

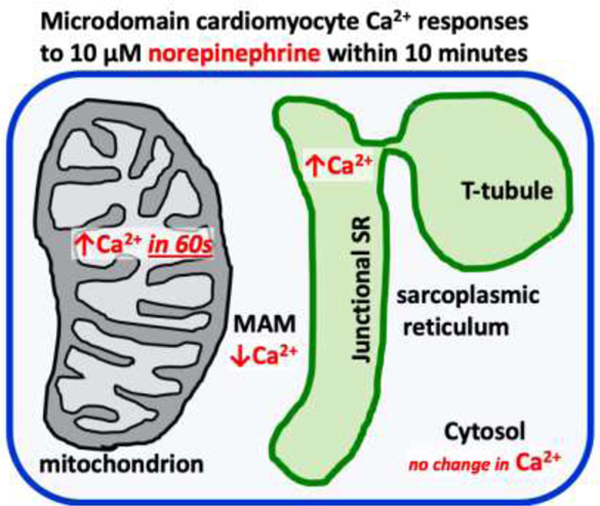
Abstract
Calcium (Ca2+), an important second messenger, regulates many cellular activities and varies spatiotemporally within the cell. Conventional methods to monitor Ca2+ changes, such as synthetic Ca2+ indicators, are not targetable, while genetically encoded Ca2+ indicators (GECI) can be precisely directed to cellular compartments. GECIs are chimeric proteins composed of calmodulin (or other protein that changes conformation on Ca2+ binding) coupled with two fluorescent proteins that come closer together after an increase in [Ca2+], and enhance Förster energy transfer (FRET) that allows for ratiometric [Ca2+] assessment. Here, adult rat ventricular myocytes were transfected with specifically targeted calmodulin-based GECIs and Ca2+ responses to a physiological stimulus, norepinephrine (NE, 10 µM), were observed in a) sarcoplasmic reticulum (SR), b) mitochondria, c) the space between the mitochondria and SR, termed the Mitochondria Associated Membrane space (MAM) and d) cytosol for 10 min after stimulation. In SR and mitochondria, NE increased the [Ca2+] ratio by 17% and by 8%, respectively. In the MAM the [Ca2+] ratio decreased by 16%, while in cytosol [Ca2+] remained unchanged. In conclusion, adrenergic stimulation causes distinct responses in the cardiomyocyte SR, mitochondria and MAM. Additionally, our work provides a toolkit-update for targeted [Ca2+] measurements in multiple cellular compartments.
Keywords: FRET imaging, calcium signaling, cellular compartments, mitochondrial calcium, Cameleon, mitochondrial calcium uniporter dominant negative
Graphical abstract
Introduction
In contractile tissues such as skeletal muscle or myocardium, large amplitude cytosolic Ca2+ cycling, responsible for the regulation of contraction and relaxation, have been well described using a variety of indicators (Paredes et al., 2008). However, assessment of calcium dynamics between different subcellular compartments is crucial for understanding the role of Ca2+ in normal and pathological cell signaling and function (Balaban, 2009; Boyman et al., 2015; Brookes et al., 2004; Chami et al., 2008; Csordás and Hajnóczky, 2009; Liu and O’Rourke, 2009; Luptak et al., 2018, 2005; Raffaello et al., 2016; Santulli et al., 2015). Assessment of these low amplitude, low frequency Ca2+ transients on the background of large oscillation of cytosolic free [Ca2+] has been particularly challenging (Berridge et al., 2003; Bers, 2008).
Ca2+ indicators fall into two major categories: a) synthetic Ca2+ indicators (such as fura-2, indo1, fluo-3, calcium green) and b) genetically-encoded Ca2+ indicators (GECI) (Palmer and Tsien, 2006). The synthetic indicators are suitable for assessment of cytosolic [Ca2+] due to their large dynamic ranges, but they cannot be targeted to specific cellular compartments and are poorly sensitive in measuring low nanomolar concentrations of calcium (Paredes et al., 2008). In contrast, GECIs can be directed to specific cellular compartments, and be used to assess nanomolar [Ca2+] ranges (Horikawa et al., 2010). Of the GECIs, the Cameleons, that utilize calmodulin and Förster resonance-energy transfer (FRET) (Miyawaki et al., 1997; Palmer et al., 2006; Palmer and Tsien, 2006), collectively have the largest detection range of Ca2+ encompassing low nanomolar (Kd~15nM) to micromolar concentrations (Kd~64μM). Cameleons are chimeric proteins that change conformation to Ca2+ binding and increase FRET between two fluorescent proteins. This allows for convenient ratiometric Ca2+ fluorescent assessment (Miyawaki et al., 1997; Palmer et al., 2006; Palmer and Tsien, 2006).
To evaluate the role of calcium in cardiomyocyte compartments, we designed Cameleon probes to allow calcium imaging in: a) sarcoplasmic reticulum (SR), b) mitochondria, c) the space between the mitochondria and SR, termed the Mitochondria Associated Membrane space (MAM), and d) cytosol. For each compartment, the response to caffeine-induced SR Ca2+ release was used to determine the appropriate sensitivity for Ca2+ detection.
In cardiomyocytes, norepinephrine (NE), a well-established physiological stimulus, induces robust inotropic and lusitropic effects associated with Ca2+ channels, phospholamban and troponin I phosphorylation (Bers, 2002; Mayourian et al., 2018). As these effects of norepinephrine are driven by alpha- and beta-adrenoceptor activation, we used alpha- and beta-adrenoceptor antagonists (prazosin and propranolol, respectively) to reveal the contribution of NE’s effect on subcellular [Ca2+] responses. To date, a complex evaluation of signaling Ca2+ responses to a single stimulus (e.g. NE) in multiple subcellular compartments has not been achieved in cardiomyocytes.
In summary, we characterized Ca2+ changes in subcellular compartments in response to NE in primary cultures of adult rat ventricular myocytes (ARVM) (Ellingsen et al., 1993; Kuster et al., 2005). Furthermore, by using an updated toolkit for targeted [Ca2+] measurements in various compartments, we show that this approach allows Ca2+ measurements in multiple cells simultaneously.
Methods
Cell Culture.
ARVMs were isolated from the hearts of adult (200 to 220 g) male Sprague-Dawley rats as described previously (Kuster et al., 2005).
Cell Treatments.
L-Norepinephrine (10 μmol/L; Sigma) was added immediately before measurements. In some experiments, DL-Propranolol (2 μmol/L; Sigma) or Prazosin (100 nmol/L; Sigma) were added 60 minutes before L-norepinephrine. Ascorbic acid (100 μmol/L; Sigma) was added to prevent oxidation of L-norepinephrine. Thapsigargin (5 μmol/L, Sigma) was added 10 minutes prior to experiments.
Adenoviral Constructs.
The plasmid encoding mitochondrial calcium uniporter (MCU) was obtained from Addgene clone 31726 (Baughman et al., 2011). The 3-threonine amino acid was changed to an alanine to match the reference sequence and a stop codon was added. Using site directed mutagenesis, amino acids D261 and E264 were both changed to glutamine, which leads to a dominant negative mitochondrial calcium uniporter (DNMCU) (Rasmussen et al., 2015). A primer of tgaagtatgttactggctgcatgatctgcc-aggaatattcccacca and its reverse complement were used for the mutagenesis reaction. The clone was confirmed by sequencing (Genewiz). An adenovirus was created using the AdEasy system and β- galactosidase was used as control as previously described (Pimentel et al., 2006). All calcium biosensor adenoviral constructs were created using the AdEasy system as well.
Cloning of Cameleon-based Ca2+ biosensors targeted to various cellular compartments.
Cameleon clones with Kd of 1uM (D1) (Palmer and Tsien, 2006), ~600nM (D3) (Palmer and Tsien, 2006), 64 uM (D4 Addgene clone 37473) (Palmer et al., 2006), ~140nM (Addgene clone 51966-yellow Cameleon-Nano 140) (Horikawa et al., 2010), and 15nM (Addgene clone 51961-yellow Cameleon-Nano 15) (Horikawa et al., 2010) were used as the reporter constructs. Together, we will refer to these as Cameleon clones. To target particular compartments, we cloned or sub-cloned the following sequences: (1) The MAM targeting sequence was cloned from total C57BL/6J mouse cardiac mRNA and consisted of the first 67 AA of the DGAT2 sequence (Stone et al., 2009). The forward sequencing primer was “gtgacgtgcattggcttcag” and reverse “cctagcaccaggaaggatagg”. (2) The mitochondrial targeting sequence with four copies of the subunit VIII of human cytochrome C oxidase (Palmer and Tsien, 2006) was sub-cloned from the mitoD3CPV (kind gift from R.Y.Tsien). (3) The SR targeting sequence was sub-cloned from the D1ER clone (kind gift from R.Y.Tsien) (Palmer and Tsien, 2006) and sub-cloned onto D3CPV and 140Kd clones with a C-terminal KDEL sequence attached on each using PCR. All cloning and sequencing primers were supplied by Fisher. All clones were sequenced to ensure the appropriate sequences were obtained.
Cell imaging.
ARVM’s were transfected for 48 hours and then imaged at 37°C using an inverted Olympus Spinning Disk confocal microscope in wide field mode. The light source was Prior Lumen Pro 220. All optical filters were obtained from Chroma: excitation 436/20 nm, dichroic 455LP, emission YFP 535/30 nm, emission CFP 480/40 nm. The cardiac myocytes were localized with light microscopy and subsequently visualized for fluorescence excited at 426–446 nm while data from CFP and YFP emission were collected using a rapid emission filter changer (Prior). All images were taken with a plan-apochromatic 20×/0.4NA (Olympus). Data were analyzed using NIS Elements software (Nikon). Ratiometric images were calculated using background-subtracted YFP emission divided by background-subtracted CFP emission. Cells were observed over a period of 20 s with image acquisition every second after caffeine (20mM) administration. Cells were observed over a period of 10 minutes with image acquisition every 20–30 s after NE (10uM) in the presence and absence of propranolol (2uM) and/or prazosin (100nM). Only rod-shaped cardiomyocytes were included in the analysis.
To prove the correct localization of the probes 100nM of ER Tracker Red (Invitrogen) and Mito Tracker Red FM (Invitrogen) were added for 15 minutes and washed immediately prior to imaging. Red localization dyes were needed as the FRET probes’ emission wavelengths overlap with green fluorescent imaging. For the localization of the SR and MAM probes, structured illumination microscope (SIM) images were acquired using the Nikon N-SIM super resolution system and a SR Apo TIRF 100× 1.49NA objective (Nikon Instruments, Melville, NY). SIM image processing was performed using the previously published 3D reconstruction methodology (Gustafsson et al., 2008) and NIS-Elements software. For the localization of the mitochondrial probe confocal imaging was performed using a LSM710 system (Zeiss, Thornwood, NY). All images were taken with a Plan-Apochromat 100×/1.4NA oil objective (Zeiss). The 488 nm argon laser line was used for imaging YFP. Emitted fluorescence was collected in the range of 507 to 549 nm. Excitation of Mito Tracker was performed using the 543 nm laser. Emission was collected in the range of 561 to 735 nm. For further confirmation of localization of the endoplasmic reticulum probe confocal imaging was performed using Leica SP5 Confocal Microscope (Leica, Buffalo Grove, IL). All images were taken with a Plan-Apochromat 63 ×/1.4NA oil objective (Leica). The 405 nm argon laser line was used for imaging YFP. Emitted fluorescence was collected in the range of 464 to 600 nm. Excitation of ER tracker red was performed using the 594 nm laser. Emission was collected in the range of 603 to 700 nm. The cytosolic probe was localized using an Olympus microscope with a Plan-Apochromatic 60×/0.85NA objective (Olympus) and emission YFP 535/30 nm.
Determination of the correct Cameleon Ca2+ affinity for the individual cellular compartments.
The correct Kd for each sub-cellular compartment was determined with caffeine-induced SR Ca2+ release. Caffeine rapidly depletes the SR of Ca2+ leading to a dramatically increased cytosolic (Bers, 2008) and mitochondrial Ca2+ levels (Pan et al., 2013). The probes were tested for caffeine responsiveness at varying Kd levels based on prior literature reports (Horikawa et al., 2010; Palmer et al., 2006; Palmer and Tsien, 2006). Caffeine (20 mM) was rapidly administered and the cells were imaged every second for a total of 20 seconds. For probes directed to cellular compartments where caffeine increases [Ca2+] (mitochondria and MAM), the lowest Kd clone that was still responsive to caffeine was used for the experiments. Conversely, for the SR probe, caffeine is expected to decrease SR [Ca2+], and the reciprocal approach was taken: the highest Kd probe that still reacted to caffeine was selected for further use. (Fig 2A).
Fig 2. Responses of the Cameleons to caffeine-induced SR calcium release in different cellular sub-compartments.

Only rod-shaped cardiomyocytes were included in the analysis. Green color represents the lowest ratio (lowest [Ca2+]), red color represents the highest ratio (highest [Ca2+]). (A) endo-/sarcoplasmic reticulum (ER), decrease in [Ca2+]; (B) mitochondria, increase in [Ca2+] within 3 seconds; and (C) MAM, increase in [Ca2+] within 3 seconds. (D) cytosol, increase in [Ca2+] within 4 seconds.
Calibration of the Cameleon probes.
For calibration, cardiomyocytes were incubated in a potassium phosphate Hepes solution (in mM): Hepes (20), KCl (140), MgCl2, (1.51), KH2PO4 (0.5), BAPTA (5) and BAPTA-AM (0.01) set at pH = 7.4 with KOH for cytosolic probe and pH = 8 for the mitochondrial probe to mimic the pH in mitochondrial matrix. The Rmin was determined after membrane permeabilization. The Rmax was reached by adding small volumes of CaCl2 (10 or 50 mM stock) in stepwise manner to generate the calibration curve (Wüst et al., 2017). After conversion of the x-axis to logarithmic scale, the dose-response curve was fitted using GraphPad Prism software and 50% effective [Ca2+] (EC50) was calculated for each probe. Fitted curves were shown with ± 95% confidence interval boundaries (Fig 4).
Fig 4. Calibration of the Cameleon probes.
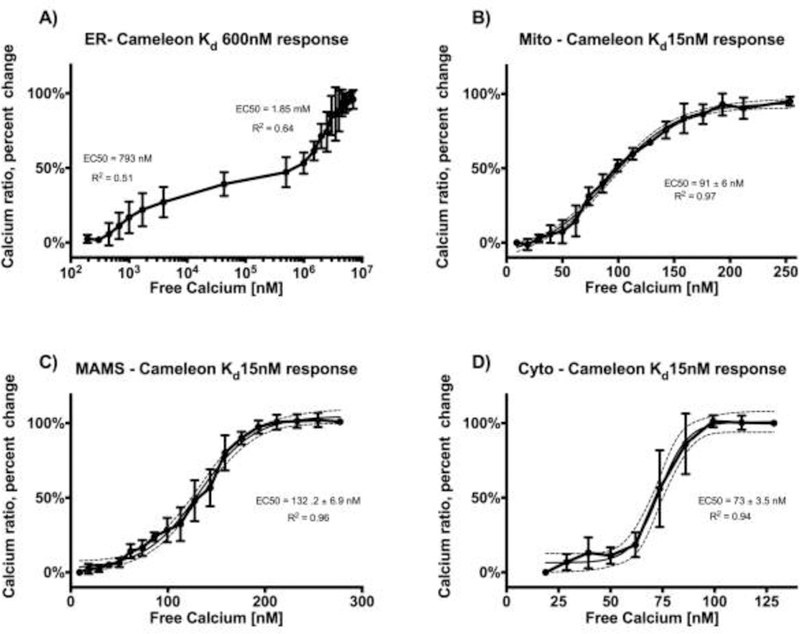
For calibration, cardiomyocytes were incubated in a potassium phosphate Hepes buffer with BAPTA 5nM and set at pH = 7.4 for cytosolic, SR and MAM probes (Panel A, C, D) and pH = 8 for mitochondrial probe (Panel B) to mimic pH in mitochondrial matrix. Ionomycin (5 uM) was added to permeabilize the membranes and to determine Rmin. Small volumes of CaCl2 were added in stepwise manner to generate the calibration curve until the Rmax was reached. After conversion of the x-axis to logarithmic scale, the dose-response curve was fitted using GraphPad Prism software and 50% effective [Ca2+] (EC50) was calculated for each probe. Fitted curves shown with ± 95% confidence interval boundaries (n=3–4 for each calibration).
Statistical analysis.
Results are presented as mean ± SEM. Comparisons between groups were performed using unpaired t-tests, Mann-Whitney non-parametric tests or 2-way or repeated measures ANOVA, as appropriate. All statistical analyses were performed using GraphPad Prism software. P-value < 0.05 was considered significant.
Results
Localization of the Cameleon probes
For SR localization, we used the calsequestrin targeting sequence from D1ER (Palmer and Tsien, 2006) in conjunction with a C-terminal KDEL retention sequence. Confocal microscopy showed overlap (Fig.1 panel A3) between the D3 ER probe (Fig.1 panel A1) and ER tracker (Fig.1 panel A2). In addition, high resolution SIM imaging confirmed the placement of the ER Tracker that binds to the sulfonylurea receptors on the outside of the SR membrane. In the image shown (Fig 1B), the Cameleon probe in the SR lumen is surrounded by the ER Tracker on the outside of the SR membrane proving appropriate localization of the Ca2+ sensing probe. Caffeine was administered and diminution of the ratio was measured. We found that the D3 probe responded appropriately to caffeine and had a dual measured Kd of 793 nM and 1.85 mM (Fig 4A).
Fig 1. Localization of the Cameleon probes to sub-cellular compartments.

(A and B): For SR localization, we utilized an N-Terminal calreticulin leader sequence coupled with a KDEL C-terminal sequence to place the probe in the ER lumen. In (A) overlap with ER tracker is shown by confocal microscopy, in (B) co-localization with ER tracker, which binds to receptors on the outside of the ER, is shown by SIM imaging. (C): For mitochondrial localization, we found that a 4× leader sequence of cytochrome C oxidase was required to lead the probe to a mitochondrial localization. Co-localization with mitotracker is shown. (D) and (E): For mitochondrial associated membrane (MAM) space, the leader sequence for DGAT2, a protein that has been reported to be present in the mitochondrial membrane space (MAM) was used. In (D) co-localization with ER tracker is shown, in (E) co-localization with mitotracker is shown.
For mitochondrial localization, we assessed the use of single to multiple copies (Filippin et al., 2005) of the targeting sequence from subunit VIII of human cytochrome C oxidase and found that a 4× leader sequence was required for successful localization of the probe to the mitochondria (Fig 1C). Similar strategies to promote mitochondrial targeting have been used previously (Wüst et al., 2017). To search for the correct Ca2+ affinity of the mitochondrial probe, a similar iterative process to the ER probe was necessary. Despite the correct localization confirmed by SIM imaging (Fig. 1C), higher Kd Cameleon probes were minimally responsive to caffeine but the Cameleon probe with an in vitro-reported Kd = 15 nM still maintained responsivity to caffeine (Fig 2B). Our calibration showed that the Kd of the probe when localized to the mitochondrial matrix of our cell type is about 6-fold higher, (EC50 91 ± 6 nM) (Fig.4B), and this value is similar to previously reported mitochondrial matrix Ca2+ levels using a different probe (Wüst et al., 2017).
Since the interplay between SR and mitochondria is important in the regulation of mitochondrial [Ca2+] (Raffaello et al., 2016), we also targeted a Cameleon probe to the SR-mitochondrial space, termed the Mitochondrial Associated Membrane (MAM) space. Using the DAGT2 leader sequence, the probe was targeted to and localized in the MAM space (Stone et al., 2009) as confirmed by SIM imaging (Fig1, D and E). The Cameleon probe with a Kd = 15 nM was reactive to caffeine in this region. The actual Kd for this probe when calibrated in our cells was found to be more than 6-fold higher (EC50 132.2 ± 6.9 nM). (Fig.4C),
Lastly, no targeting sequence was needed for cytosolic localization. Similar to the mitochondrial and MAM probes, we found that the probe with Kd 15nM was reactive to caffeine. Our calibration for the cytosolic probe revealed an estimated Kd around 60–80 nM which is consistent with previously reported values for cytosolic calcium concentrations (Boyman et al., 2013) (Fig. 4D).
NE increases free Ca2+ in the SR.
After stimulation with NE for 10 minutes, the calcium fluorescence ratio in the SR increased by 16.5 ± 2.7% vs. baseline (Fig 3A). Propranolol inhibited the NE-stimulated increase in Ca2+ ratio by 73%, whereas the alpha-adrenergic inhibitor, prazosin, caused a small, non-significant decrease. The use of both inhibitors almost completely (by 90%) prevented the Ca2+ uptake by SR after stimulation with NE.
Fig 3. Changes in free [Ca2+] to 10 uM norepinephrine (NE):
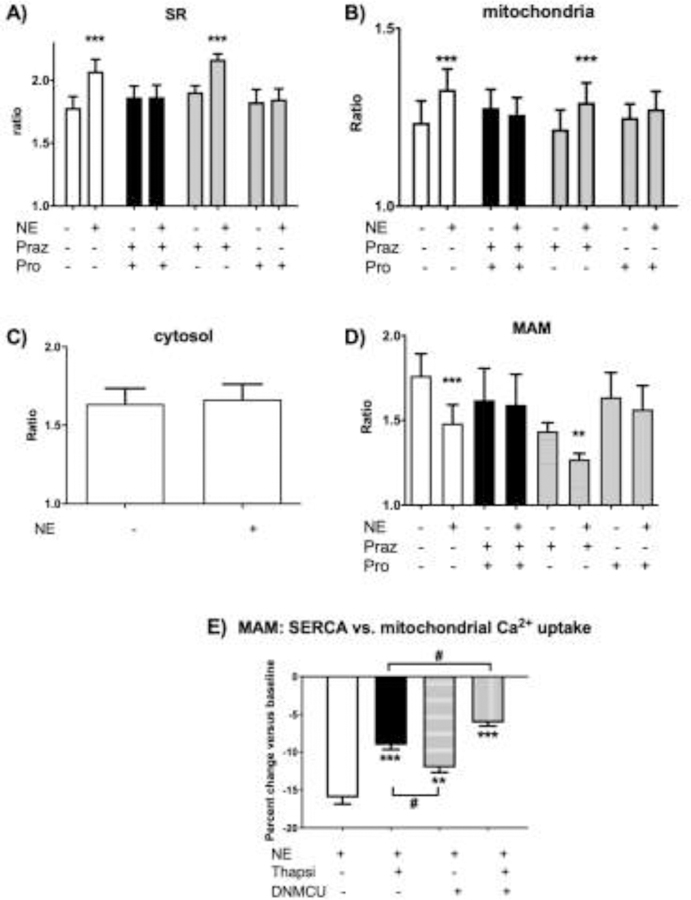
(A) SR [Ca2+] increases. Cameleon Ca2+ ratio in SR increased by 16.5 ± 2.7% compared to baseline after the stimulus. Inhibition with propranolol (Pro) reduced the signal by 89% while the reduction of the NE response by an alpha-adrenergic inhibitor, prazosin (Praz) was not statistically significant. The use of both inhibitors (Praz + Pro) completely prevents the uptake of calcium by SR after stimulation with NE.
(B) Mitochondrial [Ca2+] increases. NE increased mitochondrial free [Ca2+] by 8%. Dual adrenergic receptor inhibition (Praz + Pro) completely prevented the uptake and beta-adrenergic inhibitor propranolol (Pro) almost completely prevented the increase.
(C) Cytosolic [Ca2+] remains unchanged after norepinephrine (NE). The Ca2+ratio of the Cameleon Kd probe overexpressed in the cytosol did not change within 10 min after the stimulus with 10 uM concentration of NE.
(D) MAM [Ca2+] decreases. NE decreased Cameleon Ca2+ ratio in the mitochondrial associated membrane (MAM) space by 8%. The NE Ca2+ signal in MAM was almost completely (by 90%) prevented by dual inhibition of the beta- and alpha- receptors (Praz + Pro): a) Beta-adrenergic blockade (Pro) prevents the Ca2+ decrease by 73% while b) prazosin (Praz) alone has no significant effect.
(E) MAM [Ca2+] decrease is attenuated by inhibition of SERCA and mitochondrial Ca2+ uptake. NE decreased the calcium ratio in MAM by 16%. Inhibition with Thapsigargin 5μM (Thapsi) attenuated the Ca2+ decrease by 55%. DNMCU expression attenuated the Ca2+ decrease by 25%. Inhibition of both reduced the Ca2+ uptake from MAM by 62%. (n=5–7, ***p<0.001, ** p<0.01 vs control; #p<0.05 vs. groups as outlined).
NE increases free Ca2+ in the mitochondria.
As visualized with the mitochondria-directed probe, NE increased mitochondrial calcium ratio by 7.8 ± 1.0%. The increase was evident already at 60 seconds after NE application, and preceded the increase we observed in SR which occurred over ~ 10 min. Propranolol inhibited the NE-induced increase in mitochondrial Ca2+ by 86%. There was a small non-significant inhibition by prazosin, and the combination of propranolol and prazosin almost completely inhibited the increase (Fig 3B). This suggests mostly direct activation of calcium entry to mitochondria unrelated to SERCA activation by NE.
NE decreases free Ca2+ in the Mitochondrial Associated Membrane (MAM) space.
In contrast to the NE-stimulated increases observed in the SR and mitochondria, the calcium ratio in the MAM space decreased by 15.8 ± 1.4% (Fig 3D). Similar to the NE-stimulated increases in SR this effect was most evident after 10 min since NE application and was decreased by propranolol (73%), with only a non-significant decrease with prazosin and almost complete inhibition by the combination of propranolol and prazosin. To elucidate the contribution of both SERCA uptake and mitochondrial Ca2+ uptake to the decrease of Ca2+ in MAM space, we inhibited these with either thapsigargin or mitochondrial calcium uniporter (MCU) dominant negative viral construct (DNMCU), respectively (Fig 3E). NE decreased the calcium ratio by 16.0 ± 0.9%. Inhibition with thapsigargin attenuated the Ca2+ decrease by 55% (to 9.1 ± 0.6%, p<0.0001). DNMCU expression attenuated the Ca2+ decrease by 25% (to 12 ± 0.6%, p<0.01). Inhibition of both reduced the Ca2+ uptake from MAM further, by 62% (to 6.1 ± 0.4%, p<0.0001) (Fig 3E). Thus, both Ca2+ uptake by SERCA and mitochondrial uptake via MCU seem to be important mechanisms mediating the decrease in MAM [Ca2+] after adrenergic stimulation.
NE does not alter Ca2+ in the cytosol.
Overexpression of the probe with no leader sequence resulted in diffuse distribution in the cytosol. NE caused no change in cytosolic Ca2+ ratio over 10 minutes (Fig 3C).
Discussion
The Ca2+ transients responsible for excitation-contraction coupling have been well described using small molecule Ca2+ indicators in the cytosol (Bers, 2008). However, in addition to these large cytosolic Ca2+ transients, there are smaller Ca2+ movements within the localized regions of the cell (Berridge, 2006). Given their low amplitude relative to the much larger cytosolic contractile Ca2+ these transients have been difficult to characterize (Berridge, 2006; Berridge et al., 2003). Here we show signaling Ca2+ changes in different cellular compartments of cultured, non-beating adult rat ventricular myocyte (ARVM) stimulated by NE. ARVM’s are commonly used to study cardiac growth, apoptosis and SR stress (Ellingsen et al., 1993), representing a useful model system to test methods for Ca2+ measurements.
The development of the genetically encoded calcium indicators (GECI) can be affected by two major pitfalls: First, the subcellular targeting of the probe can be challenging and second, the response range of the probe may not match the reported in vitro values (Filippin et al., 2005; O’Malley et al., 1999). In our experiment, the targeting was particularly difficult in the case of the mitochondrial probe, where a single targeting sequence proved to be insufficient for the correct probe localization. In fact, quadrupling of the subunit VIII COX targeting sequence was necessary to ensure any mitochondrial localization. Similar approach to mitochondrial targeting of a different probe was described previously (Filippin et al., 2005; Wüst et al., 2017).
In all four subcellular localizations, the measured intracellular Kd was different from the in vitro reported Kd value (Fig 4). This difference likely results from the different molecularly dense microenvironments (O’Malley et al., 1999) and could not be accurately predicted prior to the experiments. Thus, extensive iterative trials were required to arrive at appropriate in vitro [Ca2+] sensitivities. Nevertheless, our measured Kd values are consistent with the prior literature for each cellular sub-compartment. The calibration of both cytosolic (measured EC50 73 ± 3.5 nM) and mitochondrial (measured EC50 91 ± 6 nM) probes proved that each of Kd values are within the expected range of previously reported [Ca2+] for the respective compartment (Raffaello et al., 2016; Wüst et al., 2017). Similarly, the Kd of the SR probe, with its double dissociation constant as detected by our calibration (with higher EC50 at 1.85mM) falls into the previously reported range as well (Greotti et al., 2016; Raffaello et al., 2016). The MAMs probe calibration revealed a cellular Kd higher than the cytosolic probe (EC50 132.2 ± 6.9 nM). This likely reflects a more molecularly dense environment in MAMs compared to the cytosol and also makes the probe suitable to assess higher [Ca2+] in proximity of “Ca2+ hot spots” (Giacomello et al., 2010).
In the sarcoplasmic reticulum, NE increased [Ca2+], as expected. This effect was mostly due to beta-adrenergic stimulation, although there may have been a small contribution from alpha-adrenergic stimulation as there was complete attenuation when both beta- and alpha-receptors were blocked. The beta-adrenergic rise in SR calcium was likely due to activation of influx via SERCA. A small alpha-receptor mediated effect may reflect influx via store operated channels (Bartoli and Sabourin, 2017).
In mitochondria, calcium concentrations as high as ~600nM, have been estimated in contracting cardiac myocytes during systole (Wüst et al., 2017; Zaglia et al., 2017). Since our cells were not contracting, systolic flux of calcium from the cytosol was absent, thus resulting in the lower measured [Ca2+]. Additionally, since Cameleon probes respond to free Ca2+, and much of the mitochondrial Ca2+ is buffered (Coll et al., 1982; Wei et al., 2012), our results are to be expected to be on the lower end of the previously published [Ca2+] range. The calibration of the mitochondrial probe (measured IC50 91 ± 6 nM) suggests that [Ca2+]mito in our model ranges between 60 – 150 nM, which is consistent with previously published data (Wüst et al., 2017). NE stimulated an increase in mitochondrial [Ca2+]. Similar to the SR, in mitochondria, the effect of NE was largely blocked by propranolol, with some further inhibition with the addition of prazosin, suggesting that the increase was mediated primarily by the beta-adrenergic signaling. Unlike the SR, the increase in mitochondrial Ca2+ was faster and occurred as early as 60 s after NE stimulation. This suggests a direct effect of NE on mitochondrial Ca2+ uptake, possibly through stimulation of the mitochondrial calcium uniporter (MCU) (Finkel et al., 2015; Pan et al., 2013; Wu et al., 2015).
In the Mitochondria Associated Membrane space, in contrast to mitochondria, NE decreased the [Ca2+]. As in the SR, this effect of NE was primarily mediated by beta-receptors, although a small alpha-adrenergic component may have contributed. The MAM represents space where the SR and mitochondria are in close proximity that facilitates Ca2+ transfer (Csordás and Hajnóczky, 2009; Raffaello et al., 2016; Stone et al., 2009; Wang et al., 2018). The NE-stimulated increase in mitochondrial Ca2+ occurred rapidly over about 60 seconds, whereas the NE-stimulated decrease in the MAM was slower, evolving over about 10 minutes, a time course similar to that for the NE-stimulated increase in the SR. We found that inhibition of either SR or mitochondrial uptake attenuated the NE-induced decrease in MAM calcium. This suggests that both the SR and mitochondria participate in regulation of MAM [Ca2+]. These data coupled with the differential temporal relationship suggests that the decrease in MAM [Ca2+] is related to interplay between SR and mitochondrial uptake. Our findings are consistent with the reports suggesting that MAM contains SERCA and represents a place of SR/mitochondrial cross-talk (Chami et al., 2008; Raffaello et al., 2016; Rizzuto et al., 1998).
Conclusion
In quiescent primary culture ARVM, we used genetically-targeted Cameleon probes to monitor Ca2+ in subcellular locations. Under conditions that had no effect on [Ca2+] in the cytosol, adrenergic stimulation caused directionally and temporally distinct responses in the SR, mitochondria and MAM. This targeted approach provides better understanding of the role Ca2+ plays in signaling in and between these compartments in cardiac myocytes and could be easily applicable to other primary cell culture systems as well.
Highlights:
Updated toolkit to track Ca2+ movements between cardiomyocyte sub-compartments
Norepinephrine (NE) increases Ca2+ in sarcoplasmic reticulum and mitochondria
NE decreases Ca2+ in mitochondria associated membrane space via MCU and SERCA
Ca2+ uptake appears faster in mitochondria than in sarcoplasmic reticulum
Acknowledgments
Sources of Funding
Supported by NIH grants HL-064750 (WSC), NIDDK R01 DK103750 (MMB) and the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178, WSC). Dr. Bachschmid was also funded by American Heart Association ‘‘Grant in Aid’’ 16GRNT27660006. Dr. Luptak is the recipient of an AHA Fellow-to-Faculty Award 15FTF25890062.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ivan Luptak, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
Robert Morgan, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
Dominique Croteau, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
Daniel Moverman, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
Hannah Sarnak, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
Wilson S. Colucci, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
David R. Pimentel, Myocardial Biology Unit, Boston University School of Medicine, Boston, MA.
References
- Balaban RS, 2009. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta - Bioenerg 1787, 1334–1341. 10.1016/j.bbabio.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoli F, Sabourin J, 2017. Cardiac Remodeling and Disease: Current Understanding of STIM1/Orai1- Mediated Store-Operated Ca2+ Entry in Cardiac Function and Pathology, in: Store-Operated Ca2+ Entry (SOCE) Pathways pp. 523–534. 10.1007/978-3-319-57732-6 [DOI] [PubMed]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, 2006. Calcium microdomains: Organization and function. Cell Calcium 40, 405–412. 10.1016/j.ceca.2006.09.002 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL, 2003. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol 4, 517–529. 10.1038/nrm1155 [DOI] [PubMed] [Google Scholar]
- Bers DM, 2008. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol 70, 23–49. 10.1146/annurev.physiol.70.113006.100455 [DOI] [PubMed] [Google Scholar]
- Bers DM, 2002. Cardiac Excitation-Contraction Coupling. Nature 415, 198–205. 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- Boyman L, Williams GSB, Khananshvili D, Sekler I, Lederer WJ, 2013. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol 59, 205–213. 10.1016/j.yjmcc.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman L, Williams GSB, Lederer WJ, 2015. The growing importance of mitochondrial calcium in health and disease. Proc. Natl. Acad. Sci. USA 112, 11150–11151. 10.1073/pnas.1514284112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS, 2004. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am.J.Physiol Cell Physiol 287, C817–C833. 10.1152/ajpcell.00139.2004 [DOI] [PubMed] [Google Scholar]
- Chami M, Oulès B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Bréchot P, 2008. Role of SERCA1 Truncated Isoform in the Proapoptotic Calcium Transfer from ER to Mitochondria during ER Stress. Mol. Cell 32, 641–651. 10.1016/j.molcel.2008.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll KE, Joseph SK, Corkey BE, Williamson JR, 1982. Determination of the matrix free Ca2+ concentration and kinetics of Ca2+ efflux in liver and heart mitochondria. J. Biol. Chem 257, 8696–8704. [PubMed] [Google Scholar]
- Csordás G, Hajnóczky G, 2009. SR/ER-mitochondrial local communication: Calcium and ROS. Biochim. Biophys. Acta - Bioenerg 1787, 1352–1362. 10.1016/j.bbabio.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingsen O, Davidoff AMYJ, Prasad SK, Marsh JD, Kelly RA, Smith W, Davidoff J, Springhorn P, Smith TW, 1993. Adult rat ventricular myocytes cultured in defined medium: phenotype and electromechanical function. Am. J. Physiol. Hear. Circ. Physiol 265, H747–H754. 10.1152/ajpheart.1993.265.2.H747 [DOI] [PubMed] [Google Scholar]
- Filippin L, Abad MC, Gastaldello S, Magalhães PJ, Sandonà D, Pozzan T, 2005. Improved strategies for the delivery of GFP-based Ca2+sensors into the mitochondrial matrix. Cell Calcium 37, 129–136. 10.1016/j.ceca.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Finkel T, Menazza S, Holmström KM, Parks RJ, Liu Julia, Sun J, Liu, Jie, Pan X, Murphy E, 2015. The ins and outs of mitochondrial calcium. Circ. Res 116, 1810–1819. 10.1161/CIRCRESAHA.116.305484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T, 2010. Ca2+ Hot Spots on the Mitochondrial Surface Are Generated by Ca2+ Mobilization from Stores, but Not by Activation of Store-Operated Ca2+ Channels. Mol. Cell 38, 280–290. 10.1016/j.molcel.2010.04.003 [DOI] [PubMed] [Google Scholar]
- Greotti E, Wong A, Pozzan T, Pendin D, Pizzo P, 2016. Characterization of the ER-targeted low affinity Ca2+probe D4ER. Sensors (Basel) 16, 1419 10.3390/s16091419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW, 2008. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J 94, 4957–4970. 10.1529/biophysj.107.120345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa K, Yamada Y, Matsuda T, Kobayashi K, Hashimoto M, Matsu-Ura T, Miyawaki A, Michikawa T, Mikoshiba K, Nagai T, 2010. Spontaneous network activity visualized by ultrasensitive Ca2+indicators, yellow Cameleon-Nano. Nat. Methods 7, 729–732. 10.1038/nmeth.1488 [DOI] [PubMed] [Google Scholar]
- Kuster GM, Pimentel DR, Adachi T, Ido Y, Brenner DA, Cohen RA, Liao R, Siwik DA, Colucci WS, 2005. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation 111, 1192–1198. 10.1161/01.CIR.0000157148.59308.F5 [DOI] [PubMed] [Google Scholar]
- Liu T, O’Rourke B, 2009. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr 41, 127–132. 10.1007/s10863-009-9216-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R, 2005. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 112, 2339–2346. 10.1161/CIRCULATIONAHA.105.534594 [DOI] [PubMed] [Google Scholar]
- Luptak I, Sverdlov AL, Panagia M, Qin F, Pimentel DR, Croteau D, Siwik DA, Ingwall JS, Bachschmid MM, Balschi JA, Colucci WS, 2018. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J. Mol. Cell. Cardiol 116, 106–114. 10.1016/j.yjmcc.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayourian J, Ceholski DK, Gonzalez DM, Cashman TJ, Sahoo S, Hajjar RJ, Costa KD, 2018. Physiologic, Pathologic, and Therapeutic Paracrine Modulation of Cardiac Excitation-Contraction Coupling. Circ. Res 122, 167–183. 10.1161/CIRCRESAHA.117.311589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki a, Llopis J, Heim R, McCaffery JM, Adams J. a, Ikura M, Tsien RY, 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887. 10.1038/42264 [DOI] [PubMed] [Google Scholar]
- O’Malley DM, Burbach BJ, Adams PR, 1999. Confocal Microscopy, Humana Press Inc; Totowa, New Jersey: 10.1385/159259722X [DOI] [Google Scholar]
- Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY, 2006. Ca2+Indicators Based on Computationally Redesigned Calmodulin-Peptide Pairs. Chem. Biol 13, 521–530. 10.1016/j.chembiol.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Palmer AE, Tsien RY, 2006. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc 1, 1057–1065. 10.1038/nprot.2006.172 [DOI] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, 2013. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol 15, 1464–1472. 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD, 2008. Chemical calcium indicators. Methods 46, 143–151. 10.1016/j.ymeth.2008.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel DR, Adachi T, Ido Y, Heibeck T, Jiang B, Lee Y, Melendez JA, Cohen RA, Colucci WS, 2006. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J. Mol. Cell Cardiol 41, 613–622. 10.1016/j.yjmcc.2006.05.009 [DOI] [PubMed] [Google Scholar]
- Raffaello A, Mammucari C, Gherardi G, Rizzuto R, 2016. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci 41, 1035–1049. 10.1016/j.tibs.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TP, Wu Y, Joiner MA, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song L-S, Zingman LV, Anderson ME, 2015. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 112, 9129–9134. 10.1073/pnas.1504705112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T, 1998. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca 2+ Responses. Science 280, 1763–1766. 10.1126/science.280.5370.1763 [DOI] [PubMed] [Google Scholar]
- Santulli G, Xie W, Reiken SR, Marks AR, 2015. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U. S. A 112, 11389–11394. 10.1073/pnas.1513047112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Zhou P, Han J, Walther TC, Farese RV, 2009. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem 284, 5352–5361. 10.1074/jbc.M805768200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Fernandez-Sanz C, Sheu SS, 2018. Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim. Biophys. Acta Mol. Basis Dis 1864, 1991–2001. 10.1016/j.bbadis.2017.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei A-C, Liu T, Winslow RL, O’Rourke B, 2012. Dynamics of matrix-free Ca 2+ in cardiac mitochondria: two components of Ca 2+ uptake and role of phosphate buffering. J. Gen. Physiol 139, 465–478. 10.1085/jgp.201210784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval OM, Joiner MLA, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XHT, Song LS, Anderson ME, 2015. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun 6, 6081 10.1038/ncomms7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüst RC, Helmes M, Martin JL, van der Wardt TJ, Musters RJ, van der Velden J, Stienen GJ, 2017. Rapid frequency-dependent changes in free mitochondrial calcium concentration in rat cardiac myocytes. J. Physiol 595, 2001–2019. 10.1113/JP273589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, Prando V, Coppini R, Vida V, Stølen TO, Ulrik W, Cerbai E, Stellin G, Faggian G, De Stefani D, Sandri M, Rizzuto R, Di Lisa F, Pozzan T, Catalucci D, Mongillo M, 2017. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 114, E9006–E9015. 10.1073/pnas.1708772114 [DOI] [PMC free article] [PubMed] [Google Scholar]