

Abstract
Background
Culture-independent methods such as quantitative polymerase chain reaction (qPCR) are more sensitive for detecting pathogens than conventional culture. This study aimed to test the clinical potential of a multiple target qPCR array in identifying sputum pathogens, compared to traditional culture.
Methods
Forty chronic obstructive pulmonary disease (COPD) patients provided spontaneous sputum and blood samples during an exacerbation event (n=25 patients) and in stable state (n=15 patients). Sputum was processed and analysed by microscopy, culture and sensitivity testing (MCS) to identify living microbial isolates, and multiple target qPCR (44 targets for bacterial and fungal pathogens and antibiotic resistance genes), and 16S rRNA gene sequencing.
Results
Six microbial isolates (5 bacterial, 1 fungal) were cultured from 20 exacerbation and 10 stable patient sputum samples. Four of these microbial isolates had their presence in patient sputum confirmed by qPCR. All bacterial targets detected by qPCR were further confirmed by 16S rRNA gene sequencing at a genus level. qPCR identified significantly more bacterial pathogens than culture (P<0.001). The most prevalent bacterial species identified by qPCR were Streptococcus pneumoniae (72% of patients), Pseudomonas aeruginosa (40%), Prevotella oris (32%) and Haemophilus influenzae (17%). Microbial species diversity and richness were not significantly different between samples obtained from exacerbating and clinically stable cases. 16S rRNA gene sequencing identified Pseudomonas 4408227 (P=0.022, FDR =0.043 AUC =0.72) as a significantly different bacterial OTU (operational taxonomic units) in exacerbation sputum samples compared to stable state samples.
Conclusions
Multiple target qPCR was more sensitive for detection of sputum pathogens in COPD patients than conventional culture. 16S rRNA gene sequencing confirmed the identity at a genus level of all bacterial targets detected by qPCR, as well as identifying bacterial OTUs that could potentially be used to distinguish between exacerbation and stable COPD disease states. Multiple target qPCR pathogen detection in the sputum of COPD patients warrants further investigation to determine how it may influence COPD clinical management.
Keywords: Chronic obstructive pulmonary disease (COPD), exacerbations, sputum, microbiome
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic lung disease characterized by persistent respiratory symptoms and progressive airflow limitation, due to exposure to noxious particles and gases (1). COPD exacerbations occur when there is an increase in patients’ symptoms above their usual baseline, resulting in a change in pharmacological treatment and/or hospitalisation (1). COPD exacerbations contribute to excessive morbidity and mortality worldwide.
Respiratory infections have been estimated to account for 70% of exacerbations of COPD (2,3). Bacteria are a major cause of infection in COPD exacerbations, with prevalence ranging from 26% to 81% (4). However, detection of bacteria by conventional sputum culture during exacerbations is not sensitive. Quantitative polymerase chain reaction (qPCR), a culture-independent molecular method for detection of bacteria, has identified significantly more bacterial species in exacerbations than culture-dependent methods (5,6).
It remains unclear why some patients experience more frequent exacerbations than others. The composition of the lung microbiome (the entire community of microorganisms that reside in the lung) has been reported to vary with an exacerbation event (7-9) and may play a role in altering the risk of exacerbations (10,11).
Given the low sensitivity of sputum culture in detecting bacteria during exacerbations, we evaluated the potential clinical utility of a multiple target qPCR array to detect bacteria, fungi and antibiotic resistance genes in exacerbating and stable COPD patients. We further utilized 16S rRNA gene sequencing to confirm qPCR targets as well as identify significant sputum bacterial OTUs that could potentially distinguish exacerbation and stable state in COPD patients.
Methods
Statement of ethics approval
The University of Queensland and The Prince Charles Hospital Human Research Ethics Committee has approved of the protocols (2016000248/HREC/15/QPCH/259) and informed written consent was obtained from all patients.
Patient recruitment
Patients with an exacerbation of COPD (n=25) were recruited consecutively within 4 days of admission to The Prince Charles Hospital, Brisbane, Australia. Inclusion was based on a clinical diagnosis of severe COPD exacerbation (acute deterioration in COPD symptoms requiring hospitalisation), as determined by a thoracic physician. Patients with other comorbid lung disease including predominant asthma based on a physician diagnosis, lung cancer, and interstitial lung disease were excluded. Fifteen other patients (outpatients with COPD who had stable symptoms for at least 4 weeks) were recruited as a control group (Figure 1).
Figure 1.

COPD patient cohorts (EX and ST) and sputum samples eligible for further pathogen analysis methods. COPD, chronic obstructive pulmonary disease. MCS, microscopy, culture and sensitivity testing.
Clinical data collection
Patient demographics and clinical data including smoking history, respiratory symptoms, comorbid health conditions and exacerbation history, were obtained by face-to-face interview. Prescribed treatments including steroid medication and antibiotics were recorded for each case. Lung function test results were obtained from medical records. Patients completed the COPD Assessment Test (CAT) (12) within the first 48 hours of admission to hospital [time point 1 (T1), exacerbation]. Clinically stable patients completed the CAT at a routine clinic appointment [time point 2 (T2), stable].
Sputum and blood sample collection and processing
A spontaneously expectorated sputum sample and blood samples were collected at each applicable time point (T1, T2). Details of sputum processing can be found in the supplementary file online. Microscopy, culture and sensitivity (MCS) testing was undertaken within 48 hours of each sputum sampling time. Sputum characteristics were recorded, and sputum plugs (13) were tested for total cell count and cell viability, and the remaining sputum plug fraction was stored at –80 °C for later nucleic acid extraction using the QIAamp Cador Pathogen Mini kit (QIAGEN, Hilden, Germany).
Respiratory infections qPCR
Microbial DNA Respiratory Infections qPCR array plates (QIAGEN) were used to test for 48 bacterial, fungal, and antibiotic resistance genes and control targets. Detailed methods can be found in the online supplementary file.
Microbiome analysis
16S rRNA gene sequencing was undertaken at the Australian Centre for Ecogenomics (ACE) at The University of Queensland, based on a standardised protocol (supplementary appendix, available online) (14). Amplicon 16S rRNA gene sequences were quality controlled and trimmed to 250 bases using Trimmomatic software (14). The resulting sequence data were processed through the QIIME pipeline to cluster sequence reads into operational taxonomic units (OTUs) and for taxonomic assignment of OTU reference sequences based on the Greengenes database. Rare OTUs with an abundance of less than 0.05% were removed. Microbial 16S rRNA gene sequence data were rarefied to 11,000 counts and visualized using Calypso (detailed Calypso analyses is available online in the supplementary file) (12).
Statistical analysis
Statistical analysis was performed using Statistical Package for Social Science (SPSS V.22) and using the Calypso platform. Continuous and categorical data were analysed using parametric and non-parametric tests including unpaired two-tailed t-tests, with Levene’s test for equality of variances and frequencies, Mann Whitney U test and chi square test, respectively. The significance level α was set to 0.05.
Results
Clinical and sputum characteristics, and blood inflammatory biomarkers differed between exacerbation and stable patients
We examined 40 sputum samples from 40 COPD patients (21 females, 19 males). Twenty-five patients each provided a sputum sample during an exacerbation of COPD (EX), and 15 other COPD patients each provided a sputum and blood sample during an outpatient visit while clinically stable (ST). Both cohorts (EX and ST) were older adults, and the majority were former smokers with severe GOLD class based on spirometry. Clinical characteristics were not different between exacerbation and stable patients, except for CAT score (15) which was significantly higher in the exacerbation cohort compared to stable (P=0.024), and current use of both antibiotics and oral steroids was significantly higher in exacerbation patients (P=3×10-6 and P=0.015, respectively) (Table 1).
Table 1. Clinical characteristics of exacerbation (n=25) versus stable (n=15) COPD patients.
| Clinical characteristic | Exacerbation | Stable | Total | Significance |
|---|---|---|---|---|
| Total patients (n) | 25 | 15 | 40 | – |
| Gender (n, %) | 2P=0.567 | |||
| Male | 11 [44] | 8 [53] | 19 (47.5) | |
| Female | 14 [56] | 7 [47] | 21 (52.5) | |
| Age (years) | 72.64 (7.80) | 71.00 (6.06) | 72.03 (7.16) | 1ᵉP=0.490 |
| BMI (kg/m2) | 25.34 (6.56) | 25.80 (5.27) | 25.52 (6.01) | 1ᵉP=0.819 |
| Smoking history (n, %) | 3P=0.567 | |||
| Former | 19 [76] | 12 [80] | 31 (77.5) | |
| Current | 4 [16] | 2 (13.3) | 6 [15] | |
| Never | 2 [8] | 1 (6.7) | 3 (7.5) | |
| Pack years | 54.17(30.09) | 53.54 (23.65) | 53.92 (27.41) | 1ᵉP=0.947 |
| Prior diagnosis of concurrent bronchiectasis (n, % yes) | 8 [32] | 3 [20] | 11 (27.5) | ᶠP=0.486 |
| FEV1% predicted | 41.14 (17.23) | 57.39 (28.67) | 47.55 (23.5) | 1ᵜP=0.062 |
| FEV1/VC | 0.46 (0.17) | 0.49 (0.20) | 0.47 (0.18) | 1ᵉP=0.639 |
| GOLD stage (n, %) | 4P=0.091 | |||
| 2 | 5 [20] | 7 [47] | 12 [30] | |
| 3 | 12 [48] | 2 (13.3) | 14 [35] | |
| 4 | 6 [24] | 3 [20] | 9 (22.5) | |
| CAT score | 24.10 (5.09) | 19.27 (7.94) | 22.29 (6.65) | 1ᵉP=0.024* |
| HS-CRP (mg/L) | 0.87 (0.80)' | 0.44 (0.70)' | 0.71 (0.78) | 1ᵉ'P=0.101 |
| Fibrinogen (g/L) | 4.15 [6] | 4.00 [4] | 4.10 [7] | 4P=0.329 |
| Antibiotics (n, % yes) | 19 [76] | 0 | 19 [47.5] | 2P=3×10-6** |
| Oral steroids (n, % yes) | 9 [36] | 0 | 9 (22.5) | ᶠP=0.015* |
| Colour | 4P=0.177 | |||
| Mucoid | 2 [8] | 3 [20] | 5 (12.5) | |
| Mucopurulent | 8 [32] | 6 [40] | 14 [35] | |
| Purulent | 15 [60] | 6 [40] | 21 (52.5) | |
| pH | 7.38 (0.78) | 6.55 (0.48) | 7.06 (0.79) | 1ᵜP=0.0003** |
| Total cell count (TCC) (×106) | 0.01 (0.48)' | −0.32 (0.31)' | −0.11 (0.45)' | 1ᵜ'P=0.013* |
| DNA concentration (ng/μL) | 2.13 (0.60)' | 1.95(0.72)' | 2.06 (0.65)' | 1ᵉP=0.413 |
| RNA concentration (ng/μL) | 2.10 (0.64)' | 1.95 (0.74)' | 2.04 (0.67)' | 1ᵉP=0.480 |
Categorical data represented as n (population %) or median (range). Continuous data represented as mean (standard deviation). *, P≤0.05; **, P≤0.001; 1, Independent t-test; 2, Pearson Chi square test of independence; 3, Chi square test of goodness of fit; 4, Mann Whitney U test; ᶠ, Fisher’s test; ᵉ, Levene’s test for equal variance assumed; ᵜ, Levene’s test for equal variance not assumed; ', Log10 analysis.
All patients (n=40) were able to produce a spontaneous sputum sample, but 5 patients needed a lung flute [device that produces a low frequency acoustic wave to increase mucus clearance (16)], in order to obtain a larger sputum volume. The exacerbation and stable patients had similar sputum characteristics, except that pH and total cell count (TCC ×106) were both significantly higher in the exacerbation cohort compared to the stable cohort (P=0.0003 and P=0.013) (Table 1).
Conventional sputum culture detected a restricted range of microbial pathogens
Sputum MCS was successfully performed in 20 of the 25 exacerbation patients and 10 of the 15 stable patients, and failed in 10 patients due to insufficient sputum volume (80%) and/or excessive oral contamination (70%).
Successful sputum cultures (n=20 EX, n=10 ST) reported normal respiratory flora [which typically includes Neisseria, Candida, Diphtheroids, Streptococcus and Staphylococcus (17)] or mixed bacterial species growth (with and without speciation). Only five bacterial isolates were identified at a species level, which included Acinetobacter baumannii (n=1), Haemophilus influenza (n=3), Pseudomonas aeruginosa (n=2), Staphylococcus aureus (n=2), and Stenotrophomonas maltophilia (n=1). One fungal isolate was identified at a genus level, Candida (n=6) (Figure 2). There was no statistically significant difference in the number of bacterial species cultured in sputum in the exacerbation cohort compared to the stable cohort.
Figure 2.

Quantitative visualization of qPCR microbial gene targets and culture positive targets. Forty-four bacterial, fungal and antibiotic resistance gene targets were successfully tested in 36 sputum samples using the QIAGEN’s Respiratory Infections Microbial DNA qPCR array. Samples were ordered by patient group, exacerbation cohort (n=25) and stable cohort (n=15). Highly positive (+) targets are shown in green, low positive (+/−) targets in yellow and negative (−) targets in grey. Five individual bacterial species and one fungal species were identified by culture.
Multiple target qPCR is a sensitive culture-independent technique enabling identification of sputum pathogens that may be missed by conventional culture
Sputum samples were analysed for pathogens using QIAGEN’s Respiratory Infections Microbial DNA qPCR arrays, testing for 44 microbial targets, including 38 bacterial species, 3 fungal species and 3 antibiotic resistance gene targets with internal controls (full list of target details including species available in Table S1). 36 of the 40 sputum samples showed amplification of qPCR targets (Figure 2). In the four samples from which no bacterial, fungal or antibiotic resistance gene targets were identified, the nucleic acid concentrations were below 77 ng/µL.
The total number of qPCR targets positively identified was not significantly different between exacerbation patients and stable patients. The most prevalent bacterial species in both cohorts was Streptococcus pneumoniae, positively identified in 29 (72.5%) patients (Figure 2), Pseudomonas aeruginosa in 16 (40%) patients, Prevotella oris in 13 (32.5%) patients and Haemophilus influenzae in 7 (17.5%) patients. Only one patient was positive for an individual fungal species (Pneumocystis jirovecii).
Bacteria represented on the respiratory Infections microbial DNA qPCR array (38 individual bacterial species) can be categorized into six phyla (Actinobacteria, Bacteroidetes, Chlamydiae, Firmicutes and Proteobacteria). Proteobacteria was the dominant phylum in 64% of the exacerbation patients, but only in 33% of stable patients (P=0.06) (Table 2). Firmicutes was the most abundant phylum in 20% of exacerbation patients and in 47% of stable patients (P=0.09). Bacteroidetes showed similar abundance levels in both cohorts and was the most abundant phylum in 8% of the exacerbation patients and 7% in the stable cohort.
Table 2. Dominant Respiratory Infections microbial DNA qPCR array targets (44 total). Exacerbation (n=23) and stable (n=13) patients were assigned a predominant phylum group (Proteobacteria, Firmicutes or Bacteroidetes). Phylum abundances were calculated by summing up the qPCR abundances of all bacterial targets belonging to the same phylum.
| Dominant phylum | Exacerbation | Stable | Total | Significance |
|---|---|---|---|---|
| Proteobacteria (n, %) | 16 (64.0) | 5 (33.3) | 21 (58.3) | 2P=0.060 |
| Firmicutes (n, %) | 5 (20.0) | 7 (46.6) | 12 (33.3) | ᶠ2P=0.09 |
| Bacteroidetes (n, %) | 2 (8.0) | 1 (6.6) | 3 (8.3) | ᶠ2P=1.00 |
Categorical data represented as n (population %). 2, Pearson Chi square test of independence; ᶠ, Fisher’s test.
All of the microbial isolates (5 bacterial, 1 fungal) positively identified by culture were also targets on the Respiratory Infections Microbial DNA qPCR array. Four of the 6 microbial isolates that were culture positive in patients’ sputum were confirmed by qPCR, with the exception of Acinetobacter baumannii (Figure 2) and Candida species. The patient who was culture positive for Acinetobacter baumannii had Acinetobacter calcoaceticus/and Acinetobacter rhizosphaerae species identified by qPCR (Figure 2), as well as by 16S rRNA gene sequencing (Figure 3). In the remaining patients who were culture positive for Haemophilus influenzae, Pseudomonas aeruginosa, Staphyloccus aureus and Stenotrophomonas maltophilia, these bacteria were also detected by qPCR. Six patients were positive for fungal species Candida on culture; however only 3 of these patients had Candida species detected by qPCR, under the Pan Aspergillus/Candida target. qPCR detected vastly more microbial targets (116 microbial targets total) than sputum culture (14 microbial targets total) (P<0.001).
Figure 3.

Quantitative visualisation of the culture positive targets (identified in EX and ST patient cohort’s sputum samples) and their presence confirmed by culture-independent methods qPCR (A) and 16S rRNA gene sequencing, at a species and genus level (B). Presence of bacteria are identified by primary colors and their blends; only culture positive (red), only qPCR positive targets (yellow), only 16S rRNA gene sequencing positive targets (blue), both culture and qPCR positive (red + yellow = orange), both qPCR and 16S rRNA gene sequencing positive (yellow + blue = green) and positive by culture, qPCR and 16S rRNA gene sequencing (red + yellow + blue = dark green).
Sputum 16S rRNA gene sequencing provides information about bacterial abundance in exacerbation and stable states of COPD
All 40 sputum samples (25 EX and 15 from ST patients) underwent high throughput sequencing of bacterial 16S rRNA gene encompassing the V1 to V3 regions. Microbial 16S sequence data was normalized by rarefying patients sequence reads to 11,000 per sample (this removed 2 patients for further analysis, n=24 EX, n=14 ST) and visualized using Calypso software, where sputum bacterial profiles between cohorts were assessed (Figure 4).
Figure 4.

Relative abundance of the top 20 prevalence bacterial OTUs. Patient sputum samples were categorized by exacerbation (EX) (n=24) or stability (ST) (n=14). Square size indicates relative abundance of bacterial OTUs. OTUs, operational taxonomic units.
Visualisation of beta diversity by PCoA using weighted UniFrac distances (see Supplementary Methods) revealed minimal separation of microbial communities based on disease state (EX vs. ST), indicating that sputum from patients during an exacerbation had a similar microbial community structure to sputum from stable patients (Figure S1). Further, there was no difference in microbial diversity (Shannon index, P=0.53) or microbial richness (P=0.48) between stable and exacerbation sputum samples (Figure S2).
Patients who were culture positive for the 5 detected bacterial species (Acinetobacter baumannii, Haemophilus influenzae, Pseudomonas aeruginosa, Staphylococcus aureus and Stenotrophomonas maltophilia) had these results recapitulated by 16S rRNA gene sequencing, however only at the genus level (Figure 3B), with the exception of Staphylococcus and Stenotrophomonas species (Figure 3).
Further, results from 16S rRNA gene sequencing were able to confirm all of the targets that were qPCR positive at a genus level.
16S rRNA gene sequencing identifies significant bacterial OTUs that can differentiate between COPD disease states
Differences in OTUs between exacerbation and stable states were identified by Wilcoxon rank test. The discriminatory power of bacterial OTUs to differentiate between the exacerbation and stable states is described by the area under the ROC curve (AUC), odds ratio, delta (difference in means in units of standard deviation) and fold change, and the top 10 discriminatory OTUs are listed in Figure 5. Pseudomonas 4408227 (P=0.022, FDR =0.043 AUC =0.72) was the strongest OTU associated with exacerbation.
Figure 5.

Top 10 bacterial OTUs associated with exacerbation (n=24) and stable (n=14) sputum samples. OTUs, operational taxonomic units.
Discussion
This study has demonstrated the feasibility and potential clinical utility of a comprehensive multiple target qPCR array for the detection of sputum pathogens in patients with COPD, compared to conventional culture. Furthermore, 16S rRNA gene sequencing results have identified significant sputum bacterial OTUs of interest that could potentially distinguish exacerbation and stable state in COPD patients and requires further investigation.
qPCR versus culture methodology
qPCR is a sensitive, culture-independent technique that enables the identification of potentially pathogenic microorganisms (PPMs) that may have otherwise not been detected by conventional culture (18). qPCR is therefore a potentially suitable tool to simultaneously detect multiple pathogens rapidly for clinical diagnostics (19).
Our study used the comprehensive Microbial DNA Respiratory Infections qPCR array to test COPD patients’ sputum samples during exacerbation and stable state. Results from this current study lend further support of multiple target qPCR arrays potential use in clinical practice, as they were able to identify more microbial targets that were not detected by routine culture, in approximately two-thirds of sputum samples sent for culture, and the targets that identified positive by qPCR were all confirmed by 16S rRNA gene sequencing at a genus level, with the exception of Staphylococcus species.
Our results suggest that in relation to which identification technique is more superior to detect pathogen presence in sputum, qPCR was able to recapitulate all bacterial species from culture results, with the exception of fungal species of the genus Candida, and bacterial species Acinetobacter baumannii, which was identified as positive under the qPCR target Acinetobacter calcoaceticus/Acinetobacter rhizosphaerae species, with this target further confirmed by 16S rRNA gene sequencing.
The high sensitivity of the multiple target qPCR array in comparison to conventional culture highlights the strength of this culture-independent technique, and suggests that its potential use should be further validated, with a view for future implementation into clinical practice, similar to nasopharyngeal swabs for respiratory virus identification.
Difference in microbial species identified in exacerbation versus stable COPD states
In this study, both exacerbation and stable COPD patients identified similar numbers of microbial species by three different identification techniques. The most common bacteria identified by qPCR in exacerbating and stable patients included Streptococcus pneumoniae, Pseudomonas aeruginosa, and Prevotella oris, whereas other studies have reported Haemophilus influenzae, Streptococcus pneumoniae and Staphylococcus aureus as the most prevalent bacteria (5,20-25). S. pneumoniae is usually the 2nd or 3rd most prevalent pathogen found in exacerbations, and is the most common cause of community-acquired pneumonia in the general population.
Our findings that exacerbating COPD patients had similar number of PPMs identified in sputum as stable COPD patients suggests that chronic colonization may contribute to chronic airway inflammation and disease progression, and is consistent with other studies suggesting that bacterial infection is driving an exacerbation (26).
Further support to chronic colonization of PPMs were the results of 16S rRNA gene sequencing of stable patients’ sputum, which identified Rothia mucilaginosa as significant. The bacterium Rothia mucilaginosa is associated with pneumonia and considered a part of the normal respiratory flora, but can also become an opportunistic pathogen, particularly in immunocompromised patients, therefore suggesting that stable COPD patients may be chronically colonized with Rothia mucilaginosa (27).
Additionally, our results support that the lung microbiome is influenced by the aspiration of opportunistic pathogens (28), such as Veillonella, Streptococcus and Prevotella (bacterial species identified by qPCR and confirmed by 16S rRNA gene sequencing to be significant to exacerbation and stable COPD patient sputum), which typically inhabit the oral cavity (29,30).
Culture results from exacerbation and stable cohorts were similar in the type of microbial organisms cultured. While S. pneumoniae was the most prevalent target identified by qPCR, as well as the most prevalent bacterial species identified by 16S rRNA gene sequencing, this bacterium was not detected by culture. False positives would be unlikely due to the inbuilt controls in each qPCR run. In addition, in sputum samples that were qPCR positive for Streptococcus, 16S rRNA gene sequencing further confirmed Streptococcus OTU sequences.
16S rRNA gene sequencing
Bacterial composition was profiled by sequencing the 16S rRNA gene—of which the transcribed rRNA is a component of the ribosome, which has remained highly conserved in bacteria (31). Sequencing the 16S rRNA gene in sputum samples describes the composition of natural microbial communities inhabiting an ecosystem (32), potentially identifying significant bacterial OTUs that can distinguish between exacerbation and stable COPD states.
Results from 16S rRNA gene sequencing highlighted that both exacerbation and stable sputum microbiomes were highly variable and diverse between individuals. We did not observe significant difference in species diversity or richness between exacerbation and stable cohorts. Previous studies have reported that bacterial microbiome diversity decreases with increasing COPD severity (33-35). Our results suggest that a reduction in species diversity does not necessarily disturb the microbiome.
16S rRNA gene sequencing is able to identify rare bacteria, slow-growing bacteria, and un-culturable bacteria. Further sequencing can identify potential candidates that could be used to distinguish significant exacerbation OTUs to stable OTUs. Therefore, it could be a useful complement to qPCR, particularly for COPD patients who are frequent exacerbators, allowing for more robust identification of which bacteria/s may be pathogenic in exacerbations. However further validation is needed to determine the contribution and significance of candidate bacteria identified by qPCR and 16S rRNA gene sequencing in exacerbations and stable state.
Limitations of this study
There are advantages and disadvantages of the pathogen identification techniques used in this study. Whilst qPCR may be more sensitive, a potential limitation arises from identification of DNA copies (of the 16S rRNA gene) of both viable and non-viable bacteria. However, this limitation may be offset by the short duration that DNA from lysed bacteria are known to remain in the airways, due to rapid phagocytosis by alveolar macrophages (36). Other limitations of this study include the relatively small sample size (25 exacerbation patients and 15 stable patients) and implications of exacerbation patients who received antibiotics, as this may have reduced the presence of sputum pathogens. Further 16S rRNA gene sequencing does not have the ability to resolve all species within a sample. For future studies using sputum samples as a non-invasive biofluid for microbiome studies, metagenomics sequencing should be considered, due to this methods ability to capture DNA sequences from all organisms within the sample, including viruses and fungi.
Conclusion
We have shown that a comprehensive multiple target qPCR array has potential clinical utility for the identification of sputum pathogens, in both exacerbation and stable patients with COPD, compared to conventional culture methods. Implementation of qPCR in clinical practice for pathogen detection in sputum needs to be further investigated, as it could potentially provide more rapid and comprehensive diagnostic treatment and management strategies for COPD.
Acknowledgments
We thank the patients and staff of The Prince Charles Hospital for their participation in this study. We would like to acknowledge Natalie Castrechini, PhD from QIAGEN for her technical assistance and support with the Microbial DNA qPCR Respiratory Infections Arrays.
Funding: The Prince Charles Hospital Foundation New Investigator Grant (NI2016-122).
Supplementary
Contents
Methods
Patient recruitment
Clinical data collection
Sputum and blood sample collection
Sputum sample processing
Nucleic acid extraction
Respiratory infections qPCR
Microbiome analysis
ACE PCR amplification and Amplicon sequencing protocol
Figures
Supplementary file Figure S1
Supplementary file Figure S2
Tables
Supplementary file Table S1. QIAGEN’s respiratory infections DNA qPCR array targets
Methods
Patient recruitment
Patients with an exacerbation of COPD (n=25) were recruited consecutively into this study within 4 days of admission to The Prince Charles Hospital, Brisbane, Australia. Inclusion was based on a clinical diagnosis of severe COPD exacerbation (acute deterioration in COPD symptoms, requiring hospitalization), as determined by a thoracic physician. Patients with other comorbid lung disease including predominant asthma based on a physician diagnosis, lung cancer and interstitial lung disease were excluded). For an additional independent control group, fifteen other hospital outpatients with stable COPD, defined as COPD symptoms stable for at least 4 weeks were included.
Clinical data collection
Patient demographics and respiratory data, including smoking history, respiratory symptoms, comorbid health conditions and exacerbation history, were obtained by face-to-face interview with the patients. Prescribed treatments including steroid medication and antibiotics were recorded for each case. Lung function test results were obtained from medical records. Patients completed the COPD Assessment Test (CAT) (15) within the first 48 hours of admission to hospital [time point 1 (T1), exacerbation CAT]. Clinically stable patients completed the CAT at a routine clinic appointment (time point 2, T2).
Sputum and blood sample collection
A spontaneously expectorated sputum sample and blood samples were collected at each applicable time point (T1 and T2). If a patient was unable to provide a spontaneous sputum sample at these times, sputum was collected after exhaling into an oscillating positive expiratory pressure (OPEP) device (Lung Flute, Medical Acoustics, Buffalo, NY, USA). Peripheral venous blood (10 mL from citrate and serum tubes) was collected by the hospital phlebotomists, and processed by the hospital pathology service for high sensitivity C-reactive protein (CRP) and fibrinogen, as inflammatory markers.
Sputum sample processing
The expectorated sputum sample was processed using the methods described below. If patients did not have sputum microscopy, culture and sensitivity (MCS) testing requested by their clinical team within 48 hours of each sputum sampling time for this study, then an aliquot of sputum collected for this research was sent for MCS testing at each applicable time point. The pH and weight of each sputum sample was recorded. Sputum colour was recorded in categories of increasing purulence: mucoid, mucoid purulent, purulent 1 or purulent 2 (37).
Sputum plugs [defined as sputum from lower airways with minimal saliva contamination, containing secretions, cellular debris and microorganisms (13)] were identified by visual examination and extracted from each expectorated sample. An aliquot of this plug fraction was then treated with dithiothreitol (DTT) at 1:10 dilution (Calbiochem, CA, USA) and 4× the volume of the aliquot. Plugs were dispersed on a mixing apparatus for 30–60 minutes. An equivalent volume of PBS was then added. The sputum mixture was filtered through a nylon filter to remove debris and then analysed for total cell count and cell viability using trypan blue dye exclusion. The remainder was stored frozen for later nucleic acid extraction.
Nucleic acid extraction
Nucleic acid extraction was performed using the QIAamp Cador Pathogen Mini kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instruction, using the automated QIAcube protocol. The lysis pretreatment option supplied by the Cador kit was employed to extract bacterial and fungal DNA. Briefly, up to 1.5 mL of frozen sputum plugs was added to pathogen lysis tubes and centrifuged for 5 minutes at maximum speed (>14,000 g). The supernatant was discarded and 500 µL of Buffer ATL was added to the sample before being subjected to vortex mixing at maximum speed for 10 minutes. Supernatant (200 µL) was used as the starting material for nucleic acid extraction. DNA and RNA quality and purity were assessed using the Nanodrop ND-100 spectrophotometer (Thermo Fisher Scientific, MA, USA).
Respiratory infections qPCR
Standard curves for each microbial qPCR assay were prepared as per manufacturer’s instructions using known copy numbers of microbial DNA positive control V2 (2,000, 1,000, 500 and 0 copies were generated), supplied by QIAGEN. Microbial DNA Respiratory Infections qPCR array plates (QIAGEN) were used to test for 48 bacterial, fungal, and antibiotic resistance genes and control targets. Briefly, 500 ng of DNA was added to microbial qPCR master mix and microbial DNA-free water to create the qPCR reaction mix. 10 µL of each qPCR reaction mix was loaded into a 384-well plate using customized plate loaders. The plate was sealed and centrifuged briefly before being run on a ViiA 7 real-time thermal cycler (Thermo Fisher Scientific, MA, USA). The cycle threshold (Ct) values were determined by the recommended settings from the Qiagen microbial DNA qPCR handbook for the ViiA 7 thermal cycler. The baseline was set to 8–20 cycles and threshold at 0.2. cycle threshold (Ct) values were then exported into Microsoft Excel. The ‘Respiratory Infections Microbial DNA qPCR Array Microbial Identification Data Analysis’ template (QIAGEN) was used to determine if the qPCR Ct values obtained for each target were a high positive (+), low positive (+/−) or negative (−). This was calculated using the ΔCt method, which measured the background of the assay. A No Template Control (NTC) was used to establish the lower Ct value for positive calls and the upper Ct value was used for negative calls. These Ct values were then used to determine the number of qPCR reaction copies for each positive target, using linear equations derived from the standard curves of each target. For bacteria, these qPCR reaction copies were then divided by the individual bacterial targets 16S gene copy number obtained from the ‘Ribosomal RNA database’ (38), to give a final 16S rRNA copies/500 ng total DNA input for each sample.
Microbiome analysis
16S rRNA gene sequencing was undertaken at the Australian Centre for Ecogenomics (ACE) at The University of Queensland, based on a standardised protocol detailed below (14).
ACE PCR amplification and Amplicon sequencing protocol
The 16S rRNA gene encompassing the V1 to V3 regions was targeted using the 27F (5'- AGAGTTTGATCMTGGCTCAG -3') and 519R (5'- GWATTACCGCGGCKGCTG -3') primers (14) modified to contain Illumina specific adapter sequence (27F: 5'- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAGAGTTTGATCMTGGCTCAG-3' and 519R: 5'- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGWATTACCGCGGCKGCTG-3'). The bacterial primer pair Bac_SSU_27F-519R amplifies the small subunit (SSU) ribosomal RNA of bacteria (16S) specifically the V1, V2 and V3 regions. In Escherichia coli, it amplifies the 27–519 region of the 16S gene.
Preparation of the 16S library was performed as described, using the workflow outlined by Illumina (#15044223 Rev.B). In the 1st stage, PCR products of ~466 bp were amplified according to the specified workflow with an alteration in polymerase used to substitute NEBNext® Ultra™ II Q5® Mastermix (New England Biolabs #M0544) in standard PCR conditions. Resulting PCR amplicons were purified using Agencourt AMPure XP beads (Beckman Coulter). Purified DNA was indexed with unique 8 bp barcodes using the Illumina Nextera XT 384 sample Index Kit A-D (Illumina FC-131-1002) in standard PCR conditions with NEBNext® Ultra™ II Q5® Mastermix. Indexed amplicons were pooled together in equimolar concentrations and sequenced on MiSeq Sequencing System (Illumina) using paired end sequencing with V3 300 bp chemistry in the Australian Centre for Ecogenomics according to manufacturer’s protocol.
Control reactions included in the amplicon library construction and sequencing include:
❖ Positive amplification control from a known mock community to monitor for bias in the amplicon library construction;
❖ Negative amplification control from a like processed reagent control to monitor for contamination in library construction;
❖ Single well empty chamber controls within processing plates to monitor for cross contamination within the library preparation;
❖ Negative index positions between runs to monitor for run to run bleed through designated as in line controls. Passing QC of resulting sequence is determined as 10,000 raw reads per sample prior to data processing and passing Quality Control metrics in line with Illumina supplied reagent metrics of overall Q30 for 600 bp reads of >70%.
Amplicon 16S rRNA gene sequences were quality controlled and trimmed to 250 bases using the Trimmomatic software. The resulting sequence data was processed through the QIIME pipeline for clustering sequence reads into operational taxonomic units (OTUs) and for taxonomic assignment of OTU reference sequences based on the Greengenes database. Rare OTUs with an abundance of less than 0.05% were removed. Microbial 16S rDNA data was rarefied to 11,000 counts per sample and visualized using Calypso, as per standard data filtering, normalization and transformation settings (18). Further Calypso software used the microbial 16S sequence data to provide visualisation of relative abundance, visualisation of beta diversity, as well as identification of significant OTUs between COPD exacerbation and stable patient’s sputum samples, by providing significance values, false discovery rate values and area under the curve values.
Figure S1.
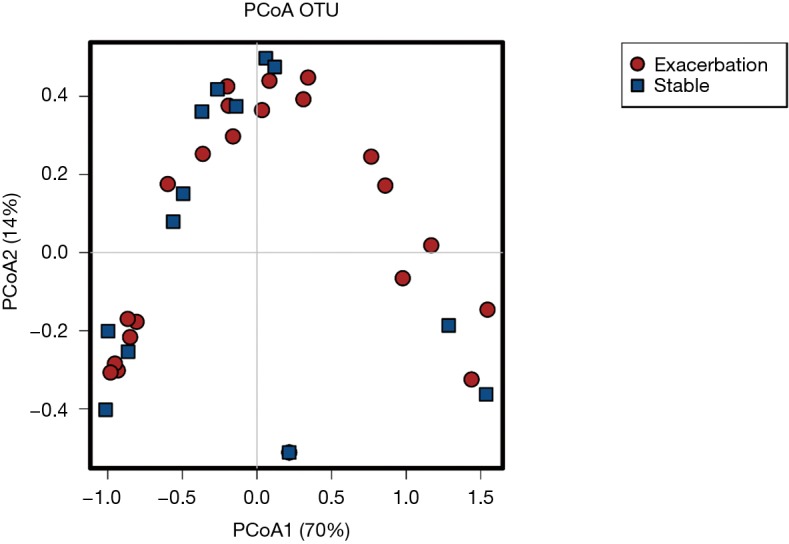
Principal coordinate analysis. Visualisation of β-diversity by PCoA using weighted UniFrac distances of bacterial 16S rRNA gene profiles from exacerbation (n=24) and stable sputum samples (n=14). The taxonomic information shows distributions of OTUs (points) coloured by exacerbation (red) or stability (blue). The proportion of the variation explained is indicated on the axes.
Figure S2.
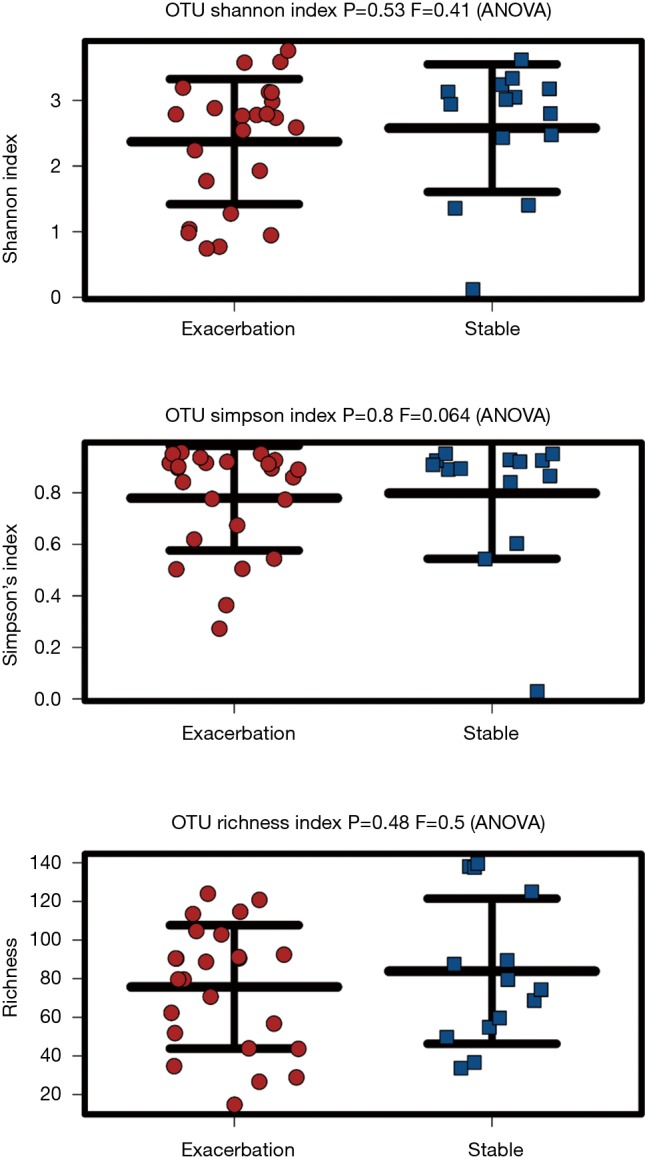
Microbial diversity assessment. From 16S rRNA gene sequencing, OTUs from exacerbation (n=24) and stable (n=14) sputum samples were assessed for microbial diversity. This was shown by Shannon and Simpson diversity index, which highlighted no statistically significant difference in species diversity between the two states (P=0.53, P=0.8) and Richness index, which highlighted no statistically significant difference in species richness between exacerbation and stable samples (P=0.48).
Table S1. QIAGEN’s respiratory infections DNA qPCR array targets.
| Respiratory infections (BAID-1404Z) array targets | Details |
|---|---|
| Name | Acinetobacter baumannii |
| Associated species | Acinetobacter baumannii |
| Accession | NZ_ABXK01000050.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 402870 |
| Description | Acinetobacter rhizosphaerae (243922) |
| Name | Acinetobacter calcoaceticus |
| Associated species | Acinetobacter calcoaceticus, Acinetobacter rhizosphaerae |
| Accession | GU385012.1 |
| Sensitivity (gene copies) | 50 |
| Green gene ID | 575203 |
| Name | Actinobacillus hominis |
| Associated species | Actinobacillus hominis |
| Accession | L06076.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 9577 |
| Name | Aspergillus flavus |
| Associated species | Aspergillus flavus |
| Accession | GU594735.1 |
| Sensitivity (gene copies) | 50 |
| Name | Aspergillus fumigatus |
| Associated species | Aspergillus fumigatus |
| Accession | KC119202.1 |
| Sensitivity (gene copies) | 20 |
| Name | Bacillus anthracis |
| Associated species | Bacillus anthracis, Bacillus weihenstephanensis, Bacillus cereus |
| Accession | NC_003997.3 |
| Sensitivity (gene copies) | 40 |
| Green gene ID | 85366 |
| Description | Bordetella bronchiseptica (518), Bordetella pertussis (520) |
| Name | Bordetella parapertussis |
| Associated species | Bordetella parapertussis, Bordetella bronchiseptica, Bordetella pertussis |
| Accession | BX640430.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 91830 |
| Description | Burkholderia vietnamiensis (60552), Burkholderia pyrrocinia (60550), Burkholderia cenocepacia (95486) |
| Name | Burkholderia cepacia |
| Associated species | Burkholderia cepacia, Burkholderia vietnamiensis, Burkholderia pyrrocinia, Burkholderia cenocepacia, Burkholderia gladioli |
| Accession | HF678361.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 165641 |
| Name | Burkholderia gladioli |
| Associated species | Burkholderia gladioli |
| Accession | AY586517.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 98973 |
| Description | Burkholderia pseudomallei (28450) |
| Name | Burkholderia mallei |
| Associated species | Burkholderia mallei, Burkholderia pseudomallei |
| Accession | NZ_AAHO01000091.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 562256 |
| Name | Chlamydia trachomatis |
| Associated species | Chlamydia trachomatis |
| Accession | NC_000117.1 |
| Sensitivity (gene copies) | 40 |
| Green gene ID | 50790 |
| Name | Chlamydophila pneumoniae |
| Associated species | Chlamydia pneumoniae |
| Accession | NC_002179.2 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 112346 |
| Name | Chlamydophila psittaci |
| Associated species | Chlamydia psittaci, Chlamydophila abortus |
| Accession | AB001816.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 2998 |
| Name | Clostridium sordellii |
| Associated species | [Clostridium] sordellii |
| Accession | FJ375837.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 353639 |
| Name | Corynebacterium diphtheriae |
| Associated species | Corynebacterium diphtheriae, Corynebacterium simulans |
| Accession | NC_002935.2 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 471201 |
| Name | Coxiella burnetii |
| Associated species | Coxiella burnetii |
| Accession | NC_002971.3 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 83477 |
| Description | Francisella novicida (264) |
| Name | Francisella tularensis |
| Associated species | Francisella tularensis, Francisella novicida |
| Accession | NC_008369.1 |
| Sensitivity (gene copies) | 40 |
| Green gene ID | 250583 |
| Name | Haemophilus influenzae |
| Associated species | Haemophilus influenzae, Haemophilus haemolyticus |
| Accession | AY613709.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 111359 |
| Name | Legionella pneumophila |
| Associated species | Legionella pneumophila |
| Accession | NC_002942.5 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 99499 |
| Name | Moraxella catarrhalis |
| Associated species | Moraxella catarrhalis |
| Accession | NR_028669.1 |
| Sensitivity (gene copies) | 40 |
| Green gene ID | 584250 |
| Description | Mycobacterium tuberculosis (1773), Mycobacterium bovis (1765) |
| Name | Mycobacterium africanum |
| Associated species | Mycobacterium africanum, Mycobacterium tuberculosis, Mycobacterium bovis |
| Accession | AF480605.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 32152 |
| Name | Mycobacterium avium |
| Associated species | Mycobacterium avium |
| Accession | NC_008595.1 |
| Sensitivity (gene copies) | 40 |
| Green gene ID | 175923 |
| Name | Mycobacterium kansasii |
| Associated species | Mycobacterium kansasii |
| Accession | AJ536035.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 77825 |
| Name | Mycobacterium tuberculosis |
| Associated species | Mycobacterium tuberculosis |
| Accession | NC_002755.2 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 54279 |
| Name | Mycoplasma pneumoniae |
| Associated species | Mycoplasma pneumoniae, Mycoplasma genitalium |
| Accession | NC_000912.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 108728 |
| Name | Neisseria meningitidis |
| Associated species | Neisseria gonorrhoeae, Neisseria cinerea, Neisseria meningitidis |
| Accession | NC_008767.1 |
| Sensitivity (gene copies) | 300 |
| Green gene ID | 187019 |
| Description | Nocardia cyriacigeorgica (135487), Nocardia abscessus (120957), Nocardia cummidelens (165807), Nocardia flavorosea (53429), Nocardia pseudobrasiliensis (45979), Nocardia fluminea (134984) |
| Name | Nocardia asteroides |
| Associated species | Nocardia asteroides, Nocardia cyriacigeorgica, Nocardia abscessus, Nocardia cummidelens, Nocardia flavorosea, Nocardia pseudobrasiliensis, Nocardia fluminea |
| Accession | AF430019.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 82196 |
| Name | Peptostreptococcus anaerobius |
| Associated species | Peptostreptococcus anaerobius |
| Accession | AY326462.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 89770 |
| Name | Pneumocystis jirovecii |
| Associated species | Pneumocystis jirovecii |
| Accession | JF442104.1 |
| Sensitivity (gene copies) | 100 |
| Name | Prevotella bivia |
| Associated species | Prevotella bivia |
| Accession | L16475.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 2217 |
| Name | Prevotella oris |
| Associated species | Prevotella oris |
| Accession | NZ_ACUZ02000034.1 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 484401 |
| Description | Proteus vulgaris(585) |
| Name | Proteus mirabilis |
| Associated species | Proteus mirabilis, Proteus vulgaris, Candidatus Hamiltonella |
| Accession | AY167953.1 |
| Sensitivity (gene copies) | 200 |
| Green gene ID | 76398 |
| Name | Pseudomonas aeruginosa |
| Associated species | Pseudomonas aeruginosa |
| Accession | NC_002516.2 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 48315 |
| Name | Rhodococcus equi |
| Associated species | Rhodococcus equi |
| Accession | AF420418.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 64672 |
| Description | Xanthomonas retroflexus(305959),Pseudomonas geniculata(86188) |
| Name | Stenotrophomonas maltophilia |
| Associated species | Stenotrophomonas maltophilia, Xanthomonas retroflexus, Pseudomonas geniculata |
| Accession | AB008509.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 7826 |
| Name | Streptobacillus moniliformis |
| Associated species | Streptobacillus moniliformis |
| Accession | AB330757.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 255526 |
| Name | Streptococcus agalactiae |
| Associated species | Streptococcus agalactiae |
| Accession | NC_004116.1 |
| Sensitivity (gene copies) | 30 |
| Green gene ID | 59530 |
| Name | Streptococcus pneumoniae |
| Associated species | Streptococcus infantis, Streptococcus oralis, Streptococcus pneumoniae, Streptococcus pseudopneumoniae, Streptococcus mitis |
| Accession | NC_003028.3 |
| Sensitivity (gene copies) | 20 |
| Green gene ID | 53325 |
| Name | Streptococcus pyogenes |
| Associated species | Streptococcus pyogenes |
| Accession | NC_004606.1 |
| Sensitivity (gene copies) | 50 |
| Green gene ID | 81122 |
| Description | Yersinia pseudotuberculosis (633) |
| Name | Yersinia pestis |
| Associated species | Yersinia pseudotuberculosis, Yersinia rohdei |
| Accession | NC_010465.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 261946 |
| Name | Staphylococcus aureus |
| Associated species | Staphylococcus aureus, Staphylococcus epidermidis |
| Accession | NZ_ACOT01000039.1 |
| Sensitivity (gene copies) | 100 |
| Green gene ID | 588044 |
| Description | Beta-lactam resistance |
| Name | mecA |
| Accession | KC243783.1 |
| Sensitivity (gene copies) | 40 |
| Description | Panton-Valentine leukocidin chain F precursor |
| Name | lukF |
| Associated species | Staphylococcus aureus |
| Accession | VFG1276 |
| Sensitivity (gene copies) | 20 |
| Virulence Factor Database ID | VFG1276 |
| Description | Immunoglobulin G binding protein A precursor |
| Name | spa |
| Associated species | Staphylococcus aureus |
| Accession | VFG1313 |
| Sensitivity (gene copies) | 200 |
| Virulence Factor Database ID | VFG1313 |
| Name | Pan Aspergillus/Candida |
| Associated species | Aspergillus |
| Accession | X70659.1 |
| Name | Pan Bacteria 1 |
| Accession | HQ640630.1 |
| Name | Pan Bacteria 3 |
| Accession | DQ889917.1 |
| Description | Positive PCR control assay |
| Name | PPC |
| Accession | MB_PPC |
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The University of Queensland and The Prince Charles Hospital Human Research Ethics Committee has approved of the protocols (2016000248/HREC/15/QPCH/259) and informed written consent was obtained from all patients.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.GOLD. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease - 2018 Report 2018. Available online: http://goldcopd.org/wp-content/uploads/2017/11/GOLD-2018-v6.0-FINAL-revised-20-Nov_WMS.pdf
- 2.Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010;363:1128-38. 10.1056/NEJMoa0909883 [DOI] [PubMed] [Google Scholar]
- 3.Miravitlles M, Anzueto A. Role of infection in exacerbations of chronic obstructive pulmonary disease. Curr Opin Pulm Med 2015;21:278-83. 10.1097/MCP.0000000000000154 [DOI] [PubMed] [Google Scholar]
- 4.Sapey E, Stockley RA. COPD exacerbations . 2: aetiology. Thorax 2006;61:250-8. 10.1136/thx.2005.041822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcha DS, Thurston SJ, Patel AR, et al. Changes in prevalence and load of airway bacteria using quantitative PCR in stable and exacerbated COPD. Thorax 2012;67:1075-80. 10.1136/thoraxjnl-2012-201924 [DOI] [PubMed] [Google Scholar]
- 6.Sethi S. Molecular diagnosis of respiratory tract infection in acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis 2011;52 Suppl 4:S290-5. 10.1093/cid/cir044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fodor AA, Klem ER, Gilpin DF, et al. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 2012;7:e45001. 10.1371/journal.pone.0045001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tunney MM, Einarsson GG, Wei L, et al. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med 2013;187:1118-26. 10.1164/rccm.201210-1937OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang YJ, Sethi S, Murphy T, et al. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol 2014;52:2813-23. 10.1128/JCM.00035-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko FW, Chan KP, Hui DS, et al. Acute exacerbation of COPD. Respirology 2016;21:1152-65. 10.1111/resp.12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw JG, Vaughan A, Dent AG, et al. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J Thorac Dis 2014;6:1532-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zakrzewski M, Proietti C, Ellis JJ, et al. Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017;33:782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson PG, Girgis-Gabardo A, Morris MM, et al. Cellular characteristics of sputum from patients with asthma and chronic bronchitis. Thorax 1989;44:693. 10.1136/thx.44.9.693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelbrektson A, Kunin V, Wrighton KC, et al. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J 2010;4:642-7. 10.1038/ismej.2009.153 [DOI] [PubMed] [Google Scholar]
- 15.Jones PW, Harding G, Berry P, et al. Development and first validation of the COPD Assessment Test. Eur Respir J 2009;34:648. 10.1183/09031936.00102509 [DOI] [PubMed] [Google Scholar]
- 16.Sethi S, Yin J, Anderson PK. Lung flute improves symptoms and health status in COPD with chronic bronchitis: A 26 week randomized controlled trial. Clin Transl Med 2014;3:29. 10.1186/s40169-014-0029-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis C. In: Baron S. editor. Medical Microbiology. 4th ed. Galveston (TX), 1996. [Google Scholar]
- 18.Shimizu K, Yoshii Y, Morozumi M, et al. Pathogens in COPD exacerbations identified by comprehensive real-time PCR plus older methods. Int J Chron Obstruct Pulmon Dis 2015;10:2009-16. 10.2147/COPD.S82752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curran T, Coyle PV, McManus TE, et al. Evaluation of real-time PCR for the detection and quantification of bacteria in chronic obstructive pulmonary disease. FEMS Immunol Med Microbiol 2007;50:112-8. 10.1111/j.1574-695X.2007.00241.x [DOI] [PubMed] [Google Scholar]
- 20.Monso E, Rosell A, Bonet G, et al. Risk factors for lower airway bacterial colonization in chronic bronchitis. Eur Respir J 1999;13:338-42. 10.1034/j.1399-3003.1999.13b20.x [DOI] [PubMed] [Google Scholar]
- 21.Rosell A, Monso E, Soler N, et al. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med 2005;165:891-7. 10.1001/archinte.165.8.891 [DOI] [PubMed] [Google Scholar]
- 22.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008;359:2355-65. 10.1056/NEJMra0800353 [DOI] [PubMed] [Google Scholar]
- 23.Bafadhel M, Haldar K, Barker B, et al. Airway bacteria measured by quantitative polymerase chain reaction and culture in patients with stable COPD: relationship with neutrophilic airway inflammation, exacerbation frequency, and lung function. Int J Chron Obstruct Pulmon Dis 2015;10:1075-83. 10.2147/COPD.S80091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barker BL, Haldar K, Patel H, et al. Association between pathogens detected using quantitative polymerase chain reaction with airway inflammation in COPD at stable state and exacerbations. Chest 2015;147:46-55. 10.1378/chest.14-0764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simpson JL, Baines KJ, Horvat JC, et al. COPD is characterized by increased detection of Haemophilus influenzae, Streptococcus pneumoniae and a deficiency of Bacillus species. Respirology 2016;21:697-704. 10.1111/resp.12734 [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson TM, Hurst JR, Perera WR, et al. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest 2006;129:317-24. 10.1378/chest.129.2.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maraki S, Papadakis IS. Rothia mucilaginosa pneumonia: a literature review. Infect Dis (Lond) 2015;47:125-9. 10.3109/00365548.2014.980843 [DOI] [PubMed] [Google Scholar]
- 28.Pragman AA, Lyu T, Baller JA, et al. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome 2018;6:7. 10.1186/s40168-017-0381-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha R, Weissenburger-Moser LA, Clarke JL, et al. Short term dynamics of the sputum microbiome among COPD patients. PLoS One 2018;13:e0191499. 10.1371/journal.pone.0191499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zakharkina T, Heinzel E, Koczulla RA, et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by T-RFLP and clone sequencing. PLoS One 2013;8:e68302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monso E. Microbiome in chronic obstructive pulmonary disease. Ann Transl Med 2017;5:251. 10.21037/atm.2017.04.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 1990;87:4576-9. 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erb-Downward JR, Thompson D, Han MK, et al. Analysis of the microbiome of the normal and COPD lung [Conference Abstract]. Am J Respir Crit Care Med 2010;181 (1 MeetingAbstracts).
- 34.Sze MA, Hogg JC, Sin DD. Bacterial microbiome of lungs in COPD. Int J Chron Obstruct Pulmon Dis 2014;9:229-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pragman AA, Kim HB, Reilly CS, et al. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One 2012;7:e47305. 10.1371/journal.pone.0047305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcha D, Thurston S, Patel A, et al. Association of airway bacterial load with inhaled corticosteroid dosage in stable COPD [Conference Abstract] Eur Respir J 2012;40. [Google Scholar]
- 37.Murray MP, Pentland JL, Turnbull K, et al. Sputum colour: a useful clinical tool in non-cystic fibrosis bronchiectasis. Eur Respir J 2009;34:361-4. 10.1183/09031936.00163208 [DOI] [PubMed] [Google Scholar]
- 38.Stoddard SF, Smith BJ, Hein R, et al. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res 2015;43:D593-8. 10.1093/nar/gku1201 [DOI] [PMC free article] [PubMed] [Google Scholar]