

Abstract
Background
Activating mutations in the pathway of phosphatidylinositol-3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) occur in 43–70% of breast cancer brain metastasis patients. To date, the treatment of these patients presents an ongoing challenge, mainly because of the lack of targeted agents that are able to sufficiently penetrate the blood–brain barrier. GDC-0068 is a pan-Akt inhibitor that has shown to be effective in various preclinical tumor models as well as in clinical trials. The purpose of this study was to analyze the efficacy of GDC-0068 in a breast cancer brain metastases model.
Methods
In in vitro studies, antitumor activity of GDC-0068 was assessed in breast cancer cells of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA)–mutant and PIK3CA-wildtype breast cancer cell lines using cell viability and apoptosis assays, cell cycle analysis, and western blots. In vivo, the efficacy of GDC-0068 was analyzed in a PIK3CA-mutant breast cancer brain metastasis orthotopic xenograft mouse model and evaluated by repeated bioluminescent imaging and immunohistochemistry.
Results
GDC-0068 decreased cell viability, induced apoptosis, and inhibited phosphorylation of proline rich Akt substrate 40 kDa and p70 S6 kinase in a dose-dependent manner in PIK3CA-mutant breast cancer brain metastatic cell lines compared with PIK3CA-wildtype cell lines. In vivo, treatment with GDC-0068 notably inhibited the growth of PIK3CA-mutant tumors and resulted in a significant survival benefit compared with sham, whereas no effect was detected in a PIK3CA-wildtype model.
Conclusions
This study suggests that the Akt inhibitor GDC-0068 may be an encouraging targeted treatment strategy for breast cancer brain metastasis patients with activating mutations in the PI3K pathway. These data provide a rationale to further evaluate the efficacy of GDC-0068 in patients with brain metastases.
Keywords: brain metastases, breast cancer, PI3K/Akt/mTOR pathway, Akt inhibition, GDC-0068
Key Points.
GDC-0068 induces apoptosis in PIK3CA-mutant breast cancer brain metastatic cell lines in vitro.
In a PIK3CA-mutant metastasis xenograft model, GDC-0068 resulted in an overall survival benefit.
Importance of the Study.
Despite the fact that multidisciplinary treatment approaches for patients with brain metastases have been optimized in recent years, patients still face a poor overall survival. Next-generation sequencing techniques have identified new molecular targets that potentially present new treatment options for brain metastases patients. Somatic genetic alterations in the PI3K pathway, including PIK3CA mutations, are present in approximately 40–70% of all patients with breast cancer brain metastases. Therefore, inhibition of the PI3K/Akt/mTOR pathway is a promising therapeutic strategy. However, most targeted agents are not able to penetrate the blood–brain barrier. In this study, we demonstrate that the pan-Akt inhibitor GDC-0068 achieves significant tumor growth inhibition in a PIK3CA-mutant xenograft model and results in a significant overall survival benefit. To this extent, Akt inhibition with GDC-0068 might be a promising treatment option in patients harboring PIK3CA mutations. Clinical trials are warranted for further validation of these results.
Brain metastases account for the most common brain tumors in adult cancer patients and represent a devastating complication of systemic cancer.1 The incidence of brain metastases is rising, which has been attributed to refined neuroimaging techniques and improved systemic therapies, which have led to an earlier detection of these lesions and a prolonged overall survival of cancer patients, respectively.1,2 Current therapeutic options for patients with brain metastases include whole-brain radiation therapy, surgical resection, and stereotactic radiosurgery.1 Although a number of improvements have been made in recent years to optimize multidisciplinary treatment strategies, affected patients still face a poor median overall survival, ranging from 3 to 18 months.3,4 Breast cancer has the second highest propensity to metastasize to the brain5 and is the most common primary tumor in female cancer patients.6 Therefore, the treatment of patients with breast cancer brain metastases (BCBM) is becoming an increasing challenge that requires effective therapeutic strategies.
Unfortunately, effective targeted treatment options for BCBM patients are still limited, as patients with brain metastases are frequently excluded from clinical trials.7 Furthermore, penetration of the blood–brain barrier presents a significant challenge for effective treatment of brain metastases, and there is still an urgent unmet need for compounds that are able to cross this physiologic barrier.8
In recent years, significant progress has been made in identifying underlying genetic drivers of brain metastases with next-generation sequencing techniques.7 The phosphatidylinositol 3-kinase (PI3K) pathway is activated in 43–70% of patients with BCBM.9–11 Activating mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) result in an activation of the PI3K/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway, which is associated with cell proliferation, cancer progression, and treatment resistance across a variety of tumor types.12 Despite the fact that many PI3K/Akt/mTOR-directed compounds have been developed over the last decade, there are no FDA-approved inhibitors targeting this pathway for BCBM patients at present.13,14
The ATP-competitive, selective pan-Akt inhibitor GDC-0068 (ipatasertib)15 has already shown promising antitumor activity in preclinical models as well as in clinical trials across various cancer types.16–21 However, the efficacy of this compound has not been investigated in the setting of BCBM thus far. As an effective blockade of the PI3K/Akt/mTOR pathway represents a rationale treatment strategy in BCBM patients with activating PIK3CA mutations, the purpose of this study was to evaluate the efficacy of GDC-0068 in a preclinical BCBM model in vitro and in vivo.
Materials and Methods
Cell Lines
In this study, we used the following human metastatic breast cancer cell lines: PIK3CA-mutant (MT) MDA-MB-361 (PIK3CA E545K; HER2+, ER/PR+)22; JIMT-1 BR-3 (PIK3CA C420R; HER2+, ER/PR−)23; MDA-MB-453 (PIK3CA H1047R),24 and PIK3CA-wildtype (WT), MDA-MB-231 BrM2 (HER2−, ER/PR−); BS-004 (HER2+, ER/PR+). JIMT-1 BR-3 was a generous gift from the laboratory of Dr Patricia Steeg (National Cancer Institute, Bethesda, Maryland), and MDA-MB-231 BrM2 was kindly provided by the laboratory of Dr Joan Massagué (Memorial Sloan-Kettering Cancer Center, New York City). MDA-MB-453 and MDA-MB-361 were purchased from American Type Culture Collection (ATCC). BS-004 was derived from a patient’s breast cancer brain metastasis with written consent. The cell lines JIMT-1 BR-3, MDA-MB-231 BrM2, and BS-004 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin-amphotericin B. MDA-MB-453 was cultured in L15 supplemented with 10% FBS and MDA-MB-361 was cultured in L15 supplemented with 20% FBS and 1% penicillin-streptomycin-amphotericin B. To validate the in vitro experiments, we used the isogenic non-tumorigenic epithelial breast cell lines MCF10A (parental, PIK3CA-WT; HER2−, ER/PR−) and PIK3CA (H1047R/+) MCF10A (heterozygous knockin of PIK3CA kinase domain activating mutation, PIK3CA-MT; HER2−, ER/PR−), which were purchased from Horizon Discovery. MCF10A and PIK3CA (H1047R/+) MCF10A were cultured in DMEM/F12 including 2.5 mM L-glutamine and 15 mM HEPES (4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid), supplemented with 5% horse serum, 10 µg/mL insulin, 0.5 µg/mL hydrocortisone, 0.1 µg/mL cholera toxin, and 0.2 ng/mL epidermal growth factor (EGF).25 For drug permeability assays, colorectal adenocarcinoma Caco-2 cells (clone C2BBe1) were obtained from ATCC. Cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. The PIK3CA mutation status of all cancer cell lines was confirmed via whole-exome sequencing. Every month, all cell lines were confirmed to be mycoplasma free and were tested throughout the course of the experiments (PCR Mycoplasma Detection Kit, abm).
The study was reviewed and approved by the human subject institutional review board of the Dana-Farber Cancer/Harvard Cancer Center, and the research performed in accordance with the Declaration of Helsinki.
Viral Vectors and Transduction
For in vivo experiments, the cell lines of MDA-MB-361 and MDA-MB-231 BrM2 were engineered to express Firefly luciferase and mCherry (FmC) by transduction with the lentiviral construct LV-pico2-Fluc-mCherry (pLV-FmC). The construct was kindly provided by Khalid Shah (Brigham and Women’s Hospital) and Dr Andrew Kung (Dana Farber Cancer Institute). Cells were transduced at a multiplicity of infection of 2 in media containing Polybrene (8 μg/mL; EMD Millipore) for 48 hours. Cells were selected with puromycin (7 μg/mL) for 3 days. To confirm successful transduction, cells were visualized by fluorescence microscopy for mCherry and afterward sorted for mCherry expression using fluorescence-activated cell sorting (FACSAria Cell-Sorting System, BD Biosciences).
Akt Inhibitor
The Akt inhibitor GDC-0068, kindly provided by Genentech, was used to investigate inhibition of the PI3K/Akt/mTOR pathway. GDC-0068 was dissolved in dimethyl sulfoxide (DMSO) for in vitro studies and in a solution of 0.5% methylcellulose and 0.2% Tween 80 in water for in vivo experiments. In in vitro experiments, the compound was added at concentrations ranging from 0.25 µmol/L up to 10 µmol/L to cell culture medium with cells plated at 50–70% confluency. Controls were incubated with 0.1% DMSO.
Cell Viability and Apoptosis Assays
Cell lines were plated in triplicates for cell viability and apoptosis assays at a density of 5000 cells per well on a 96-well plate. The next day, cells were treated with GDC-0068 in concentrations from 0.25 µmol/L to 10 µmol/L for 10 hours for apoptosis assays and 72 hours for cell viability assays. Controls were incubated with 0.1% DMSO. After treatment, cells were lysed by using the CellTiter-Glo (Promega, cell viability assays) or the Caspase-Glo 3/7 (Promega, apoptosis assays) reagent. The reagents generate a luminescent signal proportional to the number of viable cells or the caspase-3/7 activity, respectively. Luminescence was analyzed using a Synergy HT multidetection microplate reader (BioTek). The percentage of viable cells or caspase-3/7 was calculated relative to DMSO-incubated controls. Half-maximal inhibitory concentration ( IC50), the drug concentration needed to achieve 50% inhibition, was calculated by using a 9-parameter curve analysis of the results of a total of 3 independent cell viability assay experiments. For cell lines in which 50% inhibition was not achieved, the highest concentration of GDC-0068 (10 µmol/L) is displayed.
Cell Cycle Analysis
Cells were plated in triplicates at a density of 2.5 × 105 in 6-well plates and treated with GDC-0068 (control, 1 µM, 2.5 µM, 5 µM, 7.5 µM, and 10 µM) the next day for 72 hours. Floating as well as adherent cells were then harvested, washed with phosphate-buffered saline (PBS), fixed in ice-cold 70% ethanol, and stored at 4°C for 24 hours. After equilibrating at room temperature, fixed cells were washed with PBS and afterward stained with 200 µL of propidium iodide solution (500 µL 20× propidium iodide and 50 µL 200× RNAse in 9.45 mL PBS; Propidium Iodide Flow Cytometry Kit [ab139418, Abcam]) and incubated at 37°C in the dark for 30 minutes. Cell cycle analysis was conducted using the BD LSRII and BD FACSDiva Software v8.0.1 and subsequently analyzed using FlowJo software v10 (LLC).
Western Blots
All cell lines were seeded in triplicates in 100 mm well plates at a density of 2.5 × 106 cells and treated with GDC-0068 (control, 1 µM, 2.5 µM, 5 µM, 7.5 µM, and 10 µM) the following day for a total of 6 hours. Adherent cells were then harvested and lysed in radioimmunoprecipitation buffer (Thermo Fisher Scientific) containing protease and phosphatase inhibitor cocktails (Roche). A total of 20 µg of protein per lane was separated by 4–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (BioRad) and transferred to polyvinylidene difluoride membranes (BioRad) by electroblotting. Membranes were blocked with 5% nonfat dry milk in TBST (20 mM Tris pH7.5, 150 mM NaCl, 0.1% Tween 20) for 1 hour at room temperature and afterward incubated with primary antibodies: phosphorylated proline rich Akt substrate, 40kDa (p-PRAS40) (Thr246) #13175, PRAS40 #2691, p-p70 S6 kinase (Thr389) #9205, p70 S6 kinase #2708, β-actin #3700 (all from Cell Signaling Technology) at 4°C overnight. Membranes were then washed with TBST and incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies (anti-rabbit imunoglobulin G [H+L], HRP conjugate and anti-mouse imunoglobulin G [H+L], HRP conjugate, both from Promega) for 1 hour at room temperature. Afterward, membranes were washed in TBST. Signals were visualized with an electrochemiluminescence blotting substrate (Thermo Fisher Scientific). Blots were analyzed with the software Image Lab (BioRad) and cropped in Adobe Illustrator software (version CC 22.1).
Drug Permeability Assays
Caco-2 cells (clone C2BBe1) were seeded on collagen-coated 12-well plates. On day 22, transepithelial electrical resistance measurements were conducted prior to the experiment. Monolayers were equilibrated in transport buffer (Hank’s Balanced Salt Solution supplemented with 15 mM D-glucose and 10 mM HEPES, pH 7.4) for 10 minutes at 37°C with 5% CO2 and 95% relative humidity. The dose solutions containing GDC-0068 (52.4 µM), atenolol (100 µM), digoxin (5 µM), and minoxidil (10 µM) were prepared in transport buffer containing 100 µM lucifer yellow. Dose solutions were added to the donor chambers and transport buffer was added to all receiver chambers in triplicate. Aliquots of 200 μL were taken from the receiver chamber and replaced with fresh assay buffer at 2 and 30 min, and 200 μL without replacement at 60 minutes. Aliquots were taken from the donor chamber before and after the incubation (0, 30, and 60 min) without replacement. All permeability samples were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS).
The apparent permeability (Papp) and percent recovery were calculated as follows:
where dCr / dt is the slope of the cumulative concentration in receiver compartment versus time (μM/s),
Vr is the volume of the receiver compartment (1.5 mL for A-to-B, 0.5 mL for B-to-A),
Vd is the volume of the donor compartment (1.5 mL for A-to-B, 0.5 mL for B-to-A),
A is the area of the cell monolayer (1.13 cm2 for 12-well Transwell).
CN is the nominal concentration of the dosing solution (μM),
Crfinal is the cumulative receiver concentration at the end of the incubation (μM),
Cdfinal is the concentration of the donor at the end of the incubation (μM), and
Efflux ratio is defined as [Papp (B-to-A)] / [Papp (A-to-B)].
Animal Experiments
Stereotactic intracranial tumor implantation
All animal experiments were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital. For MDA-MB-361 tumors, a total of 16 mice were analyzed for imaging (8 animals per cohort) and an additional 4 mice in a separate cohort were analyzed for immunohistochemistry. For MDA-MB-231 BrM2 tumors, a total of 19 mice were analyzed for imaging (9 mice for placebo, 10 mice for treatment). Eight-week-old female athymic nu/nu mice (Charles River Laboratories) were anesthetized with pentobarbital (i.p. injection, 40–70 mg/kg). The toe-pinch reflex was used to confirm the depth of anesthesia in all animals prior to surgery. Anesthetized mice were placed into a stereotactic apparatus, with their heads stabilized by ear bars. The skin over the skull was opened, bregma was identified, and a total of 1.5 × 105 MDA-MB-361 or 7.5 × 104 MDA-MB-231 BrM2 cells in 4 µL Hank’s Balanced Salt Solution were stereotactically implanted into the right striatum (2 mm lateral from bregma, 2.5 mm deep) using a Hamilton syringe. Subsequently, the burr hole was sealed with bone wax and the wound was sutured. All mice were provided carprofen postoperatively (MediGel CPF, 2-oz cup, Clear H2O) for a total of 3 days.
Dynamic bioluminescent imaging and assessment of tumor burden
Seven days after intracranial tumor cell injection, mice received a dynamic bioluminescent imaging (BLI) baseline scan to assess their initial tumor burden. Prior to imaging, mice were anesthetized with isoflurane and injected with 4.5 mg D-luciferin in 300 µL PBS. Ten minutes after the luciferin injection, up to 5 animals at a time were imaged on the Spectral Ami HTX (Spectral Instruments Imaging).
We chose exposure times of 60 s, 1 s, and 0.5 s at 5-min intervals until the peak luminescent signal from the whole mouse body was reached. Tumor burden was measured in total flux (p/s) and analyzed by subtracting the background signal from the bioluminescent signal above the mouse’s cranium with Aura software v2.2.1.0 (Spectral Instruments Imaging). Figures were generated with ImageJ 1.52k software (National Institutes of Health) using the binning 2, 0.5 s exposure and background subtracted image and a log10 transformation of the data measured in total flux (p/s).
After the assessment of initial tumor burden, mice were randomized to GDC-0068 or sham treatment by a third-party person. Treatment was initiated the day after initial imaging (8 days post tumor cell implantation). BLI was performed on a weekly basis until 77 days post intracranial tumor implantation to monitor tumor growth in all mice, as measured by flux (p/s). The primary endpoint of this study was defined as the tumor burden reflected by BLI in the cohorts receiving treatment with GDC-0068 and sham at the last day of imaging (77 days post tumor injection). The secondary endpoint was overall survival at the end of the study.
Treatment and monitoring
GDC-0068 was administered daily via oral gavage at a dose of 100 mg/kg until the date of death of the last control mouse. Mice randomized to treatment received 100 mg/kg GDC-0068 diluted in 0.5% methylcellulose and 0.2% Tween 80 per day, and mice randomized to sham received a weight-adapted dose of 0.5% methylcellulose and 0.2% Tween 80 daily. Following intracranial tumor implantation, all mice were monitored daily and sacrificed at ≥20% weight loss and/or the onset of neurological symptoms.
Immunohistochemistry
Mouse brains were fixed in 10% formalin for 24 hours and embedded in paraffin thereafter. Five-micrometer-thick sections were deparaffinized for immunohistochemistry. Manual staining was conducted for p-S6 ribosomal protein. Sections were treated with sodium citrate (pH = 6) and heated for 10 minutes for antigen unmasking. Sections were blocked with TBST/5% normal goat serum and incubated overnight with p-S6 ribosomal protein ((Ser235/236) #4858, 1:400, Cell Signaling Technology). Sections were then alternately washed in TBS and TBST and incubated with the secondary antibody SignalStain Boost IHC Detection Reagent (HRP, Rabbit, #8114, Cell Signaling Technology) for 30 minutes at room temperature. Slides were stained with 3,3′-diaminobenzidine (DAB; Dako) and counterstained with hematoxylin.
Automated staining was conducted for p-PRAS40 (Thr246). Sections were treated with pre-diluted cell conditioning solution (CC2, Ventana), heated for 62 minutes, and blocked in inhibitor conditioned medium (Ventana). Sections were incubated with the primary antibody p-PRAS40 (Thr246) #13175, 1:100, Cell Signaling Technology) for 36 minutes and then with the secondary antibody OmniMap anti-Rb HRP (Multimer HRP, Ventana) for 12 minutes. Sections were then stained with DAB (Dako), counterstained with bluing reagent (Ventana), and post-counterstained with hematoxylin.
Statistical Analysis
Statistical analysis was conducted with GraphPad Prism v7 for in vitro data and a log-rank test was implemented for survival analysis. SAS 9.4 was used for the mixed effects models for in vivo experiments. Graphs were cropped with the Adobe Illustrator software (vCC 2018). The efficacy of GDC-0068 in vivo was evaluated by autoregressive linear mixed models. For the statistical analysis, bioluminescent data (measured in total flux [p/s]) were first log10 transformed. Linear mixed effects models with an autoregressive covariance structure within mice were conducted to compare the treatment cohorts and their signaling changes over time. In this statistical model, previously log10-transformed fold changes in flux were the dependent variable, while independent predictors were time, treatment, and the interaction of both. A P-value of <0.05 was considered statistically significant.
Results
In cell viability assays, GDC-0068 potently decreased cell viability in the PIK3CA-MT breast cancer brain metastatic cell line MDA-MB-361 and the breast cancer cell line MDA-MB-453 after 72 hours of treatment (IC50 = 2.83 µmol/L and (IC50: 0.322 µmol/L, respectively) in a dose-dependent manner (Figure 1A). In contrast, only a minor reduction was observed in PIK3CA-WT cell lines, MDA-MB-231 BrM2, and BS-004, at doses up to 10 µmol/L (Figure 1A). JIMT-1 BR-3 carrying PIK3CA C420R responded slightly better but modestly to GDC-0068 (Figure 1A). Apoptosis assays were used to assess the underlying effect of GDC-0068 on BCBM cell lines. In line with the cell viability results, GDC-0068 robustly and modestly increased caspase-3/7 activity in the PIK3CA-MT BCBM cell lines MDA-MB-361 and JIMT-1 BR-3, respectively, in a dose-dependent manner. However, there was no significant increase in caspase-3/7 activity in the 2 PIK3CA-WT BCBM cell lines in response to GDC-0068 (Figure 1B). The highest caspase-3/7 signal was obtained after 10 hours of treatment in increasing concentrations of GDC-0068. GDC-0068 led to a dose-dependent phosphorylation inhibition of PRAS40, a direct substrate of Akt kinase, and further downstream p70 S6 kinase in a dose-dependent manner in PIK3CA-MT BCBM cell lines over a treatment course of 6 hours (Figure 1C). The Akt pathway signaling was particularly sensitive to GDC-0068–induced inhibition in PIK3CA E545K MDA-MB-361. In comparison, no considerable changes in phosphorylation were observed in PIK3CA-WT BCBM cell lines treated in increasing concentrations with GDC-0068 (Figure 1C).
Fig. 1.

The effect of GDC-0068 in PIK3CA-mutant and PIK3CA-wildtype breast cancer brain metastatic cell lines in cell viability and apoptosis assays and western blots.(A), Cell viability after 72 hours and (B) apoptosis assays after 10 hours of treatment of the breast cancer brain metastatic cell lines MDA-MB-361, JIMT-1 BR-3, MDA-MB-231 BrM2, and BS-004 and the breast cancer cell line MDA-MB-453 in increasing concentrations with GDC-0068. Percentage of growth inhibition is compared with 0.1% DMSO-incubated controls. Error bars represent SEM of 3 independent experiments. (C) Western blot analysis of 3 independent experiments for PRAS40, p-PRAS40 (Thr246), p-p70 S6 kinase (Thr389), and p70 S6 kinase in JIMT-1 BR-3, MDA-MB-361, MDA-MB-231 BrM2, and BS-004 cell lines treated with GDC-0068 in the illustrated concentrations for a total of 6 hours. β-actin was used as a loading control.
In order to validate if the effect of GDC-0068 is genotype selective, cell viability and apoptosis assays were performed in the isogenic, non-tumorigenic epithelial breast cell lines PIK3CA-MT (H1047R/+) MCF10A and parental MCF10A. After 72 hours of treatment, GDC-0068 significantly decreased cell viability in PIK3CA-MT (H1047R/+) MCF10A (IC50: 0.825µmol/L) in contrast to MCF10 PIK3CA-WT cells (Figure 2A). Consistent with the in vitro findings in BCBM cell lines, treatment with GDC-0068 in increasing concentrations resulted in a marked increase of caspase-3/7 activity in PIK3CA (H1047R/+) MCF10A cells, but no such response was observed in MCF10A parental cells (Figure 2B). In these isogenic pairs, the highest apoptotic signal was observed after 24 hours of treatment.
Fig. 2.

GDC-0068 results in genotype-selective antitumor activity in isogenic epithelial breast cell lines. (A) Cell viability assays after 72 hours and (B) caspase-3/7 Glo apoptosis assays after 24 hours of treatment with GDC-0068 in increasing concentrations of the epithelial breast cell lines PIK3CA (H1047R/+) MCF10A and MCF10A. Percentage of growth inhibition is calculated relative to 0.1% DMSO-incubated controls. Error bars represent SEM of triplicates treated with the same dose of GDC-0068.
In order to elucidate the biologic effect of GDC-0068 on the cell cycle, propidium iodide staining with subsequent cell cycle analysis was conducted in all BCBM cell lines after 72 hours of treatment with the compound. A dose-dependent increase in the sub-G1 fraction (apoptotic cells) with an accompanying decrease of the G1-, S-, and G2M-phase populations was observed in both PIK3CA-MT BCBM cell lines, MDA-MB-361 and JIMT-1 BR-3 (Figure 3A and Supplementary Figure 1A). Only a minor increase in the sub-G1-phase population was seen in the PIK3CA-WT cell line MDA-MB-231 BrM2 (Figure 3B), whereas no significant changes in cell cycle were observed in BS-004 (Supplementary Figure 1B).
Fig. 3.

Treatment with GDC-0068 induces apoptosis in PIK3CA-mutant and in growth inhibition in PIK3CA-wildtype cell lines. Cell cycle changes for (A) MDA-MB-361 and (B) MDA-MB-231 BrM2, treated in increasing concentrations of the inhibitor displayed as bar graphs of different phases of the cell cycle (left panel) measured in percent and as histograms of cells incubated in 0.1% DMSO and 10 µmol/L GDC-0068 (right panel). Each phase of the cell cycle (<G1, G1, S, G2M) is gated on the histogram. Percentages of phases of the cell cycle were assessed in 3 independent experiments. Error bars represent SEM.
In permeability assays, digoxin, the positive control for P-glycoprotein efflux had an efflux ratio of 13.1. The A to B Papp of GDC-0068 (0.55 × 10−6 cm/s) was similar to that of the low permeability marker atenolol (A to B Papp 0.24 × 10−6 cm/s) and much lower than the high permeability marker minoxidil (A to B Papp 5.9 × 10−6 cm/s). GDC-0068 had an efflux ratio of 43.4, suggesting it may be a substrate of one or more efflux transporters present in Caco-2 cells. The A to B and B to A recovery for GDC-0068 were complete (101% and 99.8%, respectively).
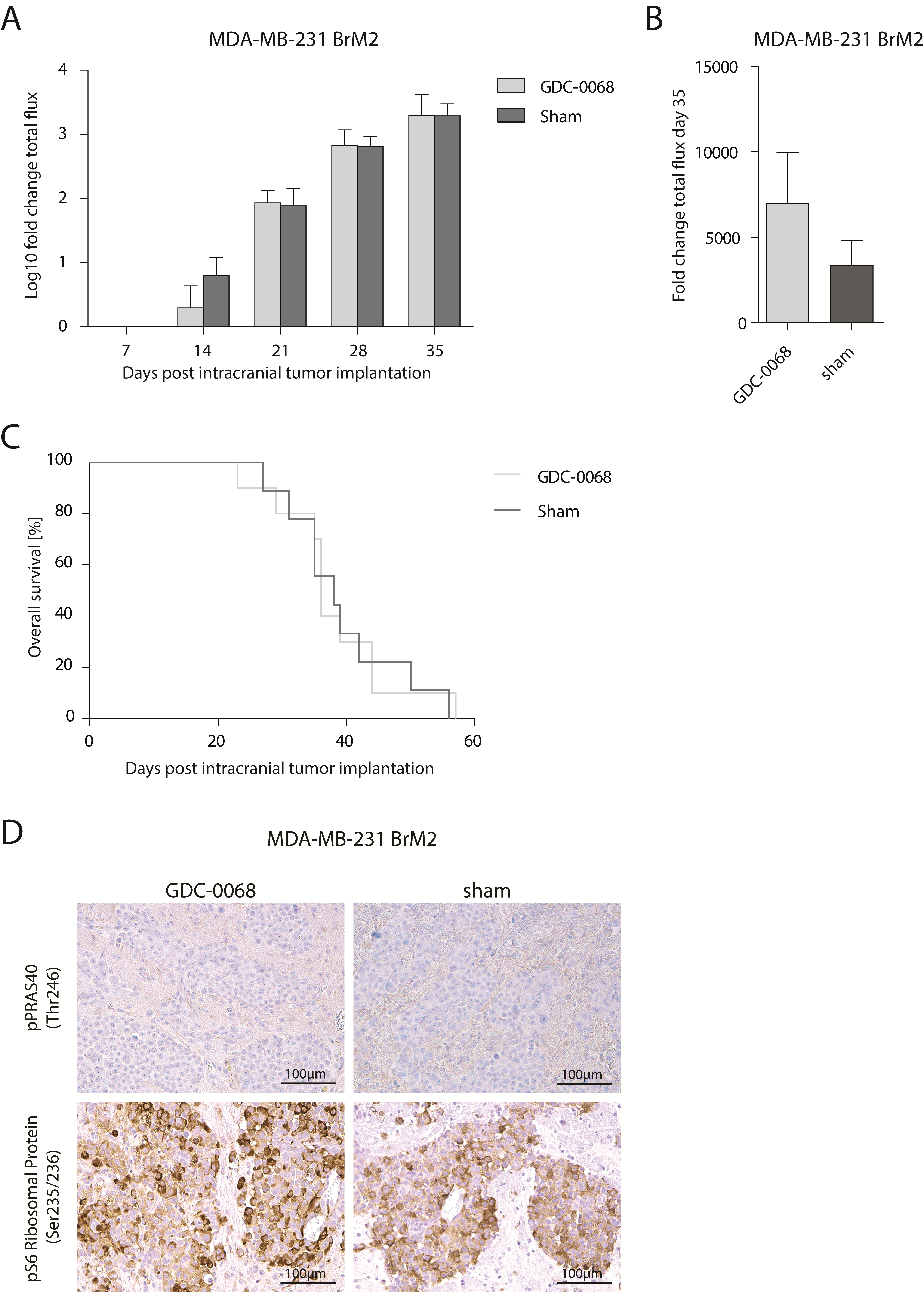
To further evaluate the antitumor activity of GDC-0068 in BCBM, the efficacy of this compound was investigated in an intracranial PIK3CA-MT (MDA-MB-361) BCBM tumor model, as well as a PIK3CA-WT model (MDA-MB-231 BrM2) in vivo. GDC-0068 was administered via oral gavage at a dose of 100 mg/kg daily until the date of death of the last mouse in the placebo group. Treatment was well tolerated and no adverse events were observed over the entire course of treatment. In the PIK3CA-MT MDA-MB-361 BCBM tumor model, GDC-0068 resulted in a significant inhibition of tumor growth measured by BLI in treated mice, whereas in contrast, sham-treated tumors continued to grow more rapidly (Figure 4A–C. No differences in tumor growth and survival were detected in MDA-MB-231 BrM2 intracranial tumors over the course of treatment (Supplementary Figure 2). Treatment with GDC-0068 led to a significant inhibition in the MDA-MB-361 tumor bearing mice compared with sham (mixed effect model, effect of treatment at day 77: P < 0.0001) (Figure 4B). Furthermore, treatment with GDC-0068 resulted in a significant survival benefit (log-rank test, P = 0.0008), with a median survival of 109 days in treated mice versus 82.5 days in mice receiving sham treatment (Figure 4D). To evaluate GDC-0068–induced phosphorylation inhibition of PI3K/Akt/mTOR pathway downstream targets, expression of p-PRAS40 and p-S6 ribosomal protein was analyzed with immunohistochemistry. In accordance with the previous in vitro and in vivo results, GDC-0068 treated tumors in MDA-MB-361 tumor bearing mice revealed reduced immunostaining of p-S6 ribosomal protein compared with tumors of the sham cohort (Figure 4E), but no such effect was seen in the PIK3CA-mutant cell line MDA-MB-231 BrM2 (Supplementary Figure 2).
Fig. 4.

GDC-0068 inhibits tumor growth in vivo in PIK3CA-mutant MDA-MB-361 tumors and results in a significant survival benefit. (A) Differences in log10-transformed fold changes in flux at days 7, 21, 35, 49, 63, and 77 of imaging. Error bars represent SEM of log10-transformed flux fold change values. (B) Comparison in mean fold changes flux values + SEM between GDC-0068 and sham cohorts at day 77 of imaging. (C) Overall survival in percent of both cohorts treated with either GDC-0068 or sham. (D) Representative BLI images with log10-transformed flux (p/s) for treated and sham mice day 77 of imaging. (E) Photomicrographs illustrating p-PRAS40 (Thr246)) and pS6 ribosomal protein (Ser235/236) expression in mice harboring MDA-MB-361 tumors, stratified by treatment with GDC-0068 and sham.
Taking everything into account, GDC-0068 effectively inhibited downstream molecular targets of the PI3K/Akt/mTOR pathway in a genotype-selective and dose-dependent manner, led to a marked inhibition in tumor growth, and resulted in a significant survival benefit in PIK3CA-MT BCBM tumor bearing mice.
Discussion
In the last decade, the identification of actionable genetic driver mutations of metastatic brain disease has revealed molecular targets that open up new treatment strategies for affected BCBM patients.10 Genomic as well as immunohistochemical studies have shown that activation of the PI3K pathway is present in 43–70% of all BCBM patients.9–11 Therefore, effective inhibition of the PI3K/Akt/mTOR pathway appears to be an appealing treatment option, which we were recently able to demonstrate in a preclinical model of BCBM.26 The ATP-competitive pan-Akt inhibitor GDC-0068 has been reported to achieve encouraging antitumor activity in a variety of preclinical cancer models and clinical trials,16–21 but has not been evaluated previously in a preclinical intracranial tumor model. A single center, proof-of-concept phase I trial is currently recruiting glioblastoma patients to investigate the combination of atezolizumab in combination with ipatasertib (GDC-0068) (NCT03673787), but there are no clinical trials evaluating the efficacy of this compound in BCBM patients at present.
In our study, we demonstrated that GDC-0068 selectively induced apoptosis in PIK3CA-MT BCBM cell lines in vitro in a dose-dependent manner. These findings are consistent with the degree of inhibition of molecular downstream targets of Akt as demonstrated in western blots. The genotype-selective effect of this compound was validated in an isogenic non-tumorigenic breast cell line with and without a PIK3CA mutation. Furthermore, although in vitro permeability assays showed that GDC-0068 might be subject to one or more efflux transporters in Caco-2 cells, we showed that treatment with GDC-0068 led to significant inhibition of tumor growth and overall survival benefit in mice harboring PIK3CA-MT intracranial tumors, but not in a PIK3CA-WT mouse model. This is in line with other studies that have shown disruption of the blood–brain barrier in the setting of brain metastases in animal models.27 Inhibition of molecular downstream targets was validated by decreased staining for p-S6 ribosomal protein in PIK3CA-MT BCBM tumors treated with GDC-0068. Of note, among the PIK3CA-MT cell lines in this study, MDA-MB-361 was more sensitive to GDC-0068 compared with JIMT-1 BR-3 in in vitro assays. This effect could be explained by the different genetic contexts of these cell lines, as the PIK3CA E545K mutation in MDA-MB-361 is a well-documented activating mutation, while JIMT-1 BR-3 harbors a PIK3CA C420R mutation.28 These different alterations might have distinct impacts on downstream signaling activation of this pathway. Also, we cannot exclude the possibility that the activity of other pathways that cross-talk with the PI3K/Akt/mTOR pathway may influence the sensitivity to treatment with GDC-0068.
Various clinical trials are currently investigating the efficacy of GDC-0068 as a single agent or in combination in patients with various tumor types such as prostate cancer (NCT01485861, NCT03072238, NCT03673787, NCT03840200), breast cancer (NCT03424005, NCT03280563, NCT03395899, NCT03337724, NCT02162719, NCT03800836, NCT03840200), ovarian cancer (NCT03840200), glioblastoma (NCT03673787), cancer of unknown primary site (NCT03498521) or in advanced solid tumors in general (NCT01362374, NCT03673787). Recently, a phase I study on GDC-0068 reported radiographic stable disease as a preliminary antitumor activity in 30% of patients with diverse solid tumors who had previously progressed on prior therapies.21 Moreover, interim results of the double-blind placebo-controlled randomized FAIRLANE trial (NCT02301988) investigating neoadjuvant GDC-0068 plus paclitaxel for early triple-negative breast cancer revealed favorable radiographic response rates for the combination of GDC-0068 and paclitaxel.29 To date, there is no clinical response data in patients with brain metastases.
A previous preclinical study by Lin et al. assessing GDC-0068 in multiple in vitro and in vivo tumor models reported that this compound is particularly potent in tumors harboring genetic alterations such as loss or decreased activity of phosphatase and tensin homolog (PTEN), mutations or amplifications of PIK3CA and HER2 overexpression, which was validated in isogenic PTEN-knockout MCF10A cells.18 Furthermore, the authors demonstrated an orally administered dose of 100 mg/kg GDC-0068 daily in tumor models, with these genetic profiles resulting in response rates ranging from tumor growth inhibition to even regression. In contrast, mutations in the oncogenes BRAF and KRAS were found to be associated with resistance to treatment with GDC-0068.18 These results are in line with our in vitro and xenograft studies of PIK3CA-MT BCBM models, showing the favorable response of the BCBM cell line MDA-MB-361 (PIK3CA-MT, HER2+; BRAF-WT, KRAS-WT) and the resistance of the PIK3CA-WT cell line MDA-MB-231 BrM2 (HER2−; BRAF-MT, KRAS-MT30).
Collectively, our preclinical data support continued investigation of the highly selective, ATP-competitive pan-Akt inhibitor GDC-0068 as a possible therapeutic option in brain metastases patients. Future studies are also needed to explore GDC-0068 in combination with other targeted therapies. Upregulation of compensatory pathways like extracellular signal-regulated kinase activation and phosphorylation of human epidermal growth factor receptor 3 (HER3) following GDC-0068 treatment has been reported in studies by Yan et al, evaluating the downstream targets of xenografts and patient samples of refractory solid tumors.31 Furthermore, concomitant blockade of EGF receptor, HER3, and the PI3K pathway has recently demonstrated superior antitumor activity in a triple-negative breast cancer model.32 These results are in line with previous studies investigating PI3K/Akt/mTOR inhibitors,25,33,34 suggesting a promising combinatorial approach with compounds targeting those pathways subject to compensatory upregulation.
Nevertheless, GDC-0068 has shown encouraging tumor growth inhibition as a single agent with a significant survival benefit in our study. Therefore, further validation of GDC-0068 is warranted in clinical trials in BCBM patients with PI3K pathway alterations.
Supplementary Material
{kind=link}
{kind=link}
Funding
This work was supported by National Institutes of Health/National Cancer Institute R01 (CA227156-01 to PKB and SLC). This work is also supported by Damon Runyon Cancer Foundation (81-15 to PKB); Susan G. Komen for the Cure Career Catalyst Award (CCR16377965 to PKB); Breast Cancer Research Foundation (ELFF-18-001 to PKB); and American Brain Tumor Association (to PKB). This work is also supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, project number: 325246018 to FMI).
Conflict of interest statement.
FMI is supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, project number: 325246018). PKB received honoraria from Genentech and Merck, consultant fees from Lilly, Tesaro, Genentech-Roche, and Angiochem, and research funding to Massachusetts General Hospital from Merck and Pfizer. DPC received honoraria from Lilly and Merck. The other authors declare no potential conflicts of interest.
Authorship statement.
Conception and design: FMI, DPC, HW, PKB
Development of methodology: FMI, HW, PKB
Acquisition of data: FMI, JKG, MS, BMK, EML, EGP JLM, NN, SPS
Analysis and interpretation of data: FMI, JKG, AG-H, SPS, SLC, HW, PKB
Writing, review, and/or revision of the manuscript: FMI, DPC, HW, PKB
Administrative, technical, or material support: FMI, PKB
Study supervision: DPC, HW, PKB
Other (interpreted the histological analysis): MM-L
References
- 1. Eichler AF, Loeffler JS. Multidisciplinary management of brain metastases. Oncologist. 2007;12(7):884–898. [DOI] [PubMed] [Google Scholar]
- 2. Soffietti R, Ducati A, Rudà R. Brain metastases. In: Aminoff MJ, Boller F, Swaab DF, eds. Handbook of Clinical Neurology. Vol 105 Elsevier; 2012:747–755.doi:10.1016/B978-0-444-53502-3.00021-5. [DOI] [PubMed] [Google Scholar]
- 3. Kocher M, Soffietti R, Abacioglu U, et al. . Adjuvant whole-brain radiotherapy versus observation after radiosurgery or surgical resection of one to three cerebral metastases: results of the EORTC 22952-26001 study. J Clin Oncol. 2011;29(2):134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murray KJ, Scott C, Greenberg HM, et al. . A randomized phase III study of accelerated hyperfractionation versus standard in patients with unresected brain metastases: a report of the Radiation Therapy Oncology Group (RTOG) 9104. Int J Radiat Oncol Biol Phys. 1997;39(3):571–574. [DOI] [PubMed] [Google Scholar]
- 5. Witzel I, Oliveira-Ferrer L, Pantel K, Müller V, Wikman H. Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res. 2016;18(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shah R, Rosso K, Nathanson SD. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World J Clin Oncol. 2014;5(3):283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han CH, Brastianos PK. Genetic characterization of brain metastases in the era of targeted therapy. Front Oncol. 2017;7:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osswald M, Blaes J, Liao Y, et al. . Impact of blood-brain barrier integrity on tumor growth and therapy response in brain metastases. Clin Cancer Res. 2016;22(24):6078–6087. [DOI] [PubMed] [Google Scholar]
- 9. Adamo B, Deal AM, Burrows E, et al. . Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res. 2011;13(6):R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brastianos PK, Carter SL, Santagata S, et al. . Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5(11):1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saunus JM, Quinn MC, Patch AM, et al. . Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J Pathol. 2015;237(3):363–378. [DOI] [PubMed] [Google Scholar]
- 12. Sadeghi N, Gerber DE. Targeting the PI3K pathway for cancer therapy. Future Med Chem. 2012;4(9):1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Gooijer MC, Zhang P, Buil LCM, et al. . Buparlisib is a brain penetrable pan-PI3K inhibitor. Sci Rep. 2018;8(1):10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin NU. Breast cancer brain metastases: new directions in systemic therapy. Ecancermedicalscience. 2013;7:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blake JF, Xu R, Bencsik JR, et al. . Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J Med Chem. 2012;55(18):8110–8127. [DOI] [PubMed] [Google Scholar]
- 16. de Bono JS, De Giorgi U, Rodrigues DN, et al. . Randomized phase II study evaluating akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res. 2019;25(3):928–936. [DOI] [PubMed] [Google Scholar]
- 17. Kim SB, Dent R, Im SA, et al. ; LOTUS investigators Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin J, Sampath D, Nannini MA, et al. . Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013;19(7):1760–1772. [DOI] [PubMed] [Google Scholar]
- 19. Morgillo F, Della Corte CM, Diana A, et al. . Phosphatidylinositol 3-kinase (PI3Kα)/AKT axis blockade with taselisib or ipatasertib enhances the efficacy of anti-microtubule drugs in human breast cancer cells. Oncotarget. 2017;8(44):76479–76491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Orditura M, Della Corte CM, Diana A, et al. . Three dimensional primary cultures for selecting human breast cancers that are sensitive to the anti-tumor activity of ipatasertib or taselisib in combination with anti-microtubule cytotoxic drugs. Breast. 2018;41:165–171. [DOI] [PubMed] [Google Scholar]
- 21. Saura C, Roda D, Roselló S, et al. . A first-in-human phase i study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017;7(1):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21(2):255–262. [DOI] [PubMed] [Google Scholar]
- 23. Jernström S, Hongisto V, Leivonen SK, et al. . Drug-screening and genomic analyses of HER2-positive breast cancer cell lines reveal predictors for treatment response. Breast Cancer (Dove Med Press). 2017;9:185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hirai M, Kataoka Y, Minami H, Mukohara T, Shimada H, Saijo N. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2009;21(2):255–262. [DOI] [PubMed] [Google Scholar]
- 25. Gustin JP, Karakas B, Weiss MB, et al. . Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc Natl Acad Sci U S A. 2009;106(8):2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ippen FM, Alvarez-Breckenridge CA, Kuter BM, et al. . The dual PI3K/mTOR-pathway inhibitor GDC-0084 achieves antitumor activity in PIK3CA-mutant breast cancer brain metastases. Clin Cancer Res. 2019. doi: 10.1158/1078-0432.ccr-18-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Do J, Foster D, Renier C, et al. . Ex vivo Evans blue assessment of the blood brain barrier in three breast cancer brain metastasis models. Breast Cancer Res Treat. 2014;144(1):93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elkabets M, Vora S, Juric D, et al. . mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5(196):196ra199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oliveira M, Saura C, Calvo I, et al. . Abstract CT041: Primary results from FAIRLANE (NCT02301988), a double-blind placebo (PBO)-controlled randomized phase II trial of neoadjuvant ipatasertib (IPAT) + paclitaxel (PAC) for early triple-negative breast cancer (eTNBC). Cancer Res. 2018;78(13 Supplement):CT041. [Google Scholar]
- 30. Jacob LS, Vanharanta S, Obenauf AC, et al. . Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res. 2015;75(18):3713–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan Y, Serra V, Prudkin L, et al. . Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res. 2013;19(24):6976–6986. [DOI] [PubMed] [Google Scholar]
- 32. Tao JJ, Castel P, Radosevic-Robin N, et al. . Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014; 7(318):ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chandarlapaty S, Sawai A, Scaltriti M, et al. . AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serra V, Scaltriti M, Prudkin L, et al. . PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30(22):2547–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.