

SUMMARY
Activation of hepatocyte cannabinoid receptor-1 (CB1R) by hepatic stellate cell (HSC)-derived 2-arachidonoylglycerol (2-AG) drives de novo lipogenesis in alcoholic liver disease (ALD). How alcohol stimulates 2-AG production in HSCs is unknown. Here, we report that chronic alcohol consumption induced hepatic cysteine deficiency and subsequent glutathione depletion by impaired transsulfuration pathway. A compensatory increase in hepatic cystine-glutamate antiporter xCT boosted extracellular glutamate levels coupled to cystine uptake both in mice and in patients with ALD. Alcohol also induced the selective expression of metabotropic glutamate receptor-5 (mGluR5) in HSCs where mGluR5 activation stimulated 2-AG production. Consistently, genetic or pharmacologic inhibition of mGluR5 or xCT attenuated alcoholic steatosis in mice via suppression of 2-AG production and subsequent CB1R-mediated de novo lipogenesis. We conclude that a bidirectional signaling operates at a metabolic synapse between hepatocytes and HSCs through xCT-mediated glutamate/mGluR5 signaling to produce 2-AG, which induces CB1R-mediated alcoholic steatosis.
Keywords: 2-arachidonoylglycerol, cannabinoid receptor, Nrf2, transsulfuration pathway, xCT
eTOC blurb
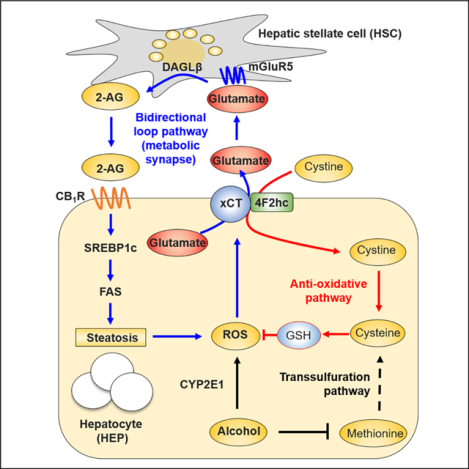
In Brief
Choi et al. show that chronic alcohol consumption induces CYP2E1-mediated ROS production by hepatocytes, which is compensated by GSH generation through xCT-mediated uptake of cystine. The parallel release of glutamate stimulates mGluR5 on hepatic stellate cells (HSCs) to produce 2-AG which, in turn, activates CB-iR on neighboring hepatocytes to induce de novo lipogenesis.
INTRODUCTION
Alcoholic liver disease (ALD) is highly prevalent worldwide and can progress from simple steatosis to alcoholic steatohepatitis (ASH), cirrhosis and hepatocellular carcinoma (Gao and Bataller, 2011). Hepatic antioxidant defense systems including glutathione (GSH) can mitigate the progression of ALD in heavy drinkers (Gao and Bataller, 2011; Seitz et al., 2018). Chronic alcohol consumption leads to disturbed methionine cycle and transsulfuration system followed by accumulation of homocysteine in endoplasmic reticulum and shortage of cysteine, resulting in decreased glutathione levels (Barak et al., 2001; Louvet and Mathurin, 2015; Tsukamoto and Lu, 2001). Hyperhomocysteinemia and the associated hepatocyte (HEP) apoptosis and lipogenic gene expression are often observed in mice, rats and humans following chronic alcohol consumption (Hultberg et al., 1993; Ji and Kaplowitz, 2003; Stickel et al., 2000). An equilibrium between alcohol-induced oxidative stress and recovery is critical for liver homeostasis. Thus, cytochrome P450 2E1 (CYP2E1)-mediated reactive oxygen species (ROS) production contributes to elevated expression of transcription factor Nrf2, which leads to upregulation of genes related to GSH synthesis (Cederbaum, 2013). The xCT transporter, an anti-porter system composed of light chain xCT (encoded by Slc7a11) and heavy chain 4F2, can be upregulated by Nrf2 to compensate GSH shortage via uptake of cystine coupled to the efflux of glutamate (Sasaki et al., 2002). However, the role of xCT in ALD has not been explored.
Endocannabinoids, including anandamide (AEA) and 2-arachidonoylglycerol (2-AG), bind to and activate the same cannabinoid receptors (CB1R and CB2R) that mediate the effects of marihuana (Pacher et al., 2006). In the liver, CB1R activation promotes steatosis and fibrosis while CB2R activation attenuates the progression of liver disease (Jeong et al., 2008; Julien et al., 2005; Louvet et al., 2011; Osei-Hyiaman et al., 2008; Teixeira-Clerc et al., 2006; Trebicka et al., 2011). Alcohol stimulates 2-AG production in cerebellar granule neurons in rats (Basavarajappa et al., 2000) and hepatic stellate cells (HSCs) in mice (Jeong et al., 2008), which has been implicated in alcohol reward and dependence (Parsons and Hurd, 2015), and alcoholic steatosis (Jeong et al., 2008), respectively. In alcoholic steatosis, paracrine stimulation of CB1R in HEPs by HSC-derived 2-AG upregulates the expression of hepatic lipogenic genes such as Srebf1 and Fasn, whereas inhibition of CB1R mitigates alcohol-induced hepatic steatosis (Jeong et al., 2008). 2-AG production is catalyzed by diacylglycerol lipase-α (DAGLα) in the brain and DAGLβ in the liver (Gao et al 2010b; Jeong et al., 2008), but the molecular mechanism by which alcohol stimulates 2-AG production in HSCs is unknown. Interestingly, the group I metabotropic glutamate receptors (mGluR1 and mGluR5) are coupled to phospholipase C-β (PLCβ) to hydrolyze phosphoinositide into inositol 1,4,5-triphosphate and diacylglycerol that can be further metabolized by DAGLs to 2-AG, and mGluR1/5 are also present in peripheral organs including the liver (Julio-Pieper et al., 2011). Notably, blockade of hepatic mGluR5 can mitigate toxin- and oxidative stress-induced liver injury in rodent models (Ferrigno et al., 2018; Jesse et al., 2009; Storto et al., 2000; Storto et al., 2003) and plasma glutamate is increased in patients with ALD (Tominaga et al., 1993), although the source of glutamate is unclear. Moreover, glutamate alone or co-administered with ethanol increases 2-AG production in neuronal cells (Basavarajappa et al., 2000; Varma et al., 2001). Based on these findings, we hypothesized that hepatic mGluR5 activation by increased glutamate may contribute to the development of ALD.
Here, we investigated the origin and metabolic impact of hepatic glutamate on the progression of ALD in both mice and patients. We found that alcohol-induced xCT-mediated excretion of glutamate from HEPs enhanced the production of 2-AG in HSCs via mGluR5 activation. Consequently, genetic or pharmacological inhibition of xCT or mGluR5 attenuated alcoholic steatosis. Diverse animal models, in vitro cell-based assays, and in situ hepatic perfusion were used to elucidate the paracrine communication between HEPs and HSCs via the xCT-glutamate-mGluR5 pathway under ethanol-induced oxidative stress conditions. These findings provide novel insights into the pathogenesis and treatment of ALD.
RESULTS
Increased hepatic xCT and glutamate coordinate antioxidant gene expression and lipid biosynthesis in alcoholic steatosis
We examined serum glutamate levels and hepatic expression of xCT mRNA and protein in chronically ethanol-fed mice and in pair-fed controls. At sacrifice, serum and hepatic glutamate levels were significantly higher in ethanol-fed than in pair-fed mice (Figure 1A), whereas the two groups had comparable levels of diet intake, body weight change, liver injury and glutamate levels in stomach and brain, where glutamate levels are the highest (Figures S1A–S1D), suggesting increased excretion and production of glutamate in the liver. Furthermore, serum glutamate levels were positively correlated with hepatic triglyceride (TG) and Fasn mRNA levels (Figure 1B).
Figure 1. Alcohol-induced increase in hepatic xCT expression and glutamate levels coordinate the upregulated gene expression of antioxidant response and lipid biosynthesis.

(A) Glutamate concentrations in serum and liver of WT mice fed with ethanol (EtOH) liquid diet or an isocaloric diet (pair) for 8 weeks (n = 8/group).
(B) Correlation of serum glutamate with liver TG or Fasn mRNA levels. Correlations were assessed by linear regression analyses (n = 8/group).
(C and D) RNA-sequencing analysis of the liver from WT mice (n = 3/group). Heatmaps show differentially expressed 525 genes (C), of which Slc7a11 mRNA was most increased (D).
(E) Representative immunofluorescent staining of xCT and Nrf2 in liver sections of WT mice. Central vein (CV).
(F) Representative Western blot analysis of xCT, Nrf2 and GCLC protein expression in isolated hepatocytes (HEPs) of WT mice.
(G) Slc7a11 mRNA expression in HEPs isolated from WT and CYP2E1 KO mice and exposed to EtOH for 24 h (3 replicates).
(H) Glutathione (GSH) concentration in liver tissues of WT mice (n = 8/group).
(I-K) RNA-sequencing analysis of the liver from WT mice (n = 3/group). Ingenuity pathway analysis of canonical pathways of the livers and heatmap representation of genes controlled by Nrf2 (I). The enriched 4 gene ontology (GO) terms (by upregulated genes), fold enrichment, FDR adjusted P value and the percentage of genes in each term (J). Heatmaps showing differentially expressed genes related with lipid biosynthetic processes (K).
Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001. Scale bars, 50 μm.
See also Figure S1.
To identify the candidate transporter for glutamate excretion in the liver, we performed RNA sequencing, immunostaining, Western blotting and qRT-PCR, and found that Slc7a11mRNA was significantly upregulated in a CYP2E1-dependent manner and xCT proteins were mainly observed in HEPs around central veins in livers of ethanol-fed compared to pair-fed mice (Figures 1C–1G). Slc7a11 mRNA was similarly increased in other murine models of ALD (Figures S1E and S1F). The xCT transporter, encoded by Slc7a11, excretes glutamate for cystine (quickly reduced to cysteine in the cells) to generate GSH, and is mainly regulated by Nrf2, a master regulator of oxidative stress (Sasaki et al., 2002). In accordance with alcohol-induced oxidative stress, hepatic GSH declined (Figure 1H) while the expression of Nrf2 and its antioxidant target genes were increased by ethanol feeding (Figures 1F and 1I). Interestingly, glutathione synthesis-related proteins (Figure 1F) and gene expression (Figure 1I; Figures S1G and S1H) along with lipid biosynthetic genes (Figures 1J and 1K)in the livers were also increased by ethanol feeding. These data are compatible with the notion that alcohol-induced oxidative stress stimulates xCT expression and elevated serum glutamate might be linked to increased hepatic lipogenesis.
Expression of glutamate biosynthetic enzymes increases in perivenous hepatocytes in alcoholic steatosis
Regarding the metabolic pathways of glutamate production, qRT-PCR analyses using whole liver tissue revealed that among multiple genes involved in glutamate biosynthesis, hepatic Oat and Aldh4a1 mRNAs were significantly increased, whereas Glud1, Glul, Gls1 and Gls2 mRNAs were decreased or not changed by ethanol feeding (Figure 2A). Moreover, freshly isolated HEPs showed similar expression of these genes (Figure 2B). Increased expression of immunostained ALDH4A1 and CYP2E1 was mostly observed around central veins along with lipid droplet accumulation, whereas the expression of glutamine synthetase (GS) was decreased around central veins in EtOH-fed mice compared with pair-fed mice (Figure 2C). However, GLS2 expression at the periportal zone was similar in pair-fed and EtOH-fed mice (Figure 2C). Furthermore, ALDH4A1 protein levels in sera and isolated HEPs were higher in ethanol-fed mice compared to pair-fed mice (Figures 2D and 2E). ALDH4A1 protein and mRNA dose-dependently increased in ethanol-treated HEPs in vitro (Figures 2F and 2G). These data suggest that glutamate might be generated by perivenous hepatocytes after ethanol exposure.
Figure 2. Increased ALDH4A1 and CYP2E1 expression in livers of liquid ethanol diet-fed mice.

(A) Graphical representation of glutamate synthesis pathways and hepatic mRNA expressions in pair-fed or EtOH-fed WT mice for 8 weeks (n = 8/group). Values represent the results from three experimental replicates.
(B) Relative mRNA expression of Gls1, G/s2, Glul, Oat, Aldh4a1, and Glud1 in freshly isolated HEPs and HSCs from WT mice (n = 4/group). Values represent the results from three experimental replicates.
(C) Representative H&E staining and immunofluorescent staining of CYP2E1, ALDH4A1, GS, and GLS2 of liver sections. Central vein (CV) and portal triad (PT)
(D) Serum ALDH4A1 levels (n = 8/group).
(E) Representative Western blot analysis of ALDH4A1 protein expression in isolated HEPs from pair-fed and EtOH-fed mouse liver.
(F) Hepatic expression of mRNAs in freshly isolated WT HEPs after 24 hour-ethanol treatment//? vitro. Values represent the results from three experimental replicates.
(G) Representative Western blot analysis of ALDH4A1 protein expression in ethanol-treated WT HEPs for 24 h. Values represent the results from two experimental replicates.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars, 50 μm.
Alcohol suppression of transsulfuration pathway stimulates xCT-derived glutamate release
Cysteine, the rate-limiting substrate for GSH synthesis, is mainly produced from methionine via the transsulfuration pathway in the liver (Lu, 1999). However, hepatic production of cysteine is often limited in various disease conditions such as insulin resistance, immune dysfunction, and cancer (Dröge, 2005). In line with this, the transsulfuration pathway in ethanol-fed mice was predicted to be suppressed by the Ingenuity Pathway Analysis (Palaga et al.) of the “Cysteine Biosynthesis/Homocysteine Degradation” pathway (Figure S2A). These features generate the possibility that chronic alcohol consumption may alter methionine cycle and transsulfuration pathway activities during GSH biosynthesis resulting in the upregulation of hepatic xCT (Figure 3A). To test this, we measured hepatic levels of methionine, S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), homocysteine, cystathionine, cysteine and GSH in ethanol-fed and pair-fed mice (Figure 3B) and in mouse livers perfused in situ with ethanol (Figures 3C and 3D). Due to CYP2E1-mediated oxidative stress (Cederbaum, 2013), concentrations of cysteine and GSH were decreased and methionine levels were increased by chronic ethanol consumption (Figure 3B). Unexpectedly, ethanol decreased homocysteine in the liver, but increased it in serum (Figure 3B), reflecting homocysteine release from HEPs as previously reported (Barak et al., 2001). Accordingly, in livers perfused with ethanol in situ, levels of SAH, cystathionine, cysteine, GSH and glutamate decreased, whereas methionine and homocysteine significantly increased compared to buffere-perfused controls (Figures 3C and 3D). Furthermore, qRT-PCR data revealed reduced expression of Bhmt and Cth that may account for homocysteine accumulation, and increased Mat1a expression might be secondary to high levels of methionine in both in vivo ethanol-fed and in situ ethanol-perfused mouse livers (Figures S2B–S2D). These data suggest that alcohol-induced oxidative stress depletes cysteine, which is required for GSH synthesis through altered methionine cycle and transsulfuration pathway, leading to increase of xCT expression and subsequent glutamate release from the liver.
Figure 3. Alcohol-induced alteration of methionine cycle and transsulfuration pathway in the liver.

(A) Graphical representation of transsulfuration pathway and xCT-mediated glutathione biosynthesis.
(B) Concentrations of methionine, SAM, SAH, homocysteine, cystathionine, cysteine, and GSH in liver tissue and serum homocysteine levels from pair-fed (n = 4) and EtOH-fed (n = 6) mice for 8 weeks.
(C and D) in situ closed liver perfusion system (C) were used for concentration measurements of methionine, SAM, SAH, homocysteine, cystathionine, cysteine, and GSH in WT mouse livers (n = 3/group) by circulating media containing 50 mM ethanol, and medium levels of ALT and glutamate (D).
(E) in vitro co-culturing HEPs with HSCs in the presence of ethanol plus sulfasalazine (SSZ) (3 replicates).
(F-H) Concentration of glutamate and cystine in media (n = 3/each) (F), and relative mRNA expression of Daglb in co-cultured HSCs (G) and those of Cnr1, Srebf1, and Fasn in HEPs (H).
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figure S2.
In co-cultures of primary HSCs and HEPs, we examined whether inhibition of hepatic xCT by sulfasalazine (SSZ) affects the ethanol-mediated glutamate release and endocannabinoid-mediated gene expression (Figure 3E). Ethanol significantly increased glutamate and decreased cystine levels in co-cultured media, and both changes were prevented by SSZ (Figure 3F). In HSCs co-cultured with HEPs, Daglb mRNA was increased with no change of Dagla mRNA (Figure 3G; Figure S2E). In co-cultured HEPs, Cnr1, Srebf1 and Fasn mRNA were increased by ethanol, while increased expression of Srebf1 and Fasn mRNA was reversed by SSZ treatment (Figure 3H). These observations led us to test whether xCT-mediated glutamate release by HEPs may stimulate DAGLβ-mediated 2-AG production in HSCs, which then stimulates lipogenic gene expression in HEPs via CB1R.
mGluR5 activation stimulates 2-AG production in HSCs, which induces lipogenic gene expression in HEPs.
Activation of Group I mGluRs (mGluR1 and mGluR5) initiates 2-AG production in the postsynaptic neuron (Suh et al., 2012; Varma et al., 2001). To explore the mechanisms of glutamate-stimulated 2-AG production by HSCs, we analyzed group I mGluR expression in HEPs and HSCs. Grm1 and Grm5 mRNA were undetectable or marginally expressed in freshly isolated HEPs, but Gmr5 mRNA and protein were clearly detectable in freshly isolated HSCs (Figures 4A and 4B) contrary to a previous study (Storto et al., 2003). Moreover, the selective mGluR5 agonist CHPG increased phosphorylation of mitogen-activated protein kinase (pMAPK) in HSCs but not in HEPs, further supporting the presence of functional mGluR5 in HSCs but not in HEPs (Figure S3A).
Figure 4. Endocannabinoid production of HSCs by mGluR5 stimulation increases lipogenic gene expression in HEPs.

(A) RT-PCR assays for Grm1 and Grm5 in brain, HEPs and HSCs (3 replicates).
(B) Representative Western blot analysis (of 2 Western blots for each band) of mGluRS protein expression in HEPs and HSCs.
(C) Relative expression of Grm5 mRNA in various hepatic cells (n = 4/each).
(D) Representative flow cytometry analysis for mGluRS expression in Neuro-2a (N2a) cells, HSCs, HEPs, Kupffer cells and liver mononuclear cells (MNCs) (3 replicates).
(E) Relative expression of Grm5 mRNA in freshly isolated HSCs from pair-fed and EtOH-fed mice (n = 4/group, 3 replicates).
(F) Representative western blotting analysis (of 2 Western blots for each band) of DAGLβ in HSCs after mGluR5 activation by 10 μM CHPG for 30 min.
(G) Relative mRNA levels of Dagla and Daglb genes in HSCs treated with 10 μM CHPG for 30 min (3 replicates).
(H) 2-arachidonoylglycerol (2-AG) levels in HSCs as in (G).
(I) Relative mRNA levels of Dagla and Daglb genes in HSCs treated with 50 μM monosodium glutamate (MSG) for 15 or 30 min (4 replicates).
(J) 2-AG levels in primary HSCs isolated from WT or mGluR5−/− mice. HSCs were incubated with vehicle or 50 μM MSG for 30 min (n = 3/each). 10 μM MPEP were treated 30 min prior to the treatment with MSG (3 replicates).
(K) qRT-PCR analysis of Srebfl and Fasn mRNA in co-cultured HEPs with HSCs. After exposure of 5 μM Rimonabant (CB1R antagonist) to WT HEPs for 30 min, HEPs were co-cultured with WT HSCs with or without 10 |jM CHPG for the indicated times (2 replicates).
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
See also Figure S3.
Grm5 mRNA was most enriched in freshly isolated HSCs compared to HEPs, Kupffer cells and liver mononuclear cells (MNCs) (Figure 4C). Similarly, flow cytometry analyses indicated that mGluR5 protein was much more abundant in HSCs than in other hepatic cells and was comparable to that in mouse Neuro-2a cells used as positive control (Figure 4D; Figure S3B). RNA-seq data from whole liver tissue samples only documented decreased expression of Grm8 mRNA whereas the expression of other mGluRs was very low (Figure S3C). In contrast, Grm5 mRNA was 15-fold higher in freshly isolated HSCs from ethanol-fed compared to pair-fed mice (Figure 4E). Immunoreactive mGluRS in HSCs was located nearfat-droplet storing HEPs that highly expressed xCT around central veins in liver sections of ethanol-fed mice (Figure S3D).
We next examined the effects of mGluR5 activation in HSCs on 2-AG production. The mGluR5 agonist CHPG significantly increased Daglb mRNA and protein expression (Figures 4F and 4G), along with elevated 2-AG content (Figure 4H), which was profoundly suppressed by the DAGLβ inhibitor KT172 (Figure S3E). Similarly, treatment of HSCs with glutamate selectively enhanced Daglb mRNA expression and increased 2-AG production, the latter being completely blocked by the mGluR5 antagonist MPEP or genetic deletion of mGluRS (Figures 4I and 4J;Figure S3F). Furthermore, CHPG treatment of HSCs slightly enhanced the expression of Srebf1 and Fasn in co-cultured HEPs, which was reversed by the CB1R antagonist rimonabant (Figure 4K). However, fibrogenic HSCs (cultured for 7 days; D7 HSCs) showed not only significant decrease of Grm5 and Slc7a11 mRNAs but also decreased 2-AG production compared with non-fibrogenic HSCs (D1 HSCs) (Figures S3G and S3H).
Inhibition of hepatic xCT attenuates alcoholic fatty liver by down regulating 2-AG-mediated lipogenesis
Based on the above data, we tested whether pharmacologic or genetic suppression of xCT might ameliorate alcoholic fatty liver by decreasing 2-AG production and CB1R-mediated lipogenesis in mice. In WT mice, adeno-associated virus (AAV)-mediated delivery of Slc7a11 shRNA resulted in a significant, ~60% knockdown of hepatic xCT expression, after which mice were placed on the liquid ethanol diet for 8 weeks (Figure 5A). For pharmacologic inhibition of xCT, SSZ treatment started at week 5 and mice were fed with ethanol diet for additional 4 weeks (Figure 5B). SSZ or shRNA treatment did not affect body weight (Figure 5B), dietary intake (Figure S4A) or ethanol-induced liver injury (Figure 5C). However, serum glutamate and hepatic 2-AG levels significantly decreased in shRNA- or SSZ-treated mice compared to their respective controls (Figures 5D and 5E). Both treatments resulted in marked attenuation of hepatic steatosis, as evidenced by H&E and Oil Red O staining (Figure 5F; Figure S4B). Accordingly, Cnr1 (but not Cnr2) mRNA and CB1R-regulated lipogenic genes were significantly decreased, whereas expression of antioxidant genes was upregulated in shRNA- or SSZ-treated mouse livers compared to controls (Figure 5G; Figure S4C). Inhibition of xCT by shRNA or SSZ did not affect Cyp2e1, Tnf and Il1b mRNA (Figure S4D). Lastly, in hepatocyte-specific xCT KO (Slc7a11 AlbCre) mice, ethanol induced similar levels of liver injury, body weight change and diet intake but reduced fat accumulation, compared to controls (Figures 5H–5J; Figures S4E and S4F). Consistently, qRT-PCR analyses revealed that Cnr1, Srebp1, and Fasn mRNAs were significantly down regulated, while Cat and Sod1 mRNAs were upregulated in Slc7a11 AlbCre mice compared with controls (Figure 5K; Figures S4G and S4H).
Figure 5. Inhibition of xCT mitigates alcoholic steatosis.

(A) Schematic for the experimental protocol. WT C57BL/6J mice were injected with AAV-Slc7a11-shRNA (2 × 1011 vg/mouse) or control and, two weeks later, fed with ethanol diet for 8 weeks (n = 5/group; 2 replicates). Western blot analysis for hepatic xCT at sacrifice.
(B) Schematic for the experimental protocol. Body weight changes of WT C57BL/6J mice treated with vehicle or sulfasalazine (SSZ; 100 mg kg−1 body weight, i.p., every day) for the last four weeks of ethanol feeding (n = 4/group; 2 replicates).
(C-E) Serum levels of ALT (C), glutamate (D), and hepatic concentration of 2-AG (E) in shRNA- or SSZ-treated mice with their corresponding controls after chronic ethanol feeding.
(F) Representative Oil Red O stainings on the liver tissue sections of shRNA- or SSZ-treated mice with their corresponding controls.
(G) qRT-PCR assays for Cnr1, Srebf1, and Fasn in each group (n = 4/group).
(H) Hepatocyte-specific xCT KO (AlbCre) mice were obtained by crossing xCTflox/flox mice with Albcre mice. Western blot analysis for hepatic xCT in WT and AlbCre mice.
(I) Serum levels of ALT in WT and AlbCre mice after ethanol diet feeding for 8 weeks (n = 4/group; 2 replicates).
(J) Representative H&E and Oil Red O stainings on the liver tissue sections of WT and AlbCre mice with their corresponding controls.
(K) qRT-PCR assays for Cnr1, Srebf1, Fasn, Cat, Sod1 and Sod2 in each group (n = 4/group).
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars, 50 μm.
See also Figure S4.
Genetic or pharmacologic inhibition of mGluR5 mitigates alcoholic steatosis
To elucidate the role of mGluR5 in the pathogenesis of alcoholic steatosis, Grm5+/+ and Grm5−/− mice were fed with 4.5% liquid ethanol diet for 8 weeks. There were no differences in diet intake and serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and glutamate between the two groups (Figures 6A and 6B; Figures S5A). However, hepatic 2-AG and Cnr1 mRNA levels were significantly lower in Grm5−/− than in Grm5+/+ mice (Figures 6C and 6D), whereas Cnr2, Cyp2e1, Tnf and Il1b mRNA levels were similar between the two groups (Figure S5B). Phosphorylated AMP-activated protein kinase (pAMPK) protein was higher whereas fatty acid synthase (Bai et al.) was much lower in the liver of Grm5−/− mice than Grm5+/+ mice (Figure 6E). Accordingly, hepatic fat accumulation was much lower in Grm5−/−than in Grm5+/+ mice (Figure 6F). In HEP/HSC co-cultures, Daglb mRNA expression in Grm5+/+ but not GrmS−/− HSCs was significantly increased after co-incubation with ethanol-treated Grm5+/+ HEPs (Figure 6G; Figure S5C), Accordingly, ethanol upregulated Srebf1 and Fasn mRNA in HEPs co-cultured with Grm5+/+ but not Grm5−/−HSCs (Figure 6G).
Figure 6. Genetic or pharmacologic inhibition of mGluR5 attenuates alcoholic steatosis.

(Pessoa-Filho et al.) Grm5−/− and Grm5+/+ mice were fed with ethanol diet for 8 weeks (n = 5/group; 3 replicates).
(A-C) Serum levels of ALT, AST (A), glutamate (B), and hepatic 2-AG concentration (C) were measured between EtOH-fed Grm5−/− and Grm5+/+ mice.
(D and E) Representative Cnr1 mRNA expression (D) and protein levels of pAMPK, AMPK and FAS (E) were assessed in liver of Grm5−/− and Grm5+/+ mice using qRT- PCR and Western blotting analysis.
(F) Representative H&E and Oil Red O stainings.
(G) Graphical representation of in vitro co-culture system. 1.5 × 105 HSCs of Grm5−/− and Grm5+/+ mice were co-cultured with 3 × 105 HEPs of Grm5+/+ mice in the absence or presence of 50 mM EtOH for 18 h. Representative Daglb mRNA expression of HSCs, and Srebf1 and Fasn mRNA expression of HEPs were assessed respectively by qRT-PCR (3 replicates).
(Buschard et al.) Graphical representation of the experimental protocol. CTEP (2 mg kg−1 body weight) was administered to WT mice per os every other day for the last four weeks of ethanol feeding (n = 7/group).
(H and I) Changes of body weight (H) and serum levels of ALT and AST (I) were measured.
(J) Representative H&E and Oil Red O stainings.
(K) Representative expression of Cnr1, Srebf1, Fasn and Scd1 mRNA levels were assessed in the liver tissues of vehicle- or CTEP-treated mice (3 replicates).
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars, 50 μm.
See also Figure S5.
Recent studies have shown that mGluR5 activation in neutrophils, which plays an important role in ALD, promotes the production of pro-inflammatory cytokines (Dai et al., 2013; Yang et al., 2017). To test any unexpected contribution of mGluR5 in immune cells and further confirm the function of mGluR5 in HSCs in the effects of alcohol, we generated chimeric mice by bone marrow transplantation (BMT) of Grm5+/+ mice to Grm5+/+ and Grm5−/− mice (referred to as HSC-Grm5+/+ and HSC-Grm5−/−, respectively) (Figure S5D). At week 6 after irradiation and BMT, we confirmed chimerism by using reporter mice or isolation of HSCs and liver MNCs, which demonstrated that MNCs, but not HSCs, originated from BM (Figure S5E). HSC-Grm5+/+ and HSC-Grm5−/− mice on ethanol diet for 8 weeks had similar liver injury, but HSC-Grm5−/− mice had reduced serum TGs, hepatic 2-AG and fat accumulation (Figures S5F–S5I). Srebf1, Fasn and Scd1 mRNAs were significantly downregulated whereas Cyp2e1, Il1b and Tnf expression was unaffected in the livers of HSC-Grm5−/− mice compared to HSC-Grm5+/+ mice (Figures S5J). The effects of alcohol in HSC-Grm5−/− mice were very similar to those observed in Grm5−/− mice (Figure 6), indicating that the presence of Grm5+/+ MNCs in HSC-Grm5−/− mice doesn’t contribute to the observed effects of alcohol.
To test the therapeutic implications of these findings, we next determined whether pharmacologic inhibition of mGluR5 could improve alcoholic steatosis after the initiation of ethanol feeding. Chronic blockade of mGluRS by CTEP from week 5 to 8 of ethanol feeding did not affect diet intake, body weight and ethanol-induced liver injury (Figures 6H and 6l; Figure S5K) but induced down regulation of Cnr1, Srebf1, Fasn and Scd1 and decreased hepatic fat accumulation (Figures 6J and 6K). Cnr2, Cyp2e1, and Il1b expression were unaffected by CTEP treatment (Figure S5L). These data are similar to those obtained in global or HSC-Grm5−/− mice, demonstrating that xCT-mediated excretion of glutamate triggered by alcohol-induced oxidative stress activates mGluR5 in HSCs that stimulates 2-AG synthesis, with the released 2-AG inducing CB1R-mediated de novo lipogenesis in HEPs.
Hepatic xCT-glutamate-endocannabinoid axis is upregulated in patients with ALD
Prompted by the above findings in mice, we measured plasma glutamate levels in patients with ALD. Plasma levels of glutamate were significantly increased in patients with alcoholic fatty liver (AFL) and ASH, whereas glutamate levels in alcoholics without liver disease (AWLD) or patients with alcoholic liver cirrhosis (ALC or ASH on LC) did not show any difference compared with healthy controls (Figure 7A; Table S1). Moreover, glutamate levels were positively correlated with serum TG levels (Figure 7B). In line with findings in mice, the expression of genes related to transsulfuration pathway and glutathione synthesis (CBS, CTH, GCLC) was decreased, whereas SLC7A11, GRM5 and CNR1 mRNA (encoding xCT, mGluR5 and CB1R, respectively), and lipogenesis-related genes (SREBF1, FASN) were significantly increased in patients with AFL compared to controls (Figure 7C;Figure S6A; Table S2). Moreover, GRM5 mRNA was positively correlated with CNR1, SREBF1 and FASN mRNA levels in patients with AFL (Figure 7D). Furthermore, IPA-predicted metabolic pathways also exhibited inhibited transsulfuration pathway as evidenced by decreased mRNA expression of CBS and CTH, whereas NRF2 target genes including SLC7A11 were increased in patients with alcoholic hepatitis (AH) compared to healthy controls (Figure S6B–S6E). Indeed, xCT and DAGLβ proteins were significantly elevated in patients with AFL compared to controls (Figure 7E). In parallel, hepatic CYP2E1, xCT, and ALDH4A1 proteins were significantly increased along with fat accumulation in patients with ALD compared with controls without ALD (Figure 7F). Notably, GRM5 mRNA was much higher in freshly isolated human HSCs (hHSCs) than hepatocytes (hHEPs) (Figure 7G). Moreover, GRM5 and DAGLB mRNAs were significantly increased, whereas GRM1 and DAGLA mRNAs remained unchanged by exposure to the mGluR agonists CHPG and DHPG (Figure 7H; Figures S6F and S6G). Ethanol also dose-dependently increased SLC7A11 mRNA in Hep3B but not HepG2 cells, where the expression of CYP2E1 is lower than that in Hep3B, suggesting CYP2E1-dependent induction of xCT (Figure S6H).
Figure 7. Increased levels of plasma glutamate and hepatic GRM5 expression is correlated with fatty liver in alcohol patients.

(A) Plasma glutamate levels were measured in healthy controls (n = 4) and alcohol patients (n = 33) with alcoholic without liver disease (AWLD; n = 8), alcoholic fatty liver (AFL; n = 6), alcoholic steatohepatitis (ASH; n = 6), alcoholic liver cirrhosis (ALC; n = 6) and ASH on liver cirrhosis (LC) (n = 7) (Table S1).
(B) Correlative analysis of plasma glutamate levels with TG concentration. Correlations were assessed by linear regression model (n = 33 for alcohol patients).
(C) qRT-PCR analysis was performed using liver biopsy samples of controls (n = 4, normal areas of nonalcoholic HCC or focal nodular hyperplasia) and patients with AFL (n = 4) (Table S2).
(D) Positive correlation of GRM5 mRNA levels with CNR1, SREBF1 and FASN mRNA levels.
(E) Representative Western blot analysis of xCT and DAGLβ protein expression, and their relative densities in controls and patients with AFL (n = 4/group),
(F) Representative immunostainings of CYP2E1, xCT and ALDH4A1 in liver tissue sections of non-tumor areas in patients with nonalcoholic or alcoholic HCC.
(G) Relative GRM5 mRNA expression in freshly isolated primary human hepatocytes (hHEPs) and human HSCs (hHSCs) from the control liver (3 replicates).
(H) Relative mRNA levels of GRM5, DAGLA and DAGLB genes in isolated primary hHSCs treated with 10 μM CHPG for 30 min (3 replicates).
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01. Scale bars, 50 μm.
DISCUSSION
A key mechanism underlying alcoholic fatty liver is an endocannabinoid-induced, CB1R-mediated increase in de novo lipogenesis in hepatocytes (Jeong et al., 2008; Trebicka et al., 2011). We have earlier demonstrated that chronic alcohol treatment upregulates the hepatic levels of the endocannabinoid 2-AG and its biosynthetic enzyme DAGLβ in HSCs, and the released 2-AG acts on neighboring hepatocytes to induce CB1R-mediated de novo lipogenesis (Jeong et al., 2008), but the metabolic trigger to induce 2-AG production in HSCs has remained unknown. Here, we provide multiple lines of evidence fora bidirectional paracrine loop between HEPs and HSCs, where alcohol-induced oxidative stress and the resulting cysteine deficiency and glutathione depletion triggers a compensatory upregulation of the cysteine/glutamate antiporter xCT in HEPs, resulting in increased glutamate secretion. Glutamate then activates the metabotropic glutamate receptor mGluR5 on neighboring HSCs to induce DAGLβ expression and 2-AG production, and the released 2-AG activates CB1R on HEPs to induce de novo lipogenesis. This bidirectional HEP-HSC-HEP loop represents a unique mechanism of AFL and it also provides novel molecular targets for its pharmacotherapy. Indeed, we demonstrate that pharmacological blockade or genetic deletion of xCT or mGluR5 mitigates alcoholic steatosis, whereas exposure of HSCs to mGluR5 agonists promotes DAGLβ expression and 2-AG production. Importantly, the key components of this bidirectional loop are similarly induced in patients with ALD. Patients with AFL have elevated circulating glutamate levels, increased expression of hepatic xCT and mGluR5, and positive correlation between mGluR5 and CB1R-mediated lipogenesis. This reinforces the translational potential of the novel therapeutic targets identified here.
As previously reported by us and others (Kim et al., 2017; Lorenz et al., 2018), we adopted in vivo mouse models of ALD, in vitro paradigms in isolated liver cells and in situ hepatic perfusion techniques to clarify the role of xCT and glutamate in mitigating alcoholic oxidative stress in hepatocytes and in dissecting the mechanism of mGluR5-mediated production of 2-AG in HSCs. Chronic consumption of 40 g or more of pure alcohol per day increases the risk of end-stage liver disease and the associated social burden (Gao and Bataller, 2011; Seitz et al., 2018). Alcohol-induced ROS generation is primarily mediated via the induction of CYP2E1, which also modulates the expression of a large repertoire of genes linked to anti-oxidative response and lipid metabolism in the context of alcoholic fatty liver (Cederbaum, 2013; Seitz et al., 2018). Ethanol-fed mice and ALD patients have reduced hepatic GSH levels along with hyper-homocysteinemia (Hultberg et al., 1993; Ji and Kaplowitz, 2003; Lee et al., 2004; Stickel et al., 2000). This leads to abnormal hepatic accumulation and elevated serum levels of homocysteine associated with decreased concentrations of cysteine and GSH, suggesting disturbed methionine cycle and transsulfuration pathway in the liver. Our findings point to a compensatory mechanism triggered by the depletion of GSH and cysteine which involves an ethanol-induced robust induction of Slc7a11 (encoding xCT) and Nfe2/2 (encoding Nrf2) in hepatocytes. Interestingly, xCT and CYP2E1 are co-localized in perivenous hepatocytes in both mice and humans, suggesting a strong relationship between CYP2E1-mediated oxidative stress and xCT induction. In this context, glutamate excretion may occur predominantly around central veins, providing a plausible explanation for the preferential accumulation of fat at the perivenous zone of hepatic lobule.
There is a striking analogy between endocannabinoid signaling in the central nervous system (CNS) and in the liver. In the CNS, endocannabinoids mediate activity-dependent synaptic plasticity by retrogradely activating presynaptic CB1R to inhibit both excitatory (glutamatergic) and inhibitory (GABAergic) synaptic transmission, after being released from post-synaptic neurons in response to calcium or metabotropic receptor activation (Augustin and Lovinger, 2018). We propose that a similar bidirectional signaling operates at a metabolic ‘synapse’ between hepatocytes, analogous to presynaptic neurons expressing CB1R, and ‘postsynaptic’ HSCs that generate 2-AG in response to metabotropic (mGluRS) receptor activation, which is then released onto neighboring hepatocytes to induce CB1R-mediated de novo lipogenesis (Jeong et al., 2008). This analogy is further supported by the cell-specific expression of metabotropic glutamate receptors: group I receptors (mGluR1 and 5) are generally expressed at postsynaptic sites, whereas the group II (mGluR2 and 3) and group III (mGluR4, 6–8) subtypes are enriched at presynaptic locations in the CNS (Niswender and Conn, 2010). Similarly, in the liver ‘postsynaptic’-like HSCs differentially express the group I mGluR5, whereas ‘presynaptic’-like hepatocytes express group II mGluRs, such as mGluR3 (Do et al., 2007). Although mGluR5 was identified in the hepatocyte fraction isolated from acetaminophen-treated mice (Storto et al., 2004), it is possible that it reflected mGluR5 in contaminating HSCs. The present findings illustrate the differential expression of mGluR5 in various types of liver cells.
Interestingly, alcohol-mediated oxidative stress did not increase liver injury after pharmacologic or shRNA-mediated disruptions of xCT. That might be due to compensation by other antioxidant genes such as Cat, Sod1 and Sod2, which are also upregulated. Therefore, pharmacologic inhibition of xCT may be a viable therapeutic strategy for ALD without further aggravating liver injury or oxidative stress. As to the source of increased glutamate in ALD, the liver is thought to generate glutamate through the glutamine-glutamate cycle, in which periportal hepatocytes metabolize glutamine to glutamate and perivenous hepatocytes convert glutamate into glutamine (Chaudhry et al., 2002). In contrast, our findings suggest that chronic alcohol consumption increases ALDH4A1-derived glutamate generation at perivenous hepatocytes along with down regulated glutamine synthetase (Glul), whereby alcohol interruption of the urea cycle might result in conversion of ornithine to glutamic-Y-semialdehyde (GSA) by ornithine aminotransferase (OAT), with the GSA being further converted into glutamate by mitochondrial ALDH4A1 (Hu et al., 1996). However, as the expression of GLS2 in periportal hepatocytes and other genes involved in glutamate generation such as G/s, P5cr2 and Prodh remained unchanged, glutamine is still useful for urea cycle activity in the periportal areas (zone 1), but it may not be a source of the alcohol-induced increase in glutamate at the perivenous areas (zone 3).
The present findings provide strong support to the emerging concept that the transmitter function of glutamate is not limited to the brain (Julio-Pieper et al., 2011). The elevated plasma glutamate in our patients with AFL and ASH was similar to that reported by others (Tominaga et al., 1993), whereas we found no change in plasma glutamate in patients with alcoholic liver cirrhosis, which may be due to the decrease in functional hepatocyte mass in cirrhosis. Plasma glutamate may therefore be a useful diagnostic marker for AFL and ASH. In the brain, inhibition of mGluR5 reduces alcohol seeking behavior in mice and alcoholic patients (Bird et al., 2008; Holmes et al., 2013). In binge alcohol drinking, mGluR1 but not mGluR5 expression is increased in the central nucleus of the amygdala and its inhibition reduces binge alcohol drinking in mice (Cozzoli et al., 2014). Thus, overactivity of group I mGluR contributes to both alcohol seeking behavior (Parsons and Hurd, 2015) and the organ toxicity of alcohol, which makes these receptors a desirable target for the treatment of both.
In the present study, mGluR5-mediated increase in DAGLβ expression and 2-AG production in HSCs is analogous to findings in the CNS and sympathetic neurons where mGluR5 activation induces 2-AG synthesis via DAGLα activation (Gregg et al., 2012; Tanimura et al., 2010; Varma et al., 2001; Won et al., 2009). They also illustrate the differential expression of DAGL isoforms in the brain where the dominant isoform is DAGLα, and the liver where DAGLβ is preferentially expressed. Accordingly, there was a greater reduction of 2-AG in the liver of Daglb−/− mice (~90%) than in Dagla−/− mice (~60%) (Gao et al., 2010a). Similarly, there was a greater increase in Daglb than Dagla gene expression in HSCs of ethanol-fed mice, as reported earlier (Jeong et al., 2008).
2-AG binds to CB1R and CB2R with similar affinity and it is a full agonist at both receptors, which play opposite roles in the pathogenesis of ALD, where CB1R-induced fat accumulation in the liver is detrimental (Jeong et al., 2008), whereas CB2R activation may mitigate alcohol-induced inflammation and steatosis (Louvet et al., 2011). In our models, which are more suitable for evaluating steatosis than inflammation, the CB1R-mediated effects of 2-AG dominate (Jeong et al., 2008), as also reflected by the protection from ALD by inhibition of mGluR5. This preferential effect of 2-AG on CB1R rather than CB2R may also reflect the fact that alcohol exposure selectively induces CB1R but not CB2R expression (Jeong et al., 2008).
In summary, our results show that oxidative stress caused by chronic ethanol intake increases hepatic excretion of glutamate, a metabo-transmitter, through the induction of xCT which leads to activation of mGluR5 and subsequent 2-AG production in HSCs, which then promotes alcoholic steatosis via CB1R-mediated lipogenesis. The inhibition of mGluR5 by CTEP, a specific antagonist of mGluR5 or the inhibition of xCT by sulfasalazine substantially prevents and reverses alcoholic steatosis via reducing hepatic 2-AG levels. Thus, this study provides novel insights into the role of glutamate in regulating hepatic lipid metabolism in ALD, and offers a potential therapeutic approach for treating ALD.
Limitations of Study
Our study reveals a functional metabolic synapse between stressed hepatocytes and neighboring HSCs in alcoholic fatty liver, linking hepatic glutamate release with metabolic mGluR5 response, to drive endocannabinoid production. However, the cellular mechanisms by which HSCs regulate mGluR5 expression in response to alcoholic liver injury are still unclear. In particular, the possible role of retinoic acid is worth exploring, in view of the fact that HSCs are the richest source of retinol in the body and an earlier study implicated retinoic acid acting via retinoic acid receptor-γ in the 2-AG-induced upregulation of CB1R in hepatocytes (Mukhopadhyay et al., 2010).
Another limitation is related to the fact that alcohol feeding in our model induces hepatic steatosis but does not replicate its progression to ASH, which in patients is often accompanied by an attenuation of hepatic fat accumulation. We also observed that the expression of mGluR5 and the subsequent 2-AG production were significantly reduced as HSCs transdifferentiated into fibrogenic myofibroblast-like cells, thereby decreasing pro-steatotic activity. Whether this is associated with a parallel increase in CB2R expression which could be the target of an antiinflammatory effect of 2-AG released by infiltrating MNCs and could also be therapeutically exploited, remains to be determined.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Won-Il Jeong (wijeong@kaist.ac.kr). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The Institutional Animal Care and Use Committee of Korea Advanced Institute of Science and Technology (KAIST, Republic of Korea) approved all animal protocols. The animals were maintained on a regular 12:12 light-dark cycle in a specific pathogen-free (SPF) animal facility under temperature-controlled (22–24°C) conditions at KAIST. Grm5+/+ and. Grm5−/− mice with C57BL/6J background were purchased from Jackson Laboratories (Stock No. 003121). Cyp2e1’/” mice were kindly provided by Dr. Bin Gao (NIAAA/NIH). Slc7a11tm1a(Knockout first allele) mice were obtained from Institute de Transgenose (France) which distributes these mice on behalf of the EMMA (European Mouse Mutant Archive, www.infrafrontier.eu; Stock No. EMMA: 10001). Protamine-FLP (flippase) mice were a kind gift from Dr. Suk-Jo Kang (KAIST). Slc7a11f/f (Slc7a11tm1c, FLPed knockout first) mice were generated by deletion of the LacZ reporter and Neomycin selection cassette by crossing with a protamine-FLP recombinase mice. These mice were further crossed and backcrossed with mice expressing the albumin-Cre mice (TG[Alb-cre]21Mgn, from Jackson Laboratories; Stock No. 003574) to obtain hepatocyte-specific xCT KO (Slc7a11f/fAlbCre) mice. Male mice aged 8 weeks were fed the Lieber-DeCarli liquid alcohol diet or control liquid diet for 8 weeks (Chen et al., 2015). In detail, mice were initially fed with Lieber-DeCarli diet (#710260, Dyets) ad libitum for 5 days to adapt to a liquid diet by gradually increasing the content of ethanol by 1% (v/v) each day until 4.5% (v/v). Then, the mice were kept on this liquid diet for a total of 8 weeks. In some experiments including pair-fed group, mice were fed with an isocaloric liquid diet adjusted with maltose dextrin for 8 weeks. Changes of body weights were monitored twice a week and diet intake was measured daily. For pharmacological inhibition of mGluR5 or xCT, mice were treated with CTEP (#B1633, APexBIO; 2 mg kg−1 body weight, p.o., every other day) or sulfasalazine (#S0883, Merck; 100 mg kg−1 body weight, i.p., every day) with corresponding vehicles, respectively, for the last 4 weeks of the experiment.
Human Patients
Human plasma samples and liver tissues were obtained from patients in various stages of ALD at Seoul Metropolitan Government Seoul National University Boramae Medical Center (Seoul, South Korea). The study conformed to the ethical guidelines of the World Medical Association Declaration of Helsinki and was approved by the Institutional Review Board of Seoul Metropolitan Government Seoul National University Boramae Medical Center (IRB No. 16-2013-45). Patients who were on excessive alcohol consumption (> 40 g/day for men and 20 g/day for women) were included in the study and were divided into 5 subgroups (alcoholic without liver disease; AWLD, alcoholic fatty liver; AFL, alcoholic steatohepatitis; ASH, alcoholic liver cirrhosis; ALC, and alcoholic steatohepatitis on liver cirrhosis; ASH on LC) according to the histological criteria. Patients with other causes of chronic liver disease such as a concurrent viral infection (hepatitis B virus and hepatitis C virus) or other diseases associated with elevated glutamate levels such as cancer, migraine, and HIV infection were excluded from the study, The clinical characteristics of the patients are listed in Table S1.
Fresh liver tissue samples were obtained from some of patients with ALD and their controls were obtained from the separate cohort of the Department of Surgery, Chungnam National University Hospital (Daejeon, South Korea) (Table S2) and were analyzed by qPCR, western blot, immunostainings and cell isolation. Authorization for the use of these tissues for research purposes and ethical approval were obtained from the Institutional Review Board of Chungnam National University Hospital (IRB number: 2016-03-020-011). Informed consents, which were approved by Institutional Review Board of Chungnam National University Hospital, were received from the entire patients who had provided the tissue. All of these patients had undergone tumor resection operation between 2017 and 2018.
Primary Cell Cultures.
The hepatocytes, hepatic stellate cells, Kupffer cells and liver mononuclear cells (MNCs) were isolated by differential centrifugation on an Opti-Prep (Sigma-Aldrich) or Percoll (Sigma-Aldrich) as previously described (Suh et al., 2012). In particular, the portal vein was cannulated and perfused with EGTA solution (5.4 mM KCl, 0.44 mM KH2PO4, 140 mM NaCl, 0.34 mM Na2HPO4, 0.5 mM EGTA, 25 mM Tricine, pH 7.2), followed by the perfusion buffer (0.075% collagenase type I in HBSS buffer with 0.02% DNase I) and the digestion buffer (0.009% collagenase type I in HBSS with 0.2% DNase I) at 37°C for 20 min. The cell suspension was filter through a 70 μm cell strainer and centrifuged at 50 ×g for 5 min at room temperature to isolate hepatocytes. Isolated hepatocytes were further purified by Percoll solutions. The supernatant were centrifuged at 650 ×g to obtain hepatic non-parenchymal cells. Half-cell pellet was suspended in 40% Percoll. The cell suspension was gently overlaid onto 70% Percoll and centrifuged at 4 °C for 25 min at 1800 ×g. Liver MNCs were collected from the interface. Then, liver MNCs were resuspended in fresh PBS after RBC lysis. The other cell pellet was then separated by 11.5% and 20% Opti-Prep gradient and centrifuged at 1800 ×g for 17 minutes at 4°C to isolate HSCs and Kupffer cell fraction, respectively. Cells were further purified via subsequent flow cytometric sorting with endogenous retinoid fluorescence for HSCs (using DAPI channel) and CD11b and F4/80 staining for Kupffer cells.
For human primary cell isolation, the liver specimen (20–50g) were obtained from fresh tumor resections provided from the cohort of the Department of Surgery, Chungnam National University Hospital (Daejeon, South Korea) (Table S2), and isolated by differential centrifugation on a 9% and 12.6% Opti-Prep as previously described (Werner et al., 2015). Briefly, the liver specimens were placed in a 100 mm dish, and a cannula was inserted in an accessible hepatic vessel. The cannula was fixed with the suture, and remaining vessels were sealed with the suture or Histoacryl (B Braun, UK). The liver tissues were then perfused with EGTA solution followed by the perfusion buffer. The remaining method was identical compared to the isolation methods of mouse cells except the Opti-prep concentration changed to 9% and 12.6%.
For in vitro experiments, after isolation of hepatocytes and HSCs, cells were then seeded on collagen-coated plates and stabilized in DMEM (#LM001–11, Welgene) or RPMI (Welgene) supplemented with 10% FBS (Welgene) and 1% penicillin-streptomycin (Thermo Fisher Scientific) overnight, respectively. After two times of PBS washing next day, both hepatocytes and HSCs were replenished with DMEM (#LM001–11, Welgene) supplemented with 1% penicillin-streptomycin. In some experiments, hepatocytes and hepatic stellate cells were co-cultured for 18 h by using 24 mm Transwell® with 8.0 pm Pore Insert (#3428, Corning). Cell were then treated with ethanol (variable concentrations, Millipore), CHPG (10μM, Tocris), DHPG (10 μM, Tocris), monosodium glutamate (50 μM, Sigma-Aldrich), MPEP hydrochloride (10 μM, Tocris), rimonabant hydrochloride (5 μM, Tocris), sulfasalazine (100pM, Sigma-Aldrich), and KT172 (2 μM, Sigma-Aldrich) in a humidified incubator with 5% CO2 at 37 °C according to the protocol for the corresponding experiments.
Cell Lines
Mouse Neuro-2a cells (ATCC) originally derived from the neuroblastoma of mouse A strain, human HEK293 T cells (ATCC) originated from human fetus, and human Hep3B cells (ATCC) and HepG2 cells (ATCC) obtained from male origin were cultured in DMDM (#LM001–05, Welgene) supplemented with 10% FBS (Welgene) and 1% penicillin-streptomycin (Thermo Fisher Scientific). All cell lines were maintained in a humidified incubator with 5% CO2 at 37 °C
METHOD DETAILS
AAV shRNA Preparation and Administration
To generate knockdown constructs, the pAAV-U6-sgRNA(shRNA)-CMV-GFP (Addgene) was digested with Sapl and ligated with annealed oligonucleotides encoding Slc7a11shRNA (5’-ACC TGG AGT TAT ACA GCT AAT TAA CTC GAG TTA ATT AGC TGT ATA ACT CCA TTT TTG-3’) or control shRNA (5’-ACC CCT AAG GTT AAG TCG CCC TCG CTC GAG CGA GGG CGA CTT AAC CTT AGG TTT TTG-3’). The shRNA constructs were transfected into HEK 293T cells, along with AAV2/8 and pAdΔF6. The steps for AAV generation, purification, and concentration were performed as previously described (Ran et al., 2015). In detail, for viral production, 293FT cells were maintained in 150 mm plates. For each transfection, 15 μg of pAAV2/8 serotype packaging plasmid, 15 μ g pAdΔF6 helper plasmid, 15 μg of pAAV carrying the construct of interest were added to 1mL of serum-free DMEM. 125 μl of PEI “Max” solution (1 mg ml−1, pH = 7.1) was then added to the mixture and incubated at room temperature for 10 min. After incubation, the mixture was added to 20 ml of warm maintenance media and applied to each dish to replace the old growth media. Cells were harvested between 48 h and 72 h post transfection by scraping and pelleting by centrifugation. The AAV2/8 (AAV2 inverted terminal repeat (ITR) vectors pseudo-typed with AAV 8 capsid) viral particles were then purified from the pellet according to a previously published protocol (Veldwijk et al., 2002). All viral preparations were then tittered by real-time qPCR using primer pair targeting AAV2 ITR (forward ITR primer, 5’-GGAACCCCTAGTGATGGAGTT; reverse ITR primer, 5’-CGGCCTCAGTGAGCGA). Then, hepatocyte-selective Slc7a11 knockdown was induced by the tail vein injection of AAV shRNA at 2 × 1011 genome copies per each mouse. Mice were allowed to recover for 2 weeks and were then placed on the Lieber-DeCarli liquid diet.
Generation of Bone Marrow Chimeras
Grm5+/+ and Grm5−/− mice were lethally irradiated with a total dose of 900 Rad. Two hours later, mice were injected in the tail vein with 3 × 106 nucleated bone marrow cells obtained by flushing the cavity of freshly dissected femurs and tibias from Grm5+/+ donors. Recipient mice received sulfamethoxazole/trimethoprim (1 mg ml−1 in drinking water) starting 1 week prior to irradiation and 2 weeks post irradiation. Chimeric mice were subjected to Lieber-DeCarli diet model experiments for 8 weeks (Xu et al., 2015).
Histological Analyses
Parts of left and medial lobes of mouse liver were extracted and fixed with 10% neutral buffered formalin or prepared using the Tissue-Tek OCT compound (Sakura, Tokyo, Japan). After deparaffinization and rehydration, paraffin sections with 5 μm thickness were stained with H&E (Sigma-Aldrich), whereas frozen sections with 10 μm thickness were subjected to Oil red O staining (Sigma-Aldrich) for visualization of lipid droplets. The histological features were observed and imaged using a light microscopy (Olympus).
Immunostaining
For immunohistochemistry analyses, paraffin-embedded tissue sections were used. Endogenous peroxidases were blocked with 0.03% H2O2 for 30 min and unspecific binding was blocked with 2.5% normal horse serum for 1 h. Tissues were incubated with primary antibody for 30 min at room temperature and further incubated with ImmPRESS HRP Anti-Rabbit IgG Polymer Detection Kit (Vector Laboratories) according to the manufacturer’s recommendation. Reactions were developed with DAB peroxidase substrate (Vector Laboratories) according to the manufacturer’s instruction and then slides were counterstained with hematoxylin. For immunofluorescence staining, paraffin-embedded tissue sections or OCT-embedded frozen liver sections were used. Tissue sections were fixed in 4% paraformaldehyde solution for 10 min and washed with PBS. Slides were blocked in PBS-GT (0.2% gelatin (v/v) and 0.1% (v/v) Triton X in PBS) for 1 h at room temperature and incubated with primary antibodies listed below in Antibodies section overnight at 4°C. After three times of PBS washing, slides were incubated with the secondary antibodies for 1 h at room temperature. After washing the slide, sections were covered with DAPI included mounting solution (Abeam) for nuclear staining. Fluorescence images were acquired using an Olympus BX51 microscope equipped with a CCD camera (Olympus, Tokyo, Japan) and computer-assisted image analysis with DP2-BSW.
Clinical Chemistry Measurements
Serum AST, ALT, TG, and total cholesterol (TC) in serum were measured by using a VetTest Chemistry analyzer (IDEXX Laboratories) according to the manufacturer’s instructions. Serum ALDH4A1 levels were measured by the ELISA kit (MyBioSource) according to the manufacturer’s instructions. Regarding liver TG, hepatic lipids were extracted from 50–100 mg of liver tissues using chloroform/methanol mixture (2:1 ratio) as previously described (Folch et al., 1957). Lipid extract was lyophilized by nitrogen gas and then resuspended in PBS with 5% albumin followed by TG measurements with a VetTest Chemistry analyzer (IDEXX Laboratories) according to the manufacturer’s instructions.
Flow Cytometry Analyses
Single-cell suspensions of hepatocytes, HSCs, Kupffer cells, liver MNCs, and neuro-2a cell (used as a positive control) were prepared and stained. Liver MNCs were labelled with fluorescence tagged antibodies under the anti-mouse CD16/CD32 (mouse Fc blocker, Clone 2.4G2) (BD Biosciences) and the Live/dead fixable aqua dead cell stain kit for 405 nm excitation (Life Technologies). Kupffer cells (CD11b+, F4/80high) were further separated by using eFlour 450-conjugated anti-mouse CD45 (Clone 30-F11) with a 1:400 dilution, FITC-F4/80 (Clone BM8) with a 1:1000 dilution (Thermo Fisher Scientific), anti-mouse APC-CD11b (Clone M1/70) (BD Biosciences) with a 1:400 dilution and HSCs were gated by endogenous retinoid fluorescence (using DAPI channel). Since the antibody used in this study binds to the C-terminal of mGluR5, Cytofix/Cytoperm (BD Biosciences) was used for intracellular staining of mGluR5. Alex 647-conjugated anti-mGluR5 antibody (Abeam) with a 1:100 dilution and isotype control antibody (Abcam) with a 1:100 dilution were used. Stained cells were read with FACS LSRII (BD Biosciences), and the result was analyzed by FlowJo software (Flow Jo LLC).
Western Blot Analyses
Total protein samples were isolated from frozen liver tissue or cultured cells using RIPA lysis buffer (30 mM Tris, pH 7.5, 150 mM NaCl, 1 mM PMSF, 1 mM Na3VO4, 10% SDS, 10% glycerol), containing protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Samples were separated in a 10% SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane (#88018, Thermo Fisher Scientific). After the membranes were blocked in 5% skim milk or 5% BSAfor 1 h at room temperature, they were incubated with primary antibodies overnight at 4°C and then with the corresponding secondary antibodies for 1 h at room temperature. The information for antibodies is listed in the KEY RESOURCES TABLE. All of the primary antibodies were used at a dilution of 1:1000 except DAGLβ (1:500) and mGluR5 (1:2000). Secondary antibodies were used at 1:2000dilution. Immunoreactive bands were detected using the ECL detection system with a Phosphorlmager (GE Healthcare). Protein expression levels were normalized to the levels of the β-actin, which was used as a loading control.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-actin (clone AC-74) | Sigma-Aldrich | Cat#: A5316; RRID: AB_476743 |
| p-AMPK (clone 40H9) | Cell Signaling Technology | Cat#:2535; RRID: AB_331250 |
| AMPK | Cell Signaling Technology | Cat#:2532; RRID: AB_330331 |
| FASN | Cell Signaling Technology | Cat#:3189; RRID: AB_2100798 |
| DAGLβ (D4P7C) | Cell Signaling Technology | Cat#:12574; RRID: AB_2797959 |
| p-p42/44 MAPK | Cell Signaling Technology | Cat#:9101; RRID: AB_331646 |
| ALDH4A1 (672) | Santa Cruz Biotechnology | Cat#:sc-100499; RRID: AB_2224476 |
| CYP2E1 | Millipore | Cat#:AB1252; RRID: AB_90517 |
| xCT | Thermo Fisher Scientific | Cat#:PA1–16893; RRID: AB_2286208 |
| GCLC | Thermo Fisher Scientific | Cat#:PA5–16581; RRID: AB_10977936 |
| NRF2 | Abcam | Cat#:ab31163; RRID: AB_881705 |
| mGluR5 | Abcam | Cat#:ab76316; RRID: AB_1523944 |
| GS | Abcam | Cat#: ab73593; RRID: AB_2247588 |
| GLS2 | Abcam | Cat#; ab113509; RRID: AB_10866157 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#:7074; RRID: AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Thermo Fisher Scientific | Cat#:31430; RRID: AB_228307 |
| Anti-rabbit IgG H&L (Alexa Fluor® 488) | Abcam | Cat#:ab150061; RRID: AB_2571722 |
| Anti-rabbit IgG H&L (Alexa Fluor® 594) | Abcam | Cat#:ab150064; RRID: AB_2734146 |
| Anti-mouse IgG H&L (Alexa Fluor® 594) | Abcam | Cat#:ab150108; RRID: AB_2732073 |
| CD16/CD32 (Clone 2.4G2) | BD Biosciences | Cat#:553141, RRID: AB_394656 |
| CD45 (Clone 30-F11)-eFluor 450 | Thermo Fisher Scientific | Cat#: 48-0451-80; RRID: AB_1518807 |
| F4/80 (Clone BM8)-FITC | Thermo Fisher Scientific | Cat#: 11-4801-82; RRID: AB_2637191 |
| CD11b (Clone M1/70)-APC | BD Biosciences | Cat#: 553312; RRID: AB_398535 |
| Anti-mGluR5 Antibody (Alexa Fluor® 647) | Abcam | Cat#:ab196929 |
| Rabbit IgG - Isotype Control (Alexa Fluor® 647) | Abcam | Cat#:ab199093 |
| Bacterial and Virus Strains | ||
| DH5α Competent Cells | Invitrogen/Fisher Scientific | LS18265017 |
| pAAV-U6-sgRNA(shRNA)-CMV-GFP | Addgene | Cat#:85451 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Acrylamide | Bio-Rad | Cat#161–0156 |
| Blotting-Grade Blocker | Bio-Rad | Cat#: 170–6404 |
| Bradford reagent | Sigma-Aldrich | Cat#:B6916 |
| BSA | Sigma-Aldrich | Cat#:A9647 |
| CHPG | Tocris | Cat#:1049 |
| Collagenase type 1 | Worthington | Cat#:LS004197 |
| CTEP (RO4956371) | APExBIO | Cat#:B1633 |
| DAPI | Abcam | Cat#:ab104139 |
| DHPG | Tocris | Cat#:0805 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#:D2650 |
| High glucose Dulbecco’s modified Eagle’s medium (DMEM) | Welgene | Cat#:LM001–05 |
| Histoacryl® | B Braun | NA |
| Low glucose DMEM | Welgene | Cat#:LM001–11 |
| Ethanol | Millipore | Cat#: 100983 |
| Fetal Bovine Serum (FBS) | Welgene | Cat#:S101–01 |
| KT172 | Sigma-Aldrich | Cat#:SML1688 |
| L-glutamic acid, monosodium salt, low toxin | Sigma-Aldrich | Cat#:RES5063G-A701X |
| MPEP hydrochloride | Tocris | Cat#:1212 |
| Optiprep™ density gradient medium | Sigma-Aldrich | Cat#:D1556 |
| Penicillin and streptomycin | Thermo Fisher Scientific | Cat#: 15240062 |
| Percoll® | Sigma-Aldrich | Cat#:P1644 |
| Protease and phosphatase inhibitor cocktail | Thermo Fisher Scientific | Cat#:78440 |
| Rimonabant hydrochloride | Tocris | Cat#0923 |
| Rosewell Park Memorial Institute (RPMI)-1640 | Welgene | Cat#:LM011–01 |
| Sapl | New England BioLabs | Cat#:R0569S |
| Sulfasalazine | Sigma-Aldrich | Cat#:S0883 |
| Triton X-100 | Sigma-Aldrich | Cat#:T9284 |
| Tween 20 | Sigma-Aldrich | Cat#:P1379 |
| TRIzol reagent | Thermo Fisher Scientific | Cat#: 15596–018 |
| Critical Commercial Assays | ||
| ALDH4A1 ELISA Kit | MyBioSource | Cat#:MBS9332194 |
| amfiRivert II cDNA Synthesis Master Mix | GenDEPOT | Cat#:R5500–100 |
| DAB peroxidase substrate | Vector Laboratories | Cat#:SK-4100 |
| ECL western blotting substrate | Thermo Fisher Scientific | Cat#:34095 |
| ImmPRESS HRP Anti-Rabbit IgG Polymer Detection Kit | Vector Laboratories | Cat#:MP-7401 |
| ReverTra Ace® qPCR RT Master Mix with gDNA Remover | Toyobo | Cat#:FSQ-301 |
| SYBR Green Realtime PCR Master Mix | Toyobo | Cat#:QPK-201 |
| Deposited Data | ||
| RNA-seq analysis of pair- and ethanol-fed mouse liver tissue | NCBI BioProject | PRJNA494040 |
| RNA-seq analysis of pair- and ethanol-fed mouse liver tissue | NCBI Gene Expression Omnibus | GEO: GSE97234 |
| Gene microarray of alcoholic hepatitis patient liver tissue | NCBI Gene Expression Omnibus | GEO:GSE28619 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | CRL-3216 |
| Human: Hep3B cells | ATCC | HB-8064 |
| Human: HepG2 cells | ATCC | HB-8065 |
| Mouse: Neuro-2a cells | ATCC | CCL-131 |
| Experimental Models: Organisms/Strains | ||
| Cyp2e1 knock-out mice | Jackson Laboratories | #002910 (provided by Dr. Bin Gao) |
| Grm5 knock-out mice | Jackson Laboratories | #003121 |
| Slc7a11 tm1a mice | EMMA mouse repository | #EM:10001 |
| Albumin-Cre mice | Jackson Laboratories | #003574 |
| C57BL/6J | Jackson Laboratories | C57BL/6J |
| Oligonucleotides | ||
| primers for qPCR | Bioneer | See Table S3 |
| control shRNA | Bionner | See STAR★METHODS |
| Slc7a11 shRNA | Bioneer | See STAR★METHODS |
| Software and Algorithms | ||
| FlowJo, Version 10 | FlowJo | www.flowjo.com |
| Ingenuity Pathway Analysis | QIAGEN | www.ingenuity.com |
| GraphPad Prism version 7 | Prism | www.graphpad.com |
| Other | ||
| Lieber-DeCarli ethanol liquid diet | Dyets | Cat#:710260 |
Quantitative PCR
For qRT-PCR assays, total RNA was isolated from liver tissues or cells with TRIzol reagent (Thermo Fisher Scientific). Total RNA was obtained and reverse-transcribed into cDNA using amfiRevert II cDNA Synthesis Master Mix (GenDEPOT) or ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo, Japan) according to the manufacturer’s instructions. qRT-PCR was performed by SYBR Green Realtime PCR Master Mix (Toyobo, Japan). The mRNA expression levels of the target genes were normalized to Actb or 18S rRNA expression. The primer pairs used in this study are listed in Table S3.
RNA-seq Library Preparation and Sequencing
Total RNA was isolated as described above from pair-fed and ethanol-fed mouse livers (n = 3/group) with TRIzol reagent (Thermo Fisher Scientific). The quality of the extracted RNA was evaluated by an Agilent 2100 Bioanalyzer RNA Nano Chip. Sequencing libraries were constructed from 1 μg of total extracted RNA using a TruSeq RNA Sample Prep Kit v2 (Illumina) according to the manufacturer’s instruction. Samples were randomly fragmented, ligated to unique adaptors, and subjected PCR for amplification. Through size selection, 200 to 400 bp of PCR product was selected and were sequenced using an Illumina HiSeq 4000 platform (Illumina) at an average of 74 million paired-end 101 bp reads.
RNA-seq Processing and Analysis
RNA-seq reads were aligned to the mouse genome (GRCm38/mm10) using TopHat2 V2.1.1 (https://ccb.jhu.edu/software/tophat/index.shtml). Differentially expressed genes were determined using the Tuxedo protocol (Trapnell et al., 2012). Then, Cufflinks V2.2.1. (https://github.com/cole-trapnell679lab/cufflinks) were used to process the aligned reads, which newly creates a minimum union set of assembled transcribed fragments with gene annotations for each replicate rather than whole transcript set during the transcript assembly. Based on the Fragments per kilobase of exon model per million mapped reads (FPKM) value, the relative abundances of transcripts were evaluated with the normalized RNA-seq fragment counts (Trapnell et al., 2010). Finally, we used Cuffdiff to calculate a difference of FPKMs between pair-fed and ethanol-fed mouse samples with three replicates per each group. Differential expression was calculated using false discovery rate adjusted P value (< 0.05). Pathway enrichment and prediction of pathway were evaluated by Ingenuity Pathway Analysis (Palaga et al.) Software (Ingenuity Systems, Qiagen; www.ingenuity.com).
Endocannabinoid Measurements
The endocannabinoid levels of mouse liver tissues, and cell lysates were measured by stable isotope dilution liquid chromatography/tandem mass spectrometry (LC-MS/MS) as described previously (Mukhopadhyay et al., 2011). In detail, liver weighing 50 mg was homogenized in 0.5 mLof ice-cold methanol/Tris buffer (50 mM, pH 8.0), 1:1, containing 7 ng of [2H4]arachidonoyl ethanolamide ([2H4]AEA) as internal standard. Homogenates were extracted three times with CHCI3-MeOH (2:1, vol/vol), dried under nitrogen flow, and reconstituted with MeOH after precipitating proteins with ice-cold acetone.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analyses were performed on an Agilent 6410 triple quadrupole mass spectrometer (Agilent Technologies) coupled to an Agilent 1200 LC system. Analytes were separated using a Zorbax SB-C18 rapid-resolution HT column. Gradient elution mobile phases consisted of 0.1% formic acid in H2O (phase A) and 0.1% formic acid in MeOH (phase B). Gradient elution (250 μL/min) was initiated and held at 10% B for 0.5 min, followed by a linear increase to 85% B at 1 min and maintained until 12.5 min, then increased linearly to 100% B at 13 min and maintained until 14.5 min. The mass spectrometer was set for electrospray ionization operated in positive ion mode. The source parameters were as follows: capillary voltage, 4,000 V; gas temperature, 350°C; drying gas, 10 L/min; nitrogen was used as the nebulizing gas. Collision-induced dissociation was performed using nitrogen. Levels of each compound were analyzed by multiple reactions monitoring. The molecular ion and fragment for each compound were measured as follows: m/z 348.3→62.1 for AEA, m/z 305.2 →91.1 for arachidonic acid, m/z 352.3→66.1 for [2H4]AEA, m/z 379.3→91.1 for 2-arachidonoylglycerol (2-AG), and m/z 326.3→62.1 for oleoylethanolamide (OEA). Analytes were quantified using MassHunter Workstation LC/QQQ Acquisition and MassHunter Workstation Quantitative Analysis software (Agilent Technologies). Levels of AEA, 2-AG, arachidonic acid, and OEA in the samples were measured against standard curves. Values are expressed as fmol or pmol/mg wet tissue.
Glutamate Measurements
Frozen liver tissue samples were weighed (~ 30 mg each), defrosted and deproteinized by homogenizing in 100% methanol. For serum (or plasma), 200 μl of each sample was mixed with 400 μl of 100% methanol and homogenized. Samples were then centrifuged at 10,000 ×g for 15 min at 4°C. Supernatants were subjected to high-performance liquid chromatography (HPLC) with autosampler. Supernatants from serum (or plasma) and media were dried using SpeedVac System and dried materials were resuspended in 50 μl of 0.1 N HCl solution, then subjected to HPLC fractionation. A ZORBAX Eclipse-AAA (4.6 × 75 mm, 3.5 μm) chromatographic column and the Agilent 1200 HPLC (Agilent) were used. Mobile phase A was 40 mM NaH2PO4 pH 7.8 (5.5 g NaH2PO4 + 1 L water, adjust to pH 7.8 with 10 N NaOH) and mobile phase B was 45:45:10 (v/v/v) acetonitrile: methanol: water. The column was run with a flow rate of 1 ml/min. Phase B increased from 0 to 57% between 0 and 9.8 min and from 57 to 100% between 9.8 and 10 min. It remained at 100% between 10 and 12 min and decreased from 100% to 0% between 12 and 12.5 min. Between 12.5 and 14 min, Phase B remained at 0%. The column temperature was 35’C. The diode array wavelength was 338 nm, and the reference wavelength was 390 nm. Amino acid standards (1 nmol/μl, Agilent) were measured as an external standard before sample measurements. Sample peak area was calculated by comparison with the area of amino acid standards of a known concentration.
Sulfur-containing Metabolite Measurements
For the measurement of methionine, the liver homogenates were deproteinized in a 3-fold volume of ice-cold methanol. For the measurement of SAM, SAH, homocysteine, GSH, cysteine, and cystine, liver tissues or cell lysates were homogenized in a 4-fold volume or 700 μl of ice-cold 80% methanol with 0.1% (v/v) perchloric acid, respectively. Culture media, human plasma, and mouse sera were deproteinized in a 2-fold volume of ice-cold 100% methanol with 0.1% (v/v) perchloric acid. After centrifugation at 10,000 ×g for 20 min, the supernatant fraction was collected and refrigerated in −70°C. Sulfur-con taining metabolite measurements were measured according to our previous methods (Kwak et al., 2015). In detail, the methionine concentrations were measured using liquid chromatography (LC) triple quadruple electrospray ionization (ESI)/mass spectrometry(MS)/MS and methionine-d3 as an internal standard. The LC-ESI/MS/MS system consisted of a Shimadzu LC-20AD XR high performance liquid chromatography (HPLC) system (Shimadzu, Tokyo, Japan) and an API 3200 QTRAP1 LC-MS/MS system (AB Sciex, Framingham, MA) equipped with a Turbo VTM Ion Source (Applied Biosystems, Foster City, CA) operated in the positive multiple reaction monitoring ion mode with the following transition: methionine m/z 150 → 104; methionine-d3 153 → 107. The sample (10 μL) was injected into a Waters Atlantis hydrophilic interaction liquid chromatography silica (2.1 mm × 150 mm, 3 μm) column with a mobile phase composed of water (solution A) and acetonitrile (solution B). The column was eluted at a flow rate of 0.25 mL/min, and developed with a gradient elution as follows: 0–2 min, 10% A; 4 min, 60% A; 9 min, 40% A; 10.5 min, 10% A. Hepatic SAM and SAH concentration was measured by an HPLC method. An HPLC system (Shimadzu, SCL-10A) equipped with an ultraviolet detector (Shimadzu, SPD-10Avp, 254 nm) and a TSK-GEL ODS-8OTM column (4.6 mm × 250mm, 5 μm; Tosoh Co., Tokyo, Japan) was used. Homocysteine, GSH, and cysteine in the liver were measured using the SBD-F method,
QUANTIFICATION AND STATISTICAL ANALYSIS
The exact value of replicates (n) is described in the figure legends and refers to the number of biological replicates. Sample-size determination by statistics was not applied in this study. Inclusion and exclusion criteria for human subjects are delineated in the “Experimental Model and Subject Details” section. For in vivo mouse experiments, any mice showing severe illness (i.e. body weight loss more than 30%) were excluded from the analysis after ethanol diet feeding. Otherwise, no animal was excluded from the study. Statistical analysis was performed by using Prism version 7.0 (GraphPad Software). Data are presented as the mean ± SEM. The researchers involved in this study were not completely blinded during animal experiments, but were blinded in human data analyses. Significance was evaluated by using the unpaired Student t test between two groups and One-way ANOVA followed by Tukey’s test or Dunnett’s test for multiple comparison (more than two groups). Adjusted P value was used to the comparison of FPKM value and GO enrichment in RNA-seq and gene expression. A P value <0.05 was considered to be significant. Detailed statistics used in this study are provided in Tables S4 and S5.
DATA AND SOFTWARE AVAILABILITY
Raw data of RNA-seq used in this study were deposited in the BioProject repository under accession numbers PRJNA494040. The accession numbers for the RNA-seq and the gene microarray from other studies used in this paper are GEO: GSE97234 and GEO: GSE28619, respectively.
Supplementary Material
Highlights.
Alcohol consumption increases xCT expression in hepatocytes (HEPs)
xCT-derived glutamate release increases expression of mGluRS in HSCs
mGluR5 stimulates 2-AG production in HSC to influence lipogenesis of HEPs via CB1R
Inhibition of xCT and mGluR5 blocks alcoholic steatosis in liver
Context and Significance.
Alcoholic fatty liver is a common consequence of excessive alcohol consumption that causes stress on hepatocytes, one of the cell types present in the liver, and can lead to liver fibrosis and cancer. Researchers at KAIST, Korea, identify here a two-ways interaction between hepatocytes and hepatic stellate cells as a driver of the disease. Hepatocytes compensate against the oxidative stress by activating the cell membrane transporter xCT to produce more antioxidant glutathione. This in turn releases glutamate, trigger the synthesis of the signaling molecule 2-arachidonoylglycerol (2-AG) in stellate cells through the mGluR5 receptor. Once released by hepatic stellate cells, 2-AG binds to its receptor, the cannabinoid receptor-1 (CB1R), on hepatocytes to stimulate fat accumulation. Components of this communication system between hepatic stellate cells and hepatocytes, including xCT, mGluR5 and CB1R, may be targeted to treat alcoholic liver disease.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MEST) (2018R1A2A1A05077608), Korea Mouse Phenotyping Project (2014M3A9D5A01073556), the Intelligent Synthetic Biology Center of Global Frontier Project (2011–0031955), and the Global Ph.D. Fellowship program (NRF-2015H1A2A1033124) through the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT and future Planning; and by intramural research funds of the National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Augustin SM, and Lovinger DM (2018). Functional Relevance of Endocannabinoid-Dependent Synaptic Plasticity in the Central Nervous System. ACS Chem Neurosci 9, 2146–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai JC, Fried M, Corazza GR, Schuppan D, Farthing M, Catassi C, Greco L, Cohen H, Ciacci C, Eliakim R, et al. (2013). World Gastroenterology Organisation global guidelines on celiac disease. J Clin Gastroenterol 47, 121–126. [DOI] [PubMed] [Google Scholar]
- Barak AJ, Beckenhauer HC, Kharbanda KK, and Tuma DJ (2001). Chronic ethanol consumption increases homocysteine accumulation in hepatocytes. Alcohol 25, 77–81. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Saito M, Cooper TB, and Hungund BL (2000). Stimulation of cannabinoid receptor agonist 2-arachidonylglycerol by chronic ethanol and its modulation by specific neuromodulators in cerebellar granule neurons. Biochim Biophys Acta 1535, 78–86. [DOI] [PubMed] [Google Scholar]
- Bird MK, Kirchhoff J, Djouma E, and Lawrence AJ (2008). Metabotropic glutamate 5 receptors regulate sensitivity to ethanol in mice. Int J Neuropsychopharmacol 11, 765–774. [DOI] [PubMed] [Google Scholar]
- Buschard K, Hansen AK, Jensen K, Lindenbergh-Kortleve DJ, de Ruiter LF, Krohn TC, Hufeldt MR, Vogensen FK, Aasted B, Osterbye T, et al. (2011). Alcohol facilitates CD1d loading, subsequent activation of NKT cells, and reduces the incidence of diabetes in NOD mice. PLoS One 6, e17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederbaum AI (2013). Nrf2 and antioxidant defense against CYP2E1 toxicity. Subcell Biochem 67, 105–130. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, and Edwards RH (2002). The glutamine commute: take the N line and transfer to the A. J Cell Biol 157, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Stärkel P, Turner JR, Ho SB, and Schnabl B (2015). Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 61, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzoli DK, Courson J, Wroten MG, Greentree DI, Lum EN, Campbell RR, Thompson AB, Maliniak D, Worley PF, Jonquieres G, et al. (2014). Binge alcohol drinking by mice requires intact group 1 metabotropic glutamate receptor signaling within the central nucleus of the amygdala, Neuropsychopharmacology 39, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S-S, Wang H, Yang N, An J-H, Li W, Ning Y-L, Zhu P-F, Chen J-F, and Zhou Y-G (2013). Plasma glutamate-modulated interaction of A2AR and mGluR5 on BMDCs aggravates traumatic brain injury-induced acute lung injury. Journal of Experimental Medicine 210, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do SH, Yun H-S, Jeong W. l., Jeong D-H, Ki M-R, Chung J-Y, Park S-J, Kim S-B, and Jeong K-S (2007). Up-regulation of Metabotropic glutamate receptor 3 (mGluR3) in rat fibrosis and cirrhosis model of persistent hypoxic condition. Molecular and cellular biochemistry 294, 189–196. [DOI] [PubMed] [Google Scholar]
- Dröge W (2005). Oxidative stress and ageing: is ageing a cysteine deficiency syndrome? Philosophical Transactions of the Royal Society of London B: Biological Sciences 360,2355–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno A, Berardo C, Di Pasqua LG, Siciliano V, Richelmi P, Nicoletti F, and Vairetti M (2018). Selective Blockade of the Metabotropic Glutamate Receptor mGluR5 Protects Mouse Livers in In Vitro and Ex Vivo Models of Ischemia Reperfusion Injury. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, and Sloane-Stanley G (1957). A simple method for the isolation and purification of total lipids from animal tissues. J biol Chem 226, 497–509. [PubMed] [Google Scholar]
- Gao B, and Bataller R (2011). Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang M-Y, Strassle BW, and Lu P (2010a). Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. Journal of Neuroscience 30, 2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, et al. (2010b). Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30, 2017–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg LC, Jung K-M, Spradley JM, Nyilas R, Suplita RL, Zimmer A, Watanabe M, Mackie K, Katona I, and Piomelli D (2012). Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-α initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. Journal of Neuroscience 32, 9457–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Spanagel R, and Krystal JH (2013). Glutamatergic targets for new alcohol medications. Psychopharmacology (Berl) 229, 539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CA, Lin WW, and Valle D (1996). Cloning, characterization, and expression of cDNAs encoding human delta 1-pyrroline-5-carboxylate dehydrogenase. J Biol Chem 271,9795–9800. [DOI] [PubMed] [Google Scholar]
- Hultberg B, Berglund M, Andersson A, and Frank A (1993). Elevated plasma homocysteine in alcoholics. Alcohol Clin Exp Res 77, 687–689. [DOI] [PubMed] [Google Scholar]
- Jeong W. i., Osei-Hyiaman D, Park O, Liu J, Bátkai S, Mukhopadhyay P, Horiguchi N, Harvey-White J, Marsicano G, and Lutz B (2008). Paracrine activation of hepatic CB 1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell metabolism 7, 227–235. [DOI] [PubMed] [Google Scholar]
- Jesse CR, Wilhelm EA, Bortolatto CF, Savegnago L, and Nogueira CW (2009). Selective blockade of mGlu5 metabotropic glutamate receptors is hepatoprotective against fulminant hepatic failure induced by lipopolysaccharide and d-galactosamine in mice. Journal of Applied Toxicology 29, 323–329. [DOI] [PubMed] [Google Scholar]
- Ji C, and Kaplowitz N (2003). Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 124, 1488–1499. [DOI] [PubMed] [Google Scholar]
- Julien B, Grenard P, Teixeira-Clerc F, Van Nhieu JT, Li L, Karsak M, Zimmer A, Mallat A, and Lotersztajn S (2005). Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 128, 742–755. [DOI] [PubMed] [Google Scholar]
- Julio-Pieper M, Flor PJ, Dinan TG, and Cryan JF (2011). Exciting times beyond the brain: metabotropic glutamate receptors in peripheral and non-neural tissues. Pharmacological reviews 63, 35–58. [DOI] [PubMed] [Google Scholar]
- Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, Lee JH, Shim YR, Yi HS, et al. (2017). Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 8, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak HC, Kim YM, Oh SJ, and Kim SK (2015). Sulfur amino acid metabolism in Zucker diabetic fatty rats. Biochem Pharmacol 96, 256–266. [DOI] [PubMed] [Google Scholar]
- Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, and Lu SC (2004). Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res 28, 173–181. [DOI] [PubMed] [Google Scholar]
- Lorenz L, Axnick J, Buschmann T, Henning C, Urner S, Fang S, Nurmi H, Eichhorst N, Holtmeier R, Bodis K, et al. (2018). Mechanosensing by betal integrin induces angiocrine signals for liver growth and survival. Nature 562, 128–132. [DOI] [PubMed] [Google Scholar]
- Louvet A, and Mathurin P (2015). Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 12, 231–242. [DOI] [PubMed] [Google Scholar]
- Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, Pecker F, Mallat A, and Lotersztajn S (2011). Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 54, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Lu SC (1999). Regulation of hepatic glutathione synthesis: current concepts and controversies. The FASEB Journal 13, 1169–1183. [PubMed] [Google Scholar]
- Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, Harvey-White J, Mordi I, Cravatt BF, and Lotersztajn S (2011). Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proceedings of the National Academy of Sciences 108, 6323–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, Jeong WI, Gao B, Duester G, Mackie K, et al. (2010). Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem 285, 19002–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, and Conn PJ (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong W.-i., Bátkai S, Marsicano G, Lutz B, and Buettner C (2008). Hepatic CB 1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. The Journal of clinical investigation 118, 3160–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, and Kunos G (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacological reviews 58, 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, and Osborne BA (2008). Notch signaling is activated by TLR stimulation and regulates macrophage functions. European journal of immunology 38, 174–183. [DOI] [PubMed] [Google Scholar]
- Parsons LH, and Hurd YL (2015). Endocannabinoid signalling in reward and addiction. Nature Reviews Neuroscience 16, 579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa-Filho M, Belo A, Alcochete AA, Rangel PH., and Ferreira ME, (2007). A set of multiplex panels of microsatellite markers for rapid molecular characterization of rice accessions. BMC plant biology 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, and Makarova KS (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, and Bannai S (2002). Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem 277, 44765–44771. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, Mathurin P, Mueller S, Szabo G, and Tsukamoto H (2018). Alcoholic liver disease. Nat Rev Dis Primers 4, 16. [DOI] [PubMed] [Google Scholar]
- Stickel F, Choi SW, Kim YI, Bagley PJ, Seitz HK, Russell RM, Selhub J, and Mason JB (2000). Effect of chronic alcohol consumption on total plasma homocysteine level in rats. Alcohol Clin Exp Res 24, 259–264. [PubMed] [Google Scholar]
- Storto M, Battaglia G, Gradini R, Bruno V, Nicoletti F, and Vairetti M (2004). Mouse hepatocytes lacking mGlu5 metabotropic glutamate receptors are less sensitive to hypoxic damage. European journal of pharmacology 497, 25–27. [DOI] [PubMed] [Google Scholar]
- Storto M, De Grazia U, Knöpfel T, Canonico PL, Copani A, Richelmi P, Nicoletti F, and Vairetti M (2000). Selective blockade of mGlu5 metabotropic glutamate receptors protects rat hepatocytes against hypoxic damage. Hepatology 31, 649–655. [DOI] [PubMed] [Google Scholar]
- Storto M, Ngomba RT, Battaglia G, Freitas I, Griffini P, Richelmi P, Nicoletti F, and Vairetti M (2003). Selective blockade of mGlu5 metabotropic glutamate receptors is protective against acetaminophen hepatotoxicity in mice. Journal of hepatology 38, 179–187. [DOI] [PubMed] [Google Scholar]
- Suh YG, Kim JK, Byun JS, Yi HS, Lee YS, Eun HS, Kim SY, Han KH, Lee KS, and Duester G (2012). CD11b+ Gr1+ bone marrow cells ameliorate liver fibrosis by producing interleukin-10 in mice. Hepatology 56, 1902–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, and Watanabe M (2010). The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 65, 320–327. [DOI] [PubMed] [Google Scholar]
- Teixeira-Clerc F, Julien B, Grenard P, Van Nhieu JT, Deveaux V, Li L, Serriere-Lanneau V, Ledent C, Mallat A, and Lotersztajn S (2006). CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nature medicine 12, 671–676. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Suzuki H, Mizuno H, Kouno M, Suzuki M, Kato Y, Sato A, Okabe K, and Miyashita M (1993). Clinical significance of measuring plasma concentrations of glutamine and glutamate in alcoholic liver diseases. Alcohol and Alcoholism 28, 103–109. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebicka J, Racz I, Siegmund SV, Cara E, Granzow M, Schierwagen R, Klein S, Wojtalla A, Hennenberg M, Huss S, et al. (2011). Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int 31, 860–870. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, and Lu SC (2001). Current concepts in the pathogenesis of alcoholic liver injury. FASEB J 15, 1335–1349. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, and Alger BE (2001). Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21, RC188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldwijk MR, Topaly J, Laufs S, Hengge UR, Wenz F, Zeller WJ, and Fruehauf S (2002). Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Molecular Therapy 6, 272–278. [DOI] [PubMed] [Google Scholar]
- Werner M, Driftmann S, Kleinehr K, Kaiser GM, Mathé Z, Treckmann J-W, Paul A, Skibbe K, Timm J, and Canbay A (2015). All-in-one: advanced preparation of human parenchymal and non-parenchymal liver cells. PloS one 10, e0138655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Y-J, Puhl HL, and Ikeda SR (2009). Molecular reconstruction of mGluR5a-mediated endocannabinoid signaling cascade in single rat sympathetic neurons. Journal of Neuroscience 29, 13603–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M-J, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, Odena G, Lu J, Tanaka N, and Matsusue K (2015). Fat-specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology 149, 1030–1041. e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Liu Y-W, Zhao L, Wang H, Yang N, Dai S-S, and He F (2017). Metabotropic glutamate receptor 5 deficiency inhibits neutrophil infiltration after traumatic brain injury in mice. Scientific reports 7, 9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data of RNA-seq used in this study were deposited in the BioProject repository under accession numbers PRJNA494040. The accession numbers for the RNA-seq and the gene microarray from other studies used in this paper are GEO: GSE97234 and GEO: GSE28619, respectively.