

Keywords: acute pancreatitis, eATP, lung injury, P2 receptors, systemic inflammation
Abstract
In the current study, we explored the role of extracellular ATP (eATP) in promoting systemic inflammation during development of acute pancreatitis (AP). Release of extracellular (e)ATP was evaluated in plasma and bronchoalveolar lavage fluid (BALF) of mice with experimental acute pancreatitis (AP). Prophylactic intervention using apyrase or suramin was used to understand the role and contribution of eATP in pancreatitis-associated systemic injury. AP of varying severity was induced in C57BL/6 mice using 1-day or 2-day caerulein, caerulein + LPS and l-arginine models. eATP was measured in plasma and BALF. Mice were treated with suramin or apyrase in the caerulein and l-arginine models of AP. Plasma cytokines, lung, and pancreatic myeloperoxidase, and morphometric analysis of pancreatic and lung histology, were used to assess the severity of pancreatitis. Plasma eATP and purinergic 2 (P2) receptors in the pancreas and lungs were significantly elevated in the experimental models of AP. Blocking the effect of eATP by suramin led to reduced levels of plasma IL-6 and TNFα as well as reduced lung, and pancreatic injury. Neutralizing eATP with apyrase reduced systemic injury but did not ameliorate local injury. The results of this study support the role of eATP and P2 receptors in promoting systemic inflammation during AP. Modulating purinergic signaling during AP can be an important therapeutic strategy in controlling systemic inflammation and, thus, systemic inflammatory response syndrome during AP.
NEW & NOTEWORTHY Released ATP from injured cells promotes systemic inflammation in acute pancreatitis
INTRODUCTION
Organ failure in acute pancreatitis (AP) remains a substantial problem and cause of mortality in severe cases (11). Despite the considerable improvement in our understanding of the pathophysiology of local pancreatic injury, the mechanisms and the contributing factors that convert local inflammation to systemic inflammation still remain obscure.
The initial injury to pancreatic parenchyma in AP is amplified by local and incoming leukocytes (25), which often leads to systemic inflammation with concomitant multiple organ dysfunction (MOD) (27). MOD is a consequence of the systemic activation of the immune system, known as systemic inflammatory response syndrome (SIRS) (22, 33). Thus, activation of the immune system has been shown to contribute to the severity of pancreatitis, and reducing this seems to reduce the severity of the disease (17, 21).
Injured acinar cells release many cellular damage-associated molecular patterns (DAMPs) (16), which interact with the immune system to induce inflammation. ATP, which is present in a high intracellular concentration in acinar cells (34), can be released from injured cells. This released extracellular ATP (eATP) has the potential to activate the immune system by binding to its receptors on immune cells (31). Conversely, eATP has been shown to be involved in guiding neutrophils to the site of injury (8), activation of macrophages (20) and maturation of dendritic cells (6). Released eATP initiates autocrine/paracrine signaling, known as purinergic signaling, by binding to either P2X or P2Y receptors. P2X receptors are ion channels, whereas P2Y are G protein-coupled receptors (28). The purinergic signaling is terminated by CD-39-mediated phosphohydrolysis of eATP, which is subsequently converted to adenosine by enzyme CD-73. Adenosine, which binds to P1 receptors, suppresses inflammation and promotes wound repair (9, 14). So far, there have been studies exploring the role of enzyme CD-39 in chronic pancreatitis and pancreatic cancer (20a, 20b), wherein CD-39 is upregulated in chronic pancreatitis and cancer and deletion of CD-39 in a mouse model led to decreased fibrosis and atropy. However, the role and contribution of eATP in AP associated systemic injury have not been investigated.
In the current study, we hypothesized that eATP, which is released by acinar cell death in AP, amplifies the local inflammation. The increase in plasma eATP and P2 receptor expression promotes systemic inflammation. Interruption of the signaling either by degradation of eATP or by blocking P2 receptors results in significantly reduced systemic inflammation.
MATERIALS AND METHODS
Materials.
Collagenase type IV was from Worthington Biochemical (Lakewood,NJ); l-arginine hydrochloride and ATP (cell culture grade, A6419) were from Sigma-Aldrich (St. Louis, MO); antibodies to p38 (9212), phosphorylated (p-)p38 (9211), and IkB (246) were from Cell signaling. Antibody to glyceraldehyde-3-phosphate dehydrogenase (GAPDH, AB2302) was from EMD Millipore. Fura 2-AM was from Thermo Scientific (F1201). ATP-γ-S tetralithium salt (C-4080) and caerulein were from Bachem (4030451).
ATP measurements.
Plasma eATP was measured using an Enliten kit (Promega, Madison WI), according to the manufacturer’s protocol.
Acinar cell preparation and treatment.
Mouse acinar cells were prepared fresh and treated according to our previously reported protocol (12).
Animal experiments.
All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Minnesota and the University of Miami. All samples were collected, frozen in liquid nitrogen, and stored at −80°C. AP was induced by caeruelin, l-arginine, or caeruelin plus LPS methods. C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA),
Preparation of plasma and tissue samples.
Mice were euthanized at the indicated time points. Blood was collected in EDTA-collated tubes. Pancreases and lungs were harvested. Serum amylase was measured using standard procedures. Myeloperoxidase (MPO) activity (i.e., the quantification of pulmonary neutrophil infiltration in lung samples) was measured via an established protocol (10).
Caerulein 7-h model.
AP was induced by seven hourly injections of 50 μg/kg ip caerulein. Animals were euthanized 1 h after the last cerulein injection. One unit of apyrase was administered (intraperitoneally) 30 min before each caeruelin injections. Suramin (10 mg/kg) was administered 1 h before the start of the first careulein injections and immediately after the fourth injections.
Caerulein + LPS model.
AP was induced by six hourly injections of 50 μg/kg ip of caerulein followed by a single injection of LPS (10 mg/kg ip), and animals were euthanized 3 h after LPS injection (11a).
l-Arginine model.
AP was induced by two hourly injections of 4.5 g/kg ip of filter-sterilized l-arginine solution (pH 7.0) in saline followed by two hourly subcutaneous injections 1.5 ml of saline. Animals were euthanized 72 h after the first injection of l-arginine. Doses of suramin (10 mg/kg) were injected at 24, 48, and 60 h after l-arginine injection and mice were sacked at 72 h.
Caerulein 2-day model.
AP was induced by eight daily injections of 50 μg/kg ip caerulein for 2 consecutive days. Animals were euthanized 1 h after the last caerulein injection on day 2.
Bronchoalveolar lavage fluid collection.
Bronchoalveolar lavage fluid (BALF) collection was performed by cannulating the trachea with a blunt 22-gauge needle and lavaging both lungs with 1 ml of sterile PBS solution. The BAL fluid was centrifuged, and the supernatant was stored at −80 C for further analysis.
Protein analysis and immunohistochemistry.
Paraffin sections (5 μm) were stained for coronin-1A (1:1,000) using standard immunohistochemistry (IHC) procedures. Quantification of IHC was performed with ImageJ software (National Institutes of Health, Bethesda, MD).
Scoring severity of pancreatitis.
Pancreatic sections were stained with hematoxylin and eosin (H&E) and assessed at ×200 magnification, over 5–10 separate fields for severity of pancreatitis by scoring for edema, inflammatory infiltrate, and necrosis.
qPCR array.
cDNA was diluted with nuclease-free water and added to RT qPCR SYBR Green Master Mix (Roche). Twenty-five microliters of the experimental cocktail were added to each well of the inflammatory gene PCR array (SA Biosciences, Frederick, MD). Real-time PCR was performed on the Roche 480 QPCR System, and SYBR Green was used as a detection dye. Data were analyzed by the 2−ΔΔCT method.
Immunoblot.
The protein concentration in the cytosolic and membrane fractions of acini was measured using Pierce’s BCA reagent (Thermo-Fisher Scientific, Waltham, MA). Samples (30 μg protein/lane) were subjected to gel electrophoresis, as described by Laemmli, using 18% ready gels and transferred to nitrocellulose membranes. Nonspecific binding was blocked by a 1-h incubation of membranes in 5% (wt/vol) nonfat dried milk in PBS. The blots then were incubated for 1 h with primary antibodies (1:1,000) in blocking buffer containing 5% (wt/vol) nonfat dried milk in 0.05% (vol/vol) Tween-20 in PBS, washed three times with 0.05% Tween-20 in PBS, and finally incubated for 1 h with a horseradish-peroxidase-labeled secondary antibody (1:3,000) in the blocking buffer. The blots were developed for visualization using the Super Signal West Dura Extended Detection Substrate.
Calcium imaging.
Fresh pancreatic acinar cells were isolated and loaded with Fura 2-AM (3 μM), for 30 min at room temperature. Cells were loaded in a cuvette and treated with ATP. Calcium signaling was recorded in a Hitachi spectrofluorometer.
ELISA.
Plasma IL-6 and tumor necrosis factor-α (TNF-α) were quantified using commercially available ELISA kits (eBioscience, Thermo Fisher Scientific, Weston, FL) according to the manufacturer’s instructions. leukotriene B4 (LTB4) levels were measured using a commercially available kit (Cayman Chemicals) according to the manufacturer’s instructions.
Real-time PCR.
Gene transcript levels were quantified by real-time PCR. Total RNA was isolated using the TRIzol method according to the manufacturer’s instructions, and quantified with the Nano-Drop spectrophotometer. One microgram of RNA was used to make cDNA using the RT kit (Applied Biosciences). PCRs were performed using gene-specific primers, and 18S was used as the internal control. A primer list is provided in Table 1.
Table 1.
List of primers used for qPCR
| Forward | Reverse | |
|---|---|---|
| Mouse | ||
| Purinergic receptor P2X, ligand-gated ion channel 1 (P2X1) | GCTCCACATCCTGCCTAAGA | TGGAAGCCACACACTGAGT C |
| P2X2 | CATCATCAATCTGGCCACTG | ACGCACCTTGTCGAACTTCT |
| P2X3 | ATTTCCTCAAAGGGGCTGAT | CACTGGTGAACACTGGGTT G |
| P2X4 | GCTGTGTGACGTCATAGTCC TC | TTCTGCATCACAGGCTGAGT |
| P2X5 | AGAGGACAAGCCACTGGAG A | GTGATGGCTTCATGTTCACG |
| P2X6 | TCCTCTGTGACCTGCTGCTA | GTAACAGGGTTAGCGGGTG |
| P2X7 | TGTGTGCATTGACTTGCTCA | CTTGCAAGACTTTTCCCAAGC |
| Purinergic receptor P2Y, G protein-coupled 1 (P2Y1) | CAAGCAGAATGGAGACACGA | CTCACTCAGGTGGCACACA C |
| P2Y2 | GGCCTGTGCATATGTGAGTG | GGCAACAGCACGTACTTGAA |
| P2Y4 | GGGGACAAGTATCGAAACCA | CACCCTCATAAGCAGGGAA G |
| P2Y6 | CAGTGTTCTGGACCCCATTC | GACTCTCTGCCTCTGCCACT |
| P2Y10 | GCTTGCTAAAGGGGCTTTCT | TACTGTTGCTGCCCATCTTG |
| P2Y12 | GGCTTTGGGAACTTATGCAA | AGGTGGTATTGGCTGAGGT G |
| P2Y13 | CAGCTGAGTCTCTTCCAAAA CA | CAGCTGTGTCATCCGAGTGT |
| P2Y14 | TCCTCCAGACACACTGATGC | CAGGAATCTCAAAGGCAAG C |
| Human | ||
| P2X1 | GCGTAATAAGAAGGTGGGCGTTA | GCCGCTCGAGGTCTGGTA |
| P2X2 | CAGGTTTGCCAAATACTACAAGATCA | AACTTCCCGGCCTGTCCAT |
| P2X3 | TGTAGGGTGGGTTTTCTTGC | GGTTACCACCGAGGACTCAA |
| P2X4 | CATCATCCCCACTATGATCAACA | AGCACGGTCGCCATGC |
| P2X5 | CGCTGGGGAAGCGGTTA | GCACCAGGCAAAGATCTCACA |
| P2X6 | CACTGCCGCTATGAACCACAA | CGAAGGTCCCTCCAGCCTT |
| P2X7 | ATCGGCTCAACCCTCTCCTAC | CTGGAGTAAGTGTCGATGAGGAAG |
| P2Y1 | CGTGCTGGTGTGGCTCATT | GGACCCCGGTACCTGAGTAGA |
| P2Y2 | CGGTGGACTTAGCTCTGAGG | GCCTCCAGATGGGTCTATGA |
| P2Y4 | TGTCCTTTTCCTCACCTGCAT | TGCCCGAAGTGGGTGG |
| P2Y6 | CCTGCCCACAGCCATCTT | GGCTGAGGTCATAGCAGACAGTG |
| P2Y11 | GTTGGTGGCCAGTGGTGTG | TTGAGCACCCGCATGATGT |
| P2Y12 | AACTGGGAACAGGACCACTG | TAAATGGCCTGGTGGTCTTC |
| P2Y13 | AGGGCTCATAGCCTTTGACA | GATCGTATTTGGCAGGGAGA |
| P2Y14 | TCTTTTACGTGCCCAGCTCT | GGCTCATCACAAAGTCAGCA |
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software. Statistical analysis was performed using Student’s t test (comparing two groups; correction was made using Sidak-Bonferroni method. For comparing more than two groups, one-way ANOVA was used. Data are represented as means ± SE. P < 0.05 was considered significant.
RESULTS
eATP and P2 receptors are elevated in experimental models of AP.
To establish the presence of eATP in AP, we used four different mouse models of experimental AP of varying severity (Fig. 1A). There was a significant increase in plasma eATP (Fig. 1B) in all these models, which correlated positively with the severity of the model. The physiological circulating levels of eATP were in nanomolar range (178 ± 97 nM, n = 6); however, in pathological conditions, this level went up to micromolar range. We could detect mean (±SE) circulating plasma eATP concentrations of 14.6 (±2.4, n = 4) μM in caerulein (7 h), 36 (±6.7, n = 7) μM in caerulein (2 days), 60 (±15, n = 4) μM in l-arginine, and 180 (±63, n = 5) μM in the caerulein + LPS model in our experiments.
Fig. 1.

Acute pancreatitis (AP) leads to increase in plasma extracellular (e)ATP and overexpression of purinergic 2 (P2) receptors in pancreas and lungs. A: schematics of different mouse models of experimental AP used. B: elevated plasma eATP in different models of experimental AP (n = 3–6). C: time-dependent increase in plasma eATP in caerulein (7 h)-induced AP (n = 6). D: increased eATP in bronchoalveolar fluid (BALF) of caerulein (7 h)- and l-arginine-induced AP. E: increased mRNA expression of P2 receptors in pancreas in caerulein (7 h)-induced AP (n = 4). F: increased mRNA expression of P2 receptors in lungs in caerulein (7 h)-induced AP (n = 4). P2X receptors are ion channels; P2Y receptors are G protein-coupled receptors *P < 0.05, **P < 0.01****P <0.0001.
We further investigated change in plasma eATP over time after the first injection of caerulein in caeruelin (7 h) AP model. Maximum plasma eATP was seen at 24 h (Fig. 1C) after initiation of injury. Since lung is one of the major organs to be affected as a result of AP, we measured the presence of eATP in BALF in caerulein (7 h) AP and l-arginine experimental AP models. Both models showed elevated BALF eATP compared with the control animals (Fig. 1D). To further characterize the involvement of purinergic signaling in pancreatic and lung injury, we measured the change in P2 receptor mRNA levels by qPCR. In pancreas, the highest change was observed in P2X2 and P2X6 receptors, followed by P2Y2 and P2X1 (Fig. 1E). In the lungs, the highest expression was of P2X4 and P2X7 receptors followed by P2X6 and P2X2 (Fig. 1F).
To investigate whether paracrine eATP signaling by itself can promote inflammation, we evaluated migration of the neutrophils (which arrive first to the site of injury in acute inflammation) into the peritoneal cavity and the increase in plasma IL-6 and TNF-α in response to ATP. Mice were injected with ATP-γ-S (4 mg/kg ip). The neutrophil populations within the peritoneal cavity, plasma IL-6, and TNF-α levels were measured. Figure 2, A and B, show a significant increase of neutrophils in the peritoneal cavity and an increase in plasma TNF-α and IL-6 (Fig. 2, C and D) in response to injected ATP. This suggests that eATP can act as a chemoattractant for neutrophils and a promoter of inflammation. We further examined the role of ATP in promoting a cytokine storm in AP by injecting a dose of nonhydrolyzable ATP in mice after the last caerulein (7 h) injection, and plasma TNF-α and IL-6 levels were measured. Notably, eATP further increased the plasma levels of TNF-α and IL-6 (Fig. 2, E and F) over caerulein injection, implying the role of eATP in amplifying immune response and promoting cytokine storm build-up in AP.
Fig. 2.

Extracellular (e)ATP induces neutrophil migration and promotes inflammation. A: C57B/6 mice were injected with ATP-γ-S (4 mg/kg ip). Peritoneal exudate cells (PECs) were harvested 3 h later, stained with Ly6G and assayed by flow cytometry. B: quantification of flow data. eATP was able to elevate plasma interleukin-6 (IL-6; C) and plasma tumor necrosis factor-α (TNF-α; D) levels. Effect of ATP-γ-S (4 mg/kg ip) addition to caerulein injection on plasma IL-6 (E) and plasma TNFα levels (F). ATP was injected after 7th injection of caerulein, and mice were euthanized 1 h after ATP injection. *P < 0.05; n = 6 in each group.
Degrading eATP-using apyrase reduces lung injury but not local injury in the caerulein (7 h) model.
To evaluate the contribution of eATP in pancreatitis-associated acute lung injury, we used the ATP degrading ectoenzyme apyrase. Pretreatment with apyrase reduced the pancreatitis-associated lung injury, as seen by improved lung histology (Fig. 3A) and significant reduction in myeloperoxidase activity, as a measure of neutrophil sequestration in tissue (Fig. 3B). Total leukocyte numbers in lungs (detected by staining for coronin-1A) decreased significantly upon apyrase treatment (Fig. 3, C and D), further confirming the contribution of eATP in attracting leukocytes and promoting lung injury.
Fig. 3.

Neutralizing extracellular (e)ATP reduced systemic inflammation but not local injury in caerulein (7 h)-induced acute pancreatitis (AP). AP was induced by 7 hourly injections of 50 μg/kg ip caerulein. Animals were euthanized 1 h after the last caerulein injection. One unit of apyrase was administered (ip) 30 min before each caeruelin injections. A: representative hematoxylin-eosin (H&E) sections of lung tissue showing decreased lung damage in apyrase-treated vs. AP animals. B: decreased lung myeloperoxidase (MPO) upon apyrase treatment. C: representative coronin-1 staining on lung tissue, showing decreased total leukocyte infiltration in apyrase-treated vs. AP animals (arrow indicates coronin-1A-positive leukocytes). D: quantification of coronin-1 staining in lung sections in both groups (values are AU and multiples of 103). Decreased plasma interleukin-6 (IL-6; E) and plasma tumor necrosis factor-α (TNF-α; F) levels upon apyrase treatment. G: representative H&E sections of pancreatic tissue showing no effect of apyrase treatment. H: plasma amylase levels remain unchanged upon apyrase treatment. *P < 0.05, **P < 0.01; n = 4–6 in each group.
We measured plasma TNF-α and IL-6, which are involved in systemic inflammation and organ damage in AP (32, 35) and found significant decreases in plasma IL-6 (Fig. 3E) and TNF-α (Fig. 3F) in apyrase-treated animals, suggesting a reduction in systemic inflammation. Although apyrase was effective in reducing systemic inflammation, the treatment did not improve pancreatic injury, as evidenced by the unchanged pancreatic morphology (Fig. 3G), plasma amylase (Fig. 3H), pancreatic MPO (data not shown), and total leukocyte migration (data not shown). This suggests that apyrase may not be an effective therapeutic strategy in reducing local injury.
Degrading eATP-using apyrase reduces lung injury but not local injury in the l-arginine AP model.
The findings from caerulein (7 h) AP were further evaluated in the l-arginine-induced, severe model of AP. In this model also, treatment with apyrase reduced the pancreatitis-associated lung injury, as suggested by reduced alveolar thickness (Fig. 4A) and reduced lung inflammation, as suggested by decreased lung MPO (Fig. 4B). Migration of total leukocytes in lungs was also reduced significantly upon apyrase treatment (Fig. 4, C and D), which corroborated with the reduced lung MPO. Plasma TNF-α (Fig. 4E) and IL-6 (Fig. 4F) levels were significantly decreased with apyrase treatment. Similar to the caerulein (7 h) model, treatment with apyrase did not improve pancreatic injury (Fig. 4, G and H) in the l-arginine induced, severe pancreatitis model.
Fig. 4.

Neutralizing plasma extracellular (e)ATP reduced systemic inflammation, but not local injury, in l-arginine-induced acute pancreatitis (AP). AP was induced by 2 hourly injections of 4.5 g/kg ip filter-sterilized l-arginine solution (pH 7.0) in saline, and 5 U ip of apyrase was injected at 24, 48, and 60 h. A: representative hematoxylin-eosin (H&E) sections of lung tissue, showing reduced lung damage upon apyrase treatment. B: decreased lung myeloperoxidase (MPO) upon apyrase treatment. C: decreased coronin-1 staining in lung tissue in apyrase-treated vs. l-arginine-induced animals (arrow indicates coronin-1-positive leukocytes). D: quantification of coronin-1 staining in lung sections in both groups (values are multiples of 104). Plasma interleukin-6 (IL-6; E) and plasma tumor necrosis factor-α (TNF-α; F) levels decreased upon apyrase treatment. G: representative H&E sections of pancreatic tissue with l-arginine- and apyrase-treated groups showing unchanged pancreatic morphology with apyrase treatment. H: plasma amylase levels remain unchanged upon apyrase treatment. *P < 0.05, **P < 0.01; NS, not significant; n = 4–6 in each group.
Overall, the results from both the caerulein and l-arginine models suggest that degrading eATP reduces systemic inflammation but not local pancreatic injury. One of the reasons for the ineffectiveness of apyrase in reducing local injury could be presence of a high number of active proteases in pancreas during AP.
Blocking P2 by suramin reduces systemic injury as well as local injury in caerulein (7 h) -induced AP.
Since eATP acts via interacting with P2 receptors, we next sought to evaluate the contribution and effect of P2 receptors in AP, for which we used suramin, a small-molecule P2 inhibitor. Blocking P2 receptors with suramin reduced lung injury, as seen by the reduced alveolar thickness (Fig. 5A) and lung MPO (Fig. 5B). Improvement in lung injury by suramin treatment was also reflected in the decreased number of coronin-1A-positive leukocytes, (Fig. 5, C and D). We found that blocking P2 receptors also decreased experimental pancreatic injury, as seen by improved morphology (Fig. 5E), decreased histopathological features like pancreatic edema, inflammation, necrosis (Fig. 5 F), total coronin-1A-positive leukocytes (Fig. 5, H and I), and decreased plasma amylase as well as pancreatic MPO (Fig. 5J). Suramin further reduced the circulating levels of IL-6 and TNF-α, (Fig. 5, K and L), indicating that P2 receptor blocking can help in reducing cytokine storm and SIRS.
Fig. 5.

Blocking purinergic 2 (P2) receptors by suramin reduced systemic and local injury in caerulein (7 h) model of acute pancreatitis (AP). AP was induced by 7 hourly injections of 50 μg/kg ip caerulein. Animals were euthanized 1 h after the last caerulein injection. Suramin (10 mg/kg) was given 1 h before start of careulein injections and immediately after 4th injections. A: representative hematoxylin-eosin (H&E) sections of lung tissue from mice with caerulein- (7 h) and suramin-treated groups. B: decreased lung myeloperoxidase (MPO) with suramin treatment. C: decreased coronin-1A staining in lungs upon suramin treatment. D: quantification of coronin-1A staining of lung sections, showing significant decreased total leukocyte infiltration in lungs (values are multiples of 104). E: representative H&E sections showing improvement of pancreatic morphology upon suramin treatment in caerulein (7 h) model. F: overall histopathological score of pancreas (combination of edema, infiltration, and necrosis scores) in caerulein- (7 h) and suramin-treated groups. G: decreased plasma amylase in suramin treatment. H: decreased total leucocyte infiltration in pancreas as measured by coronin-1 staining. I: quantification of coronin-1 staining in pancreatic sections, showing decreased leukocyte population after suramin treatment (values are multiples of 104). J: decreased pancreatic MPO upon suramin treatment. Decreased plasma interleukin-6 (IL-6; K) and plasma tumor necrosis factor-α (TNFα; L) upon suramin treatment. *P < 0.05, *P < 0.01; n = 4–6 in each group.
Blocking P2 by suramin reduces systemic injury as well as local injury in l-arginine induced AP.
The effects of suramin were further studied in the severe l-arginine-induced experimental AP model. In this model, suramin was able to significantly reduce lung injury and lung inflammation, as indicated by reduced alveolar thickness (Fig. 6A). It also significantly reduced the migration of total leukocytes in lungs, as measured by coronin-1A staining (Fig. 6, B and C). Suramin was effective in reducing pancreatic injury in this model as well (Fig. 6E), as seen by improved pancreatic morphology. There was significant reduction in pancreatic edema, inflammation, and necrosis (Fig. 6F). We also observed reduced plasma TNFα in the suramin-treated animals (Fig. 6D).
Fig. 6.

Blocking purinergic 2 (P2) receptors by suramin reduced both systemic and local injury and inflammation in an l-arginine model of acute pancreatitis (AP). AP was induced by 2 hourly injections of 4.5 g/kg ip filter-sterilized l-arginine solution (pH 7.0) in saline, and suramin (10 mg/kg) was injected at 24, 48, and 60 h. A: representative histology of hematoxylin-eosin (H&E) sections of lung tissue, showing decreased lung damage upon suramin treatment. B: decreased coronin-1 staining upon suramin treatment. C: quantification of coronin-1 staining of lung sections showing overall decrease in total leucocyte infiltration in lungs (values are multiples of 103). D: representative histology of H&E sections of pancreatic tissue from mice with l-arginine- and suramin-treated groups. E: decreased overall histopathological score of pancreas (combination of edema, infiltration, and necrosis scores) upon suramin treatment. F: decreased plasma tumor necrosis factor-α (TNF-α) upon suramin treatment. *P < 0.05, *P < 0.01; n = 4–6 in each group.
Overall, the results above from the two different models suggest that suramin is effective in reducing systemic as well as local injury.
P2 receptors are present on acinar cells, and eATP treatment induces activation of inflammatory pathways and cell death.
The presence of P2 receptors on either mouse or human pancreatic acinar cells is not well established. A qPCR array was done to measure the levels of different P2 receptors on human and mouse acini. We were able to detect the transcripts of four P2X receptors and four P2Y receptors, namely P2X1, P2X2, P2X3, P2X6, P2Y2, P2Y4, P2Y6, P2Y12 in mouse acini and P2X3, P2X4, P2X5, P2X6, P2Y2, P2Y4, P2Y11 in human acini. Interestingly, we did not detect clear presence of P2X7 receptors on either human or mouse pancreatic acinar cells. Activation of P2X7 receptor induces pores in plasma membrane and increases calcium levels in many cell types. This was verified by measuring calcium levels in mouse acinar cell in response to eATP. Acinar cells did not show any change in intracellular calcium levels in response to eATP, whereas macrophages, which are known to have P2X7, showed a change in intracellular calcium in response to eATP (Fig. 7A), implying the absence of the P2X7 receptor on acinar cells. This rules out involvement of calcium signaling in response to ATP.
Fig. 7.

Effect of extracellular (e)ATP on mouse acinar cells. A: no changes in intracellular calcium levels after eATP (100 μM) treatment. B: list of most upregulated inflammatory genes vs. controls. PCR array was performed using SYBR Green dye according to manufacturer instructions. C: representative protein blot of p38 phosphorylation and IκBα degradation in response to eATP treatment. D: eATP treatment induced secretion of tumor necrosis factor-α (TNFα) in dose-dependent manner. E: secretion of leukotriene B4 (LTB4) in medium in response to ATP (100 μM) treatment; data were normalized to total protein content in each well. F: eATP treatment induces acinar cell death. %Lactate dehydrogenase (LDH) activity was measured in medium supernatant and normalized by total protein content. Fresh acinar cells were incubated with ATP for 3 h and TNF-α, LTB4, and LDH activities were measured in supernatant. *P <0.05; n=5–6 in each group.
To get a comprehensive list of inflammatory genes that are changed in pancreatic acini by ATP treatment, we performed a PCR array for inflammatory genes. We found prominent change in gene expressions of nucleotide-binding oligomerization domain-containing protein-1 and -2, pyrin domain-containing 3, caspase-1, and IL-1β (Fig. 7B), suggesting that eATP activates the nucleotide-binding domain and leucine-rich repeat-containing protein-3 inflammosome pathway in pancreatic acinar cells. ATP further induced change in gene expression of chemokines C-C motif ligand (CCL)-12 and CCL-5 as well as increased expression of chemokine receptors CCR-4, -5, -6, and -8 in pancreatic acinar cells. Notably, CCL-12 and CCL-5 are involved in recruiting leukocyte trafficking in inflamed tissue.
eATP was also able to activate the MAPK pathway in acini, as seen by the increase in phosphorylation of P38 in a concentration-dependent manner. P38 phosphorylation was observed at as low as 10 μM ATP. eATP also induced the activation of NF-κB, as measured by IκBα degradation; however, the robust activation was seen only at a higher concentration of eATP (1 mM; Fig. 7C). Activation of NF-κB at a higher ATP concentration has been reported in endothelial cells (1). Thus, the data suggested involvement of non-inflammasome-dependent pathways as well in acinar cells in response to eATP.
To further understand the inflammatory effect of eATP on acinar cells, we measured secretion of TNFα and LTB4, and release of lactate dehydrogenase (LDH) in response to ATP. Freshly isolated pancreatic acinar cells were treated with 100 μM ATP for 3 h, and TNF-α, LTB4, and LDH levels were measured in the media supernatant. ATP treatment caused secretion of proinflammatory mediator TNF-α in a dose-dependent manner (Fig. 6D). eATP also increased the release of LTB4 (Fig. 7E) and LDH (Fig. 7F). Overall, these results suggest that eATP promotes inflammation and cell death in pancreatic acinar cells.
DISCUSSION
Understanding the connecting link between the local pancreatic injury and concomitant evolving systemic inflammation and SIRS has long been a major challenge in the field of pancreatitis. The underlying molecular mechanism of this process is still not clear. In this study, we have identified eATP as DAMP, which is released in AP, and have examined its role in pancreatitis-induced local and systemic inflammation.
To get a clear picture of eATP in AP, we used four different mouse models of AP, which vary in their levels of severity. In all the models, there was an increase in plasma eATP, which directly correlated with severity of injury. Inflamed lungs showed elevated eATP levels in BALF and high expression of P2 receptors, especially P2X7, in our experiments. P2X7 are expressed in respiratory epithelial cells and most immune cells. A longer exposure to eATP has been shown to cause P2X7-dependent cytochrome c release by mitochondria and apoptosis (15).
In physiological conditions, eATP is kept in check by enzyme CD-39, present on endothelial cells (19) neutrophils, macrophages, and Treg (2). However, during a pathological state proinflammatory cytokines like TNF-α and oxidative stress (26) can inhibit CD-39 ATPase activity, which in turn might aid in building up pathological concentrations in conditions like AP. We found that CD-39 is expressed only on ductal cells but not on acini, implying a low CD-39/acini ratio. Therefore, local eATP might take longer to become degraded in the event of large-scale acinar cell death.
Necrotic acinar cells are likely to be the main source of eATP. The early event in AP is the reduced ATP production by mitochondria; however, there is still a significant amount of ATP present that, if released, can activate purinergic signaling. Other sources that may contribute to the pathological pool of eATP, could be activated immune cells (18), stressed epithelial cells (15) or endothelial cells (5), even cells undergoing apoptosis (13), which could release ATP via pannexin channels.
In AP, both pancreas and lungs showed high expression of more than one isotype of P2 receptors, suggesting involvement of more than one receptor isotype in pathogenesis. In the future, it will be interesting to decipher the role of each receptor. An advantage of using suramin in this study was that we were able to inhibit all P2 receptors, which could have not been possible using single knockout mouse model.
Removing eATP from systemic circulation by ectoenzyme apyrase or blocking P2 receptors using suramin reduced the systemic cytokines such as IL-6 and TNF-α and lung MPO, suggesting the role of purinergic signaling in promoting systemic injury. Upon protein sequence analysis of apyrase by PeptideCutter (ExPASy), we found the presence of 82 proteolytic sites for trypsin and chymotrypsin on apyrase. Thus, apyrase is highly vulnerable to proteolysis by active trypsin and chymotrypsin present in inflamed pancreas.
Suramin, a drug used for treating sleeping sickness, was better in reducing both local and systemic inflammation by blocking P2 receptors. However, this does not exclude the possibility that the protective effect of suramin may partially be also because of other mechanisms as well. The presence of P2 receptors either on mouse or on human pancreatic acini is not well established. One study (23) using analysis on a single acinus reported the presence of transcripts for P2X1, P2X4, P2Y2, and P2Y4 receptors on rat pancreatic acini so far. In the present study, we investigated and found transcripts of eight P2 receptors, namely P2X1, P2X2, P2X3, P2X6, P2Y2, P2Y4, P2Y6, and P2Y12, on mouse acinar cells and seven P2 transcripts, namely P2X3, P2X4, P2X5, P2X6, P2Y2, P2Y4, and P2Y11 on human acinar cells. The presence of these receptors indicates the involvement of ATP-mediated signaling in the pancreas.
In terms of the underlying mechanisms, through in vitro studies using acinar cells and ATP, we found that eATP promotes production of proinflammatory cytokines like TNF-α, LTB4, and chemokines, thus amplifying local inflammation. Notably, LTB4 has been linked to neurogenic pancreatitis and pain (29).
We also found that ATP treatment of acinar cells led to activating the p38 MAPK pathway and NF-κB, two major inflammatory pathways involved in promoting pancreatic inflammation. Whether eATP directly or indirectly activates these pathways is matter of future research.
Gene array data showed that eATP induces expression of pattern recognition pathways. Notably, a study by Hoque et al. (16) has shown that NLPR-3−/− and P2X7−/− mice have less local injury than wild type. Since we could not the detect P2X7 receptor on pancreatic acini, it is likely that the reduced pancreatic injury may have been due to reduced immune activation. Apart from P2X7, P2X4 (7) and cathepsin B (24) are also reported to be involved in inflammasome formation, and it is likely that the absence of P2X7, P2X4, or cathepsin B can promote inflammasome activation in pancreatic acini. Although our current PCR data suggest the possibility of pyroptosis mediated acinar cell death, nevertheless, it is quite likely that necroptosis could also operate in parallel.
Although we have identified eATP as a mediator of systemic inflammation, it is likely that additional mechanism(s) or mediator(s) may operate at the same time. Furthermore, a thorough investigation of role of other DAMPs and their precise roles in MOD will strengthen our understanding and will help in developing a better therapy for patient care.
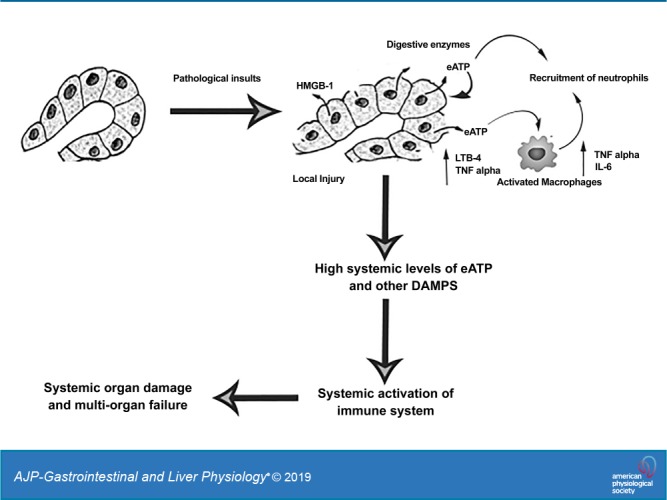
Nonetheless, the present data increase our understanding of multiorgan dysfunction and helps to define the function of purinergic signaling in AP (Fig. 8). The study also opens a new avenue to develop drug targeting against P2 receptors that link local inflammation to respiratory failure
Fig. 8.

Schematic working model of extracellular (e)ATP in promoting inflammation in acute pancreatitis (AP). HMGB1, high-mobility group protein-1; DAMPs, damage-associated molecular patterns.
GRANTS
This work was supported, in whole or in part, by National Institute of Diabetes and Digestive and Kidney Diseases Grants RO1 DK-058694, DK-092145, and R01 DK-093047 (to A. K. Saluja) and RO1 DK-111834 (to V. Dudeja).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.K.D. and A.K.S. conceived and designed research; A.D., H.C., J.G., S.I., and R.K.D. performed experiments; A.D., H.C., J.G., and V.D. analyzed data; A.D., V.D., and R.K.D. interpreted results of experiments; A.D. and H.C. prepared figures; V.D. drafted manuscript; R.K.D. and A.K.S. edited and revised manuscript; A.K.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Current address of A. Dixit: Dept. of Biomedical Engineering, Univ. of Minnesota, Minneapolis, MN 55455. This work was started at the Department of Surgery, University of Minnesota and was concluded at the University of Miami, Miami, FL.
REFERENCES
- 1.von Albertini M, Palmetshofer A, Kaczmarek E, Koziak K, Stroka D, Grey ST, Stuhlmeier KM, Robson SC. Extracellular ATP and ADP activate transcription factor NF-kappa B and induce endothelial cell apoptosis. Biochem Biophys Res Commun 248: 822–829, 1998. doi: 10.1006/bbrc.1998.9055. [DOI] [PubMed] [Google Scholar]
- 2.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med 19: 355–367, 2013. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodin P, Burnstock G. ATP-stimulated release of ATP by human endothelial cells. J Cardiovasc Pharmacol 27: 872–875, 1996. doi: 10.1097/00005344-199606000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol 16: 177–192, 2016. doi: 10.1038/nri.2016.4. [DOI] [PubMed] [Google Scholar]
- 7.Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K, He Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol 45: 932–943, 2013. doi: 10.1016/j.biocel.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314: 1792–1795, 2006. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 9.Chiang B, Essick E, Ehringer W, Murphree S, Hauck MA, Li M, Chien S. Enhancing skin wound healing by direct delivery of intracellular adenosine triphosphate. Am J Surg 193: 213–218, 2007. doi: 10.1016/j.amjsurg.2006.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawra R, Ku YS, Sharif R, Dhaulakhandi D, Phillips P, Dudeja V, Saluja AK. An improved method for extracting myeloperoxidase and determining its activity in the pancreas and lungs during pancreatiti. Pancreas 37: 62–68, 2008. doi: 10.1097/MPA.0b013e3181607761. [DOI] [PubMed] [Google Scholar]
- 11.Dervenis C, Johnson CD, Bassi C, Bradley E, Imrie CW, McMahon MJ, Modlin I. Diagnosis, objective assessment of severity, and management of acute pancreatitis. Santorini consensus conference. Int J Pancreatol 25: 195–210, 1999. [DOI] [PubMed] [Google Scholar]
- 11a.Ding SP, Li JC, Jin C. A mouse model of severe acute pancreatitis induced with caerulein and lipopolysaccharide. World J Gastroenterol 9: 584–589, 2003. doi: 10.3748/wjg.v9.i3.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixit AK, Sarver AE, Yuan Z, George J, Barlass U, Cheema H, Sareen A, Banerjee S, Dudeja V, Dawra R, Subramanian S, Saluja AK. Comprehensive analysis of microRNA signature of mouse pancreatic acini: overexpression of miR-21-3p in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 311: G974–G980, 2016. doi: 10.1152/ajpgi.00191.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461: 282–286, 2009. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7: 759–770, 2008. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homolya L, Steinberg TH, Boucher RC. Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. J Cell Biol 150: 1349–1360, 2000. doi: 10.1083/jcb.150.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas 41: 353–357, 2012. doi: 10.1097/MPA.0b013e3182321500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Liu Y, Daniluk J, Gaiser S, Chu J, Wang H, Li ZS, Logsdon CD, Ji B. Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 144: 202–210, 2013. doi: 10.1053/j.gastro.2012.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11: 201–212, 2011. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaczmarek E, Koziak K, Sévigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem 271: 33116–33122, 1996. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- 20.Kawamura H, Kawamura T, Kanda Y, Kobayashi T, Abo T. Extracellular ATP-stimulated macrophages produce macrophage inflammatory protein-2 which is important for neutrophil migration. Immunology 136: 448–458, 2012. doi: 10.1111/j.1365-2567.2012.03601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a.Künzli BM, Berberat PO, Giese T, Csizmadia E, Kaczmarek E, Baker C, Halaceli I, Büchler MW, Friess H, Robson SC. Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. Am J Physiol Gastrointest Liver Physiol 292: G223–G230, 2007. doi: 10.1152/ajpgi.00259.2006. [DOI] [PubMed] [Google Scholar]
- 20b.Künzli BM, Nuhn P, Enjyoji K, Banz Y, Smith RN, Csizmadia E, Schuppan D, Berberat PO, Friess H, Robson SC. Disordered pancreatic inflammatory responses and inhibition of fibrosis in CD39-null mice. Gastroenterology 134: 292–305, 2008. doi: 10.1053/j.gastro.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malla SR, Kärrman Mårdh C, Günther A, Mahajan UM, Sendler M, D’Haese J, Weiss FU, Lerch MM, Hansen MB, Mayerle J. Effect of oral administration of AZD8309, a CXCR2 antagonist, on the severity of experimental pancreatitis. Pancreatology 16: 761–769, 2016. doi: 10.1016/j.pan.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am 75: 257–277, 1995. doi: 10.1016/S0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- 23.Novak I, Nitschke R, Amstrup J. Purinergic receptors have different effects in rat exocrine pancreas. Calcium signals monitored by fura-2 using confocal microscopy. Cell Physiol Biochem 12: 83–92, 2002. doi: 10.1159/000063784. [DOI] [PubMed] [Google Scholar]
- 24.Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol 195: 1685–1697, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poch B, Gansauge F, Rau B, Wittel U, Gansauge S, Nüssler AK, Schoenberg M, Beger HG. The role of polymorphonuclear leukocytes and oxygen-derived free radicals in experimental acute pancreatitis: mediators of local destruction and activators of inflammation. FEBS Lett 461: 268–272, 1999. doi: 10.1016/S0014-5793(99)01470-2. [DOI] [PubMed] [Google Scholar]
- 26.Robson SC, Kaczmarek E, Siegel JB, Candinas D, Koziak K, Millan M, Hancock WW, Bach FH. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J Exp Med 185: 153–164, 1997. doi: 10.1084/jem.185.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sathyanarayan G, Garg PK, Prasad H, Tandon RK. Elevated level of interleukin-6 predicts organ failure and severe disease in patients with acute pancreatitis. J Gastroenterol Hepatol 22: 550–554, 2007. doi: 10.1111/j.1440-1746.2006.04752.x. [DOI] [PubMed] [Google Scholar]
- 28.Schwiebert E. (Editor) Extracellular nucleotide and nucleoside: Receptor, and physiology and pathophysiological effects. San Diego, CA: Academic Press, 2003. [Google Scholar]
- 29.Shahid RA, Vigna SR, Layne AC, Romac JM, Liddle RA. Acinar cell production of leukotriene B4 contributes to development of neurogenic pancreatitis in mice. Cell Mol Gastroenterol Hepatol 1: 75–86, 2015. doi: 10.1016/j.jcmgh.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal 2: pe6, 2009. doi: 10.1126/scisignal.256pe6. [DOI] [PubMed] [Google Scholar]
- 32.Viedma JA, Pérez-Mateo M, Domínguez JE, Carballo F. Role of interleukin-6 in acute pancreatitis. Comparison with C-reactive protein and phospholipase A. Gut 33: 1264–1267, 1992. doi: 10.1136/gut.33.9.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med 26: 711–715, 2008. doi: 10.1016/j.ajem.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 34.Yegutkin GG, Samburski SS, Jalkanen S, Novak I. ATP-consuming and ATP-generating enzymes secreted by pancreas. J Biol Chem 281: 29441–29447, 2006. doi: 10.1074/jbc.M602480200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Neuhöfer P, Song L, Rabe B, Lesina M, Kurkowski MU, Treiber M, Wartmann T, Regnér S, Thorlacius H, Saur D, Weirich G, Yoshimura A, Halangk W, Mizgerd JP, Schmid RM, Rose-John S, Algül H. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest 123: 1019–1031, 2013. doi: 10.1172/JCI64931. [DOI] [PMC free article] [PubMed] [Google Scholar]