stimulation induces human Tregs in humanized NSG mice
As a critical, accessible system permitting predictive in vivo immunology research, we recently made use of humanized NOD Scid Il2rg KO (NSG) mice and established human Treg induction experiments in vivo [17]. In order to dissect the translational relevance of beta3-adrenergic stimulation or cold exposure in supporting Treg induction [22], here we applied the human beta3-receptor agonist mirabegron mimicking cold exposure to humanized NSG mice in vivo.
To this end, humanized NSG mice received mirabegron application for 3 d (1 mg/kg, i.p.), which resulted in a significant enhancement of thermogenic genes including Ucp1 and Cidea in adipose tissue of humanized mice (Figure 1A). Of note, flow cytometric analyses revealed significantly increased frequencies of human CD4+CD127lowCD25high Tregs upon beta3-adrenergic stimulation in humanized mice (CD25hiCD127low [% of CD3+CD4+]: vehicle = 1.998 ± 0.5289 (n = 5) vs. mirabegron = 4.946 ± 1.006 (n = 5), p = 0.0319, Figure 1B, C).
Figure 1.

Beta3-adrenergic stimulation enhances Tregs in humanized mice. A) Gene expression analysis of thermogenic markers in murine VAT after vehicle or mirabegron treatment in vivo. B) Representative flow cytometric analysis of ex vivo Tregs in humanized mice after vehicle or mirabegron treatment in vivo. Tregs are gated as CD8A−CD11B−CD14−CD19−eF450−CD45+CD3+CD4+CD127lowCD25hi. C) Summary plot of (B) with 5 mice per group from two independent experiments. D) Representative flow cytometric analysis of in vitro induced Tregs isolated from humanized mice after in vivo treatment with vehicle or mirabegron. Tregs are gated as live CD3+CD4+CD127lowCD25hiFOXP3hi. E) Summary plot of (D). F) Dose-response titration of mirabegron administered during human in vitro Treg induction. Two independent experiments. * = p < 0.05, ** = p < 0.01, *** = p < 0.01, **** = p < 0.001; one-way ANOVA with mirabegron vs. vehicle.
Moreover, we used established protocols [15], [17], [19] to dissect human Treg induction potential in vitro. Accordingly, we demonstrated that purified naïve human CD4+ T cells from mirabegron-treated humanized NSG animals had significantly improved Treg-induction capacities in vitro (FOXP3hi [% of CD25hiCD127low]: vehicle = 14.18 ± 1.36 (n = 5) vs. mirabegron = 21.26 ± 1.235 (n = 5), p = 0.0049, Figure 1D, E).
3.2. Beta3-adrenergic stimulation enhances human Treg induction potential in vitro
In line with the observations obtained in humanized NSG mice, we used human naïve CD4+ T cells from healthy individuals to study the impact of beta3-adrenergic stimulation on human Treg induction in vitro. Media supplemented with low doses of mirabegron significantly enhanced human Treg induction in vitro (FOXP3hi [% of CD25hiCD127low]: vehicle = 46.68 ± 1.115 vs. mirabegron [0.0001 nM] = 53.12 ± 0.9243, p = 0.0013; [0.001 nM] = 56.9 ± 0.498, p < 0.0001; [0.1 nM] = 59.35 ± 0.6081, p < 0.0001; [1 nM] = 54.18 ± 0.47, p = 0.0001, Figure 1F).
3.3. Short-term cold exposure in vivo increases human Treg induction potential
Next, to understand the importance of cold exposure for human Treg induction in vivo we addressed T cell samples from the FREECE study cohort. Specifically, after a 10 h overnight fast, healthy individuals were subjected to an individualized short-term cold exposure protocol for 2.5 h in the absence of muscle shivering. Blood samples were collected prior and after individualized cold exposure (For subject biometrics please see Table 1). Highly pure human naïve CD4+ T cells were sort-purified before (T0) and after (T2) cold exposure in vivo to dissect Treg induction potential in vitro (staining examples are provided in Figure 2A).
Figure 2.

Short-term cold exposure in vivo improves human Treg induction in vitro. A) Representative flow cytometric analysis of in vitro induced Tregs before (T0) and after (T2) in vivo cold exposure. B) Paired analyses of (A). Each dot represents the mean of 3–4 replicates and an individual experiment performed with paired samples. C) Fold change of in vitro induced Tregs normalized to individual T0. * = p < 0.05, ** = p < 0.01, *** = p < 0.01, **** = p < 0.001; paired t-test.
Cold exposure significantly enhanced Treg induction in both male and female subjects while the effect was more prominent on naïve CD4+ T cells from female individuals (Figure 2B, left panel and Figure 2C, left panel, FOXP3hi [% of CD25hiCD127low]: males T0 = 41.93 ± 5.523 vs. T2 = 44.09 ± 5.689 (n = 9), p = 0.0457); Figure 2B, right panel and Figure 2C, right panel, FOXP3hi [% of CD25hiCD127low]: females T0 = 38.96 ± 3.116 vs. T2 = 47.58 ± 3.415 (n = 13), p < 0.0001). When female and male subjects of the FREECE study were sub-divided based on the presence or absence of obesity, it became clear that the Treg-enhancing impact of cold exposure was evident in lean, overweight and obese individuals when looking at Treg induction from naïve CD4+T cells (Supplemental Figure 1, FOXP3hi [% of CD25hiCD127low]: females lean+overweight: T0 = 44.14 ± 5.808 vs. T2 = 51.14 ± 4.752 (n = 5), p = 0.0110; females obese: T0 = 35.72 ± 3.332 vs. T2 = 45.35 ± 4.752 (n = 8), p = 0.0020); males lean+overweight: T0 = 35.96 ± 9.363 vs. T2 = 36.29 ± 9.131 (n = 4), p = ns; males obese: T0 = 46.69 ± 6.661 vs. T2 = 50.32 ± 6.678 (n = 5), p = 0.0491). These findings therefore suggest that from an immune-metabolic perspective, cold exposure could be used to positively impinge on Treg induction also in states of overweight and obesity. In addition, we found a negative correlation between Treg induction potential and plasma leptin levels of individuals (Supplemental Figure 2). These results are in line with studies indicating that leptin can exert pro-inflammatory characteristics that promote inflammation [35], [36], [37]. Accordingly, additional observations have demonstrated an inhibitory effect of leptin on Treg proliferation, thereby further supporting the pro-inflammatory role of leptin [38].
3.4. Short-term cold exposure in vivo enhances human Tregs ex vivo
Given the increase seen in human de novo Treg induction potential upon human cold exposure in vivo, especially in female individuals, we next assessed human Treg frequencies prior and post cold exposure ex vivo (Figure 3, staining examples are provided in Figure 3A).
Figure 3.

In vivo cold increases ex vivo Treg frequencies in human peripheral blood. A) Representative flow cytometric analysis of ex vivo Tregs in human peripheral blood before (T0) and after (T2) in vivo cold exposure. B) Paired analyses of (A). Each dot represents an individual experiment performed with paired samples. C) Fold change of (A) ex vivo Tregs normalized to individual T0. D) RT-qPCR analysis of human CD4+ T cells isolated before (T0) and after (T2) in vivo cold exposure. * = p < 0.05, ** = p < 0.01, *** = p < 0.001; paired t-test.
Short-term cold exposure resulted in a significant enhancement of human Tregs in peripheral blood, especially in female individuals (Figure 3B, C, FOXP3hiCD25hiCD127low [% of CD3+CD4+]: males T0 = 5.574 ± 0.3362 vs. T2 = 6.02 ± 0.3752 (n = 9), p = 0.0061; females T0 = 4.72 ± 0.3012 vs. T2 = 5.211 ± 0.3344 (n = 16), p < 0.0001). In Supplemental Figure 3 subjects were grouped according to their sex and BMI and as observed for the in vitro Treg induction depicted in Supplemental Figure 2, the positive effect of cold showed a trend towards increased ex vivo Treg frequencies in all sub-groups and was more pronounced in the obese sub-cohort (FOXP3highCD25highCD127low [% of CD3+CD4+] of T0 vs. T2: lean+overweight females: T0 = 3.74 ± 0.385 vs. T2 = 4.152 ± 0.339 (n = 6), p = 0.0705; obese females: T0 = 5.309 ± 0.2996 vs. T2 = 5.846 ± 0.3745 (n = 10), p = 0.0012; lean+overweight males: T0 = 5.52 ± 0.5368 vs. T2 = 6.013 ± 0.6048 (n = 4), p = 0.0532; obese males: T0 = 5.618 ± 0.4827 vs. T2 = 6.026 ± 0.5359 (n = 5), p = 0.09. Accordingly, the numerical increase in human Tregs post cold exposure was accompanied by a significantly increased abundance of Treg signature genes such as FOXP3 (Figure 3D, FOXP3 expression [15-ΔCq]: T0 = 9.056 ± 0.1014; vs. T2 = 9.324 ± 0.07054 (n = 5); p = 0.0495) or IKZF2 (Figure 3D, IKZF2 expression [15-ΔCq]: T0 = 7.812 ± 0.1387; vs. T2 = 8.952 ± 0.1559 (n = 5); p = 0.0030.
3.5. Short-term cold exposure directly impacts human adipose tissue Tregs
Given the critical role of adipose tissue Treg cells in safeguarding tissue homeostasis and function, we next asked whether a short-term cold exposure stimulus can impinge on adipose tissue Tregs. To this end, biopsies from subcutaneous adipose tissue (SAT) were taken prior and post cold exposure.
First, to validate the relevance of Treg analyses from SAT we investigated FOXP3/CD4 mRNA ratios in SAT vs. VAT from lean healthy individuals in the steady state. Here, we identified higher ratios of human FOXP3/CD4 mRNA in SAT when compared to their VAT (FOXP3/CD4 ratios in SAT = 0.6465 ± 0.02387 (n = 20) vs. VAT = 0.5464 ± 0.04857 (n = 11); p = 0.0462, Figure 4A). These findings are in line with the assumption that SAT is less prone towards obesity-induced inflammation when compared to VAT [1].
Figure 4.
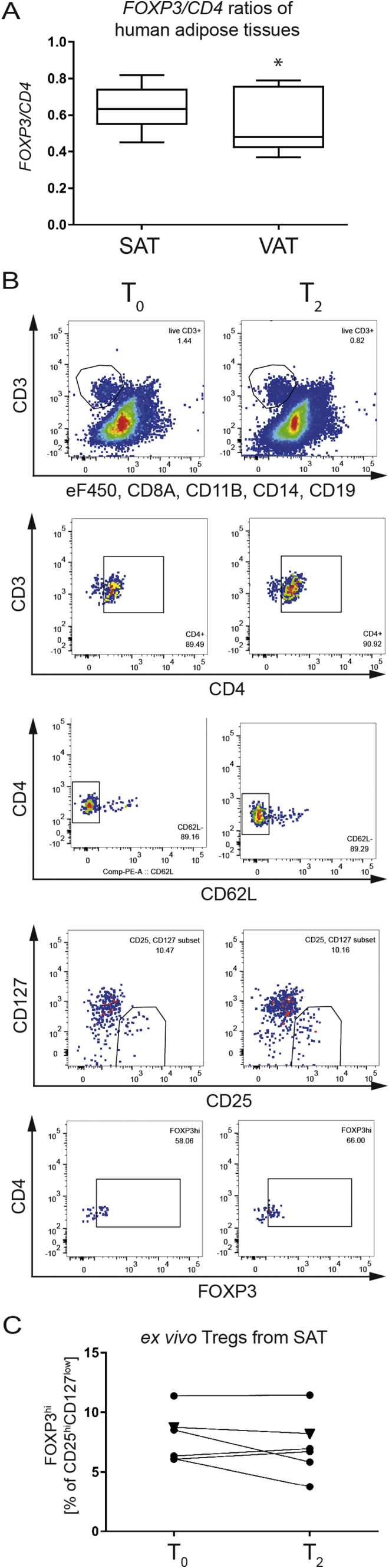
Human adipose tissue harbors FOXP3+Tregs that can be targeted by cold exposure or beta3-adrenergic stimulation. A) FOXP3/CD4 gene expression ratios in human adipose tissue biopsies. SAT = subcutaneous adipose tissue, VAT = visceral adipose tissue. B) Representative flow cytometric analysis of human SAT biopsies obtained before (T0) and after (T2) in vivo cold exposure. Each dot represents an individual experiment with paired samples. C) Paired analyses of (B). * = p < 0.05; (A) unpaired t-test.
Next, we purified CD4+ T cells in SAT biopsies from individuals prior (T0) and 2.5 h post cold exposure (T2) based on live CD8a−CD11b−CD14−CD19−CD3+CD4+T cells (see Figure 4B for staining example). In the SAT tissue context, the frequencies of locally residing Tregs after 2.5 h of cold exposure did not yet show any significant changes (Figure 4B, C). These findings are in line with previous data from the murine setting in which cold exposure supported a local increase in adipose tissue Tregs earliest after 24 h post cold exposure [22].
3.6. Human Treg induction by cold exposure involves BORCS6/C17orf59 signaling
Mechanistically and in accordance with findings in the murine setting [22], we observed BORCS6 mRNA to be significantly induced in human CD4+ T cells upon cold exposure in vivo (Figure 5A). BORCS6 encodes for C17orf59, a Ragulator-interacting protein that blocks mTORC1 activity by interacting with Ragulator at the lysosome [32]. In line with these results, inhibition of mTORC1 was demonstrated in previous studies to support Treg induction [15], [19], [22]. To validate the results seen for BORCS6 mRNA expression in human CD4+ T cells following cold exposure, we used immunofluorescence for C17orf59 in CD4+ T cells together with confocal microscopy. We found C17orf59 protein expression to be significantly upregulated following cold exposure in vivo (CD4+C17orf59+T cells per high power field: T0 = 1.0 ± 0.33; T2 = 3.1 ± 0.23 (n = 10 each); p < 0.0001; Figure 5B, C).
Figure 5.

In vivo cold exposure increases BORCS6/C17orf59 abundance in human CD4+T cells. A) RT-qPCR analysis of BORCS6 abundance in human CD4+ T cells before (T0) and after (T2) in vivo cold. B) Magnetically enriched human CD4+ T cell cytospins were stained for C17orf59 abundance before (T0) and after (T2) in vivo cold. C) Quantification of (B): CD4+ C17orf59+ per high power field. * = p < 0.05, *** = p < 0.001; paired t-test.
4. Discussion
Here, we provide first direct, translational evidence that short-term cold exposure enhances human FOXP3+ Tregs and their induction from naïve CD4+ T cells in vivo. We used humanized mice, human in vitro Treg induction together with ex vivo and in vitro analyses of T cells from the FREECE study to demonstrate an important role of low dose beta3-adrenergic stimulation or short-term cold exposure in supporting human FOXP3+Tregs. These findings highlight an important layer of immune-metabolic crosstalk in humans. Specifically, given the recent interest in cold exposure as one critical determinant of energy expenditure together with the identification of cold-inducible BAT in adult humans [25], [26], the results obtained here support the concept that metabolic targeting of energy expenditure and adipose tissue will be accompanied by a significant impact on immune regulation that is mainly executed by immune-suppressive Tregs.
Metabolically, cold exposure triggers the release of catecholamines via the sympathetic nervous system (SNS) and thereby, induces beta3-adrenergic signaling. Catecholamine release in adipose tissues plays a substantial role in inducing metabolic activity upon cold exposure and the SNS is critically involved in integrating afferent signals related to metabolism and environment and responding to them via efferent signaling. With regard to the environmental stimulus cold, this includes signaling with noradrenaline and adrenaline (i.e. catecholamines), as it was shown that mice lacking both signaling molecules (due to a knockout of the synthesizing enzyme dopamine beta-hydroxylase) cannot respond to thermal stress in an adequate manner [39]. These mice are unable to increase Ucp1 expression in BAT upon cold exposure and when being exposed to prolonged environmental cold, the constant heat loss leads to lethargy, shivering and ultimately to death due to hypothermia [39]. From an immunometabolic perspective, in previous experiments in the murine setting we aimed to assess the question whether the observed Treg-adipose-crosstalk in BAT includes a functional programming of BAT-residing T cells. When using UCP1 KO mice as a BAT loss-of-function model, we observed that local Treg induction was reduced [22]. Specifically, these results suggest that the local adipose tissue environment can co-opt local T cell differentiation programs and thereby influence tissue function. These findings are also in line with the concept that Treg induction by sympathetic drive is critically involved in the regulation of normal adipose tissue function. In addition, to mechanistically dissect these findings, we had studied mice lacking all three beta-adrenergic receptors (betaless mice) [40]. These animals presented with significantly reduced Treg frequencies in inguinal lymph nodes and subcutaneous fat depots. As one potential means to integrate sympathetic tone with reduced Treg abundance seen in betaless mice when compared to WT mice, we observed distinctly reduced proliferation of Tregs in subcutaneous fat of betaless mice. These results therefore underline an impairment of Treg induction in the absence of beta-adrenergic receptors in vivo [22]. Overall, our previous findings in the murine setting support the concept that Treg induction and adipose tissue function are adjusted according to the degree of sympathetic tone and environmental temperature [22].
Immunologically, the investigation of FOXP3/CD4 mRNA ratios in SAT vs. VAT from lean healthy individuals in the steady state revealed higher ratios of human FOXP3/CD4 mRNA in SAT when compared to their VAT. However, as a potential limitation it is important to consider that FOXP3 mRNA levels may not directly correlate with protein expression levels and therefore changes in FOXP3 alone cannot be solely equated to changes in Treg frequency.
Moreover, the Treg-enhancing impact of cold exposure can contribute critically to supporting adipose tissue and metabolic functions as well as to the reduction of local inflammatory responses [22], [23], [41], [42], [43], [44], [45]. In line with these assumptions, it is important to note that the beneficial effects of cold exposure on Treg induction and frequencies ex vivo were likewise evident in obese individuals suggesting a broader window of potential intervention for immune-metabolic targeting of adipose tissue function and inflammation by enhancing Tregs.
Future studies will be required to dissect potential differences in female vs. male subjects in response to the beneficial effect of cold exposure on Tregs also in the context of discussed alterations in pro-inflammatory characteristics of male vs. female adipose tissue [46], [47].
Here, we have focused our analyses on the direct isolation of Tregs from human adipose tissue biopsies prior and following cold exposure using cell sorting and multi-color flow cytometry. The time frame of two hours of cold exposure, however, might be rather short for the identification of local tissue-specific changes in Treg frequencies in human SAT, given recent insights and kinetics from murine adipose tissue analyses following cold exposure where a significant change in local Tregs was first observed 24 or 48 h post cold acclimation [22], [23]. However, from an immunological perspective, more rapid changes are expected to be visible in peripheral blood; in fact we identified a significant increase in Tregs purified from peripheral blood individuals after this short-term cold exposure. Mechanistically, and in line with murine data upon cold exposure [22], we found BORCS6 mRNA levels to be significantly increased in human CD4+ T cells following cold exposure. BORCS6 was recently identified to encode for C17orf59 that functions as a critical player in interacting with Ragulator at the lysosome [32], [48], [49]. mTORC1 activation requires its co-localization with the Rag–Ragulator complex at lysosomal surfaces. Overexpression of C17orf59 has been demonstrated to disrupt the Rag–Ragulator complex, thereby interfering with Rag lysosomal localization [32], [48], [49]. Thus, C17orf59 overexpression results in reduced mTORC1 activity. Strong mTOR signaling was shown to limit successful Treg induction [14], [15] and consequently, more BORCS6 mRNA expression and higher C17orf59 abundance improves Treg induction due to reduced mTOR signaling. Established mTORC1 inhibitors such as rapamycin or everolimus have shown a direct enhancement of FOXP3+Tregs due to blockade of mTORC1 activity [14], [15]. The increased BORCS6 mRNA abundance in human CD4+ T cells exposed to short-term cold is in line with a concept in which physiological levels of beta3-adrenergic stimulation can exert mTORC1-inhibiting activity, thereby directly supporting the induction of human FOXP3+Tregs.
5. Conclusion
In sum, here we demonstrate that a short-term cold stimulus induces human Treg induction in vitro and in vivo. Mechanistically, we identify BORCS6 encoding the Ragulator-interacting protein C17orf59 to be significantly induced in human CD4+ T cells upon short-term cold exposure. These novel insights into the molecular underpinnings of human Treg induction suggest an important role of Tregs in linking sympathetic tone and the environmental stimulus cold with immune and adipose function. Moreover, these discoveries shed new light on potential approaches toward tailored anti-inflammatory concepts to support human adipose tissue homeostasis.
Author contributions
MB performed in vivo and in vitro experiments, gene expression analyses, analyzed and interpreted data and wrote manuscript. IS performed in vivo and in vitro experiments. VKS performed dose-response titration. VBO analyzed human SAT/VAT gene expression. LM recruited subjects to and performed the FREECE study. MatBl is PI of the Leipzig Obesity Cohort. HH is PI of the FREECE study. MHT supported conception of the manuscript. CD analyzed data, conceptualized and wrote the manuscript.
Funding
C.D. is supported by a Research Group at Helmholtz Zentrum München, the German Center for Diabetes Research (DZD) and received support through a membership in the CRC1054 of the Deutsche Forschungsgemeinschaft (B11). H.H. received funding through the Helmholtz cross-program topic Metabolic dysfunction and the ZIEL – Institute of Food and Health. B.W. is supported by WE 4656/2 and DFG-CRC1181 (B02). This work was supported in part by funding to M.H.T. from the Alexander von Humboldt Foundation, the Helmholtz Alliance ICEMED & the Helmholtz Initiative on Personalized Medicine iMed by Helmholtz Association, funding by the European Research Council ERC (AdG HypoFlam no. 695054), the Helmholtz cross-program topic “Metabolic Dysfunction” and Initiative and Networking Fund of the Helmholtz Association.
Acknowledgements
The authors thank all study participants. Anette-G. Ziegler, Institute of Diabetes Research, Helmholtz Zentrum München, Germany, for providing human samples from the Munich Bioresource project. The FREECE study team for study participant recruitment, sampling and subject care.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.08.002.
Conflicts of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Rosen E.D., Spiegelman B.M. What we talk about when we talk about fat. Cell. 2014;156(1–2):20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fontenot J.D., Gavin M.A., Rudensky A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunology. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 3.Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 4.Bennett C.L., Brunkow M.E., Ramsdell F., O'Briant K.C., Zhu Q., Fuleihan R.L. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA-->AAUGAA) leads to the IPEX syndrome. Immunogenetics. 2001;53(6):435–439. doi: 10.1007/s002510100358. [DOI] [PubMed] [Google Scholar]
- 5.Brunkow M.E., Jeffery E.W., Hjerrild K.A., Paeper B., Clark L.B., Yasayko S.A. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature Genetics. 2001;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 6.Lahl K., Loddenkemper C., Drouin C., Freyer J., Arnason J., Eberl G. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. Journal of Experimental Medicine. 2007;204(1):57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker M.R., Kasprowicz D.J., Gersuk V.H., Benard A., Van Landeghen M., Buckner J.H. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. Journal of Clinical Investigation. 2003;112(9):1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan M.E., van Bilsen J.H., Bakker A.M., Heemskerk B., Schilham M.W., Hartgers F.C. Expression of FOXP3 mRNA is not confined to CD4+CD25+ T regulatory cells in humans. Human Immunology. 2005;66(1):13–20. doi: 10.1016/j.humimm.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 9.Liu W., Putnam A.L., Xu-Yu Z., Szot G.L., Lee M.R., Zhu S. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. Journal of Experimental Medicine. 2006;203(7):1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruprecht C.R., Gattorno M., Ferlito F., Gregorio A., Martini A., Lanzavecchia A. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. Journal of Experimental Medicine. 2005;201(11):1793–1803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wing J.B., Tanaka A., Sakaguchi S. Human FOXP3(+) regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. 2019;50(2):302–316. doi: 10.1016/j.immuni.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 12.von Boehmer H., Daniel C. Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nature Reviews Drug Discovery. 2013;12(1):51–63. doi: 10.1038/nrd3683. [DOI] [PubMed] [Google Scholar]
- 13.Daniel C., Weigmann B., Bronson R., Boehmer H.V. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. Journal of Experimental Medicine. 2011;208(7):1501–1510. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniel C., Wennhold K., Kim H.J., von Boehmer H. Enhancement of antigen-specific Treg vaccination in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(37):16246–16251. doi: 10.1073/pnas.1007422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer S., Bruno L., Hertweck A., Finlay D., Leleu M., Spivakov M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(22):7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottschalk R.A., Corse E., Allison J.P. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. Journal of Experimental Medicine. 2010;207(8):1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serr I., Furst R.W., Achenbach P., Scherm M.G., Gokmen F., Haupt F. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nature Communications. 2016;7:10991. doi: 10.1038/ncomms10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haxhinasto S., Mathis D., Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. Journal of Experimental Medicine. 2008;205(3):565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serr I., Scherm M.G., Zahm A.M., Schug J., Flynn V.K., Hippich M. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Science Translational Medicine. 2018;10(422) doi: 10.1126/scitranslmed.aag1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S.E. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalin S., Becker M., Ott V.B., Serr I., Hosp F., Mollah M.M.H. A stat6/pten Axis links regulatory T cells with adipose tissue function. Cell Metabolism. 2017;26(3):475–492. doi: 10.1016/j.cmet.2017.08.008. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medrikova D., Sijmonsma T.P., Sowodniok K., Richards D.M., Delacher M., Sticht C. Brown adipose tissue harbors a distinct sub-population of regulatory T cells. PLoS One. 2015;10(2):e0118534. doi: 10.1371/journal.pone.0118534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J., Bostrom P., Sparks L.M., Ye L., Choi J.H., Giang A.H. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58(7):1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cypess A.M., White A.P., Vernochet C., Schulz T.J., Xue R., Sass C.A. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nature Medicine. 2013;19(5):635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X.Y., Lin S.G., Wang X.M., Liu Y., Zhang B., Lin Q.X. Evidence for coexistence of three beta-adrenoceptor subtypes in human peripheral lymphocytes. Clinical Pharmacology and Therapeutics. 2007;81(5):654–658. doi: 10.1038/sj.clpt.6100154. [DOI] [PubMed] [Google Scholar]
- 28.Malik M., van Gelderen E.M., Lee J.H., Kowalski D.L., Yen M., Goldwater R. Proarrhythmic safety of repeat doses of mirabegron in healthy subjects: a randomized, double-blind, placebo-, and active-controlled thorough QT study. Clinical Pharmacology and Therapeutics. 2012;92(6):696–706. doi: 10.1038/clpt.2012.181. [DOI] [PubMed] [Google Scholar]
- 29.Takasu T., Ukai M., Sato S., Matsui T., Nagase I., Maruyama T. Effect of (R)-2-(2-aminothiazol-4-yl)-4'-{2-[(2-hydroxy-2-phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta3-adrenoceptor agonist, on bladder function. Journal of Pharmacology and Experimental Therapeutics. 2007;321(2):642–647. doi: 10.1124/jpet.106.115840. [DOI] [PubMed] [Google Scholar]
- 30.Hanssen M.J., Hoeks J., Brans B., van der Lans A.A., Schaart G., van den Driessche J.J. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nature Medicine. 2015;21(8):863–865. doi: 10.1038/nm.3891. [DOI] [PubMed] [Google Scholar]
- 31.Cypess A.M., Weiner L.S., Roberts-Toler C., Franquet Elia E., Kessler S.H., Kahn P.A. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metabolism. 2015;21(1):33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schweitzer L.D., Comb W.C., Bar-Peled L., Sabatini D.M. Disruption of the rag-ragulator complex by c17orf59 inhibits mTORC1. Cell Reports. 2015;12(9):1445–1455. doi: 10.1016/j.celrep.2015.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kloting N., Fasshauer M., Dietrich A., Kovacs P., Schon M.R., Kern M. Insulin-sensitive obesity. American Journal of Physiology Endocrinology and Metabolism. 2010;299(3):E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 34.Weir J.B. New methods for calculating metabolic rate with special reference to protein metabolism. Journal of Physiology. 1949;109(1–2):1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reis B.S., Lee K., Fanok M.H., Mascaraque C., Amoury M., Cohn L.B. Leptin receptor signaling in T cells is required for Th17 differentiation. The Journal of Immunology. 2015;194(11):5253–5260. doi: 10.4049/jimmunol.1402996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matarese G., Procaccini C., De Rosa V., Horvath T.L., La Cava A. Regulatory T cells in obesity: the leptin connection. Trends in Molecular Medicine. 2010;16(6):247–256. doi: 10.1016/j.molmed.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Lord G.M., Matarese G., Howard J.K., Baker R.J., Bloom S.R., Lechler R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 38.De Rosa V., Procaccini C., Cali G., Pirozzi G., Fontana S., Zappacosta S. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26(2):241–255. doi: 10.1016/j.immuni.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 39.Thomas S.A., Palmiter R.D. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997;387(6628):94–97. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- 40.Bachman E.S., Dhillon H., Zhang C.-Y., Cinti S., Bianco A.C., Kobilka B.K. βAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297(5582):843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- 41.Guillot X., Martin H., Seguin-Py S., Maguin-Gate K., Moretto J., Totoson P. Local cryotherapy improves adjuvant-induced arthritis through down-regulation of IL-6/IL-17 pathway but independently of TNFalpha. PLoS One. 2017;12(7):e0178668. doi: 10.1371/journal.pone.0178668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillot X., Tordi N., Mourot L., Demougeot C., Dugue B., Prati C. Cryotherapy in inflammatory rheumatic diseases: a systematic review. Expert Review of Clinical Immunology. 2014;10(2):281–294. doi: 10.1586/1744666X.2014.870036. [DOI] [PubMed] [Google Scholar]
- 43.Lange U., Uhlemann C., Muller-Ladner U. Serial whole-body cryotherapy in the criostream for inflammatory rheumatic diseases. A pilot study. Medizinische Klinik (Munich) 2008;103(6):383–388. doi: 10.1007/s00063-008-1056-5. [DOI] [PubMed] [Google Scholar]
- 44.Miller E., Mrowicka M., Malinowska K., Zolynski K., Kedziora J. Effects of the whole-body cryotherapy on a total antioxidative status and activities of some antioxidative enzymes in blood of patients with multiple sclerosis-preliminary study. The Journal of Medical Investigation. 2010;57(1,2):168–173. doi: 10.2152/jmi.57.168. [DOI] [PubMed] [Google Scholar]
- 45.Miller E., Kostka J., Wlodarczyk T., Dugue B. Whole-body cryostimulation (cryotherapy) provides benefits for fatigue and functional status in multiple sclerosis patients. A case-control study. Acta Neurologica Scandinavica. 2016;134(6):420–426. doi: 10.1111/ane.12557. [DOI] [PubMed] [Google Scholar]
- 46.Karastergiou K., Smith S.R., Greenberg A.S., Fried S.K. Sex differences in human adipose tissues - the biology of pear shape. Biology of Sex Differences. 2012;3(1):13. doi: 10.1186/2042-6410-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeffery E., Wing A., Holtrup B., Sebo Z., Kaplan J.L., Saavedra-Pena R. The adipose tissue microenvironment regulates depot-specific adipogenesis in obesity. Cell Metabolism. 2016;24(1):142–150. doi: 10.1016/j.cmet.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang T., Wang R., Wang Z., Wang X., Wang F., Ding J. Structural basis for Ragulator functioning as a scaffold in membrane-anchoring of Rag GTPases and mTORC1. Nature Communications. 2017;8(1):1394. doi: 10.1038/s41467-017-01567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filipek P.A., de Araujo M.E.G., Vogel G.F., De Smet C.H., Eberharter D., Rebsamen M. LAMTOR/Ragulator is a negative regulator of Arl8b- and BORC-dependent late endosomal positioning. The Journal of Cell Biology. 2017;216(12):4199–4215. doi: 10.1083/jcb.201703061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.