

ABSTRACT
Activation of Toll-like receptor 9 (TLR9) is known to foster innate and adaptive immune responses and thus improve immune-mediated control of malignant disease. Lefitolimod is a potent TLR9 agonist without chemical modification developed for immunotherapeutic strategies. Modulation of the tumor microenvironment (TME) is a crucial requirement for the response to various immunotherapies: Immunogenic (“hot”) tumors, characterized by their T cell-infiltrated TME, respond better compared to non-immunogenic (“cold”) tumors. It has been speculated that the mode-of-action of lefitolimod provides the necessary signals for activation of immune cells, their differentiation into anti-tumor effector cells and their recruitment into the TME. We investigated the effect of lefitolimod on TME, and its potency to induce synergistic anti-tumor effects when combined with immune checkpoint inhibitory antibodies (CPI) in a murine model. Indeed, we could show that treatment with single-agent lefitolimod beneficially modulated the TME, via infiltration of activated CD8+ cells and a shift in the macrophage population toward M1 phenotype. The result was a pronounced anti-tumor effect correlated with the magnitude of infiltrated immune cells and tumor-specific T cell responses. In line with this, lefitolimod led to persistent anti-tumor memory in the EMT-6 model after tumor re-challenge. This was accompanied by an increase of tumor-specific T cell responses and cross-reactivity against different tumor cells. Lefitolimod clearly augmented the limited anti-tumor effect of the CPI anti-PD1 in an A20 and anti-PD-L1 in a CT26 model. These properties of potent immune surveillance reactivation render lefitolimod an ideal candidate as therapeutic agent for immuno-oncology, e.g. improving CPI strategies.
KEYWORDS: MGN1703, cancer immunotherapy, tumor microenvironment, immune checkpoint inhibitors, anti-tumor memory
Introduction
Toll-like receptors (TLR) belong to the group of pattern recognition receptors (PRR) and recognize pathogen-associated molecular patterns (PAMP). PAMP can be subdivided into different classes of molecules like lipopolysaccharides or pathogen-specific nucleic acids and are ubiquitously present in pathogens but are essentially absent in vertebrates. TLR play a key role in immediate immune responses by enabling immune cells to fight pathogens via the innate immune system. This is followed by the induction of antigen-specific effector T cells as well as memory T cells of adaptive immunity. Therefore, TLR agonists are attractive candidates for the development of therapeutic immune modulators to treat a broad range of diseases like cancer, asthma, allergies, or infections.1–3
TLR9 is predominantly expressed by plasmacytoid dendritic cells (pDC) and B cells4 and recognizes non-methylated CG-motifs as PAMP, which are present in pathogenic DNA, but underrepresented in human/vertebrate nuclear DNA. TLR9 is known to broadly activate both the innate and adaptive immunity. 5Therefore, TLR9-triggered immune activation can re-activate immune surveillance to recognize tumor-specific antigens on cancer cells of tumor patients. This translates into the induction of an immune response resulting in the elimination of tumor cells. Synthetic DNA-molecules containing non-methylated CG-motifs function as TLR9 agonists by mimicking the DNA of pathogens and trigger a wide range of immunological activities and are being used for immunotherapeutic approaches.6,7
Members of the dSLIM® family (dSLIM: double Stem Loop Immunomodulator), a new family of TLR9 agonists, consist of dumbbell-shaped, covalently closed DNA molecules devoid of any chemical or other artificial modifications of the DNA.8,9 Protection against nucleolytic degradation is achieved by the covalently closed structure avoiding accessible 3ʹ ends.10 Lefitolimod (MGN1703) belongs to this group of TLR9 agonists exhibiting a specific immunomodulatory sequence and structure.10 Currently, lefitolimod is under evaluation for the maintenance treatment of metastatic colon carcinoma11,12 and data from an exploratory Phase 2 trial in ES-SCLC have recently been published.13
Intracellular signaling triggered by TLR9 results in up-regulation of two pathways: (a) activation of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) inducing the production of pro-inflammatory cytokines and acquisition of antigen-presenting function, and (b) activation of interferon (IFN) regulatory factor 7 (IRF7) leading to type I IFN (e.g. IFN-alpha) production.14,15 IFN-alpha is crucial for the link of the stimulated innate response to the adaptive arm of the immune system.16 The immunomodulatory potential of lefitolimod has been extensively studied in vitro in human PBMC and subpopulations thereof: Lefitolimod activates TLR9-expressing pDC and B cells. IFN-alpha is secreted by pDC and orchestrates cell signaling like cytokine production and expression of co-stimulatory molecules necessary for the activation of dendritic cells, cytotoxic T-lymphocytes, NK-cells, monocytes, and B-cells, which are known to be important in tumor recognition and killing.8,9,17 This mode-of-action (MoA) was confirmed by (i) use of lefitolimod variants devoid of CG-motifs and therefore unable to recognize TLR9, (ii) using human PBMC depleted of TLR9-positive pDC and (iii) blocking of the type I interferon pathway.8,18 Immune activation by lefitolimod was also confirmed in mice and cynomolgus monkeys.
It was speculated that the MoA of lefitolimod starts upstream of the initiation points of immune checkpoint inhibitors (CPI)19 so that a treatment strategy combining both agents, lefitolimod and CPI may result in enhanced anti-tumor effects. Here, we investigate the effects of lefitolimod on the tumor microenvironment (TME) in a murine model, on its potency to induce synergistic anti-tumor effects when combined with CPI as well as its potency to induce long-term immune memory in mouse tumor models.
Results
Impact of lefitolimod on the tumor microenvironment
The syngeneic colon carcinoma model CT26 known as tumor model for microsatellite-stability (MSS),20,21 was used to evaluate lefitolimod’s ability to modulate the tumor microenvironment (TME) and thus induce anti-tumoral effects in vivo (Figure 1(a)). Intratumoral (itu) injection of lefitolimod into established tumors resulted in an increased infiltration of CD3+ T cells into the tumor (Figure 1(d,e)). The subpopulation of CD8+ T cells was increased in the tumor center of the lefitolimod-treated mice (Figure 1(f,g)) and consequently correlated with an inhibition of tumor growth (Figure 1(b,c); Figure S1(a)). The increase of CD8+ T cells within the tumor observed via immunohistopathology was confirmed by flow cytometry. The CD8+ T cells showed an up-regulation of the cytolytic effector Granzyme B, and are so-called activated CD8+ T cells with cytolytic function (Figure 1(h–k)). Moreover, lefitolimod led to an increase of anti-tumoral M1 macrophages and a decrease of pro-tumoral M2 macrophages inside the TME (Figure 2). A higher ratio of M1/M2 correlated with lower tumor volume (Figure S1(b)).
Figure 1.
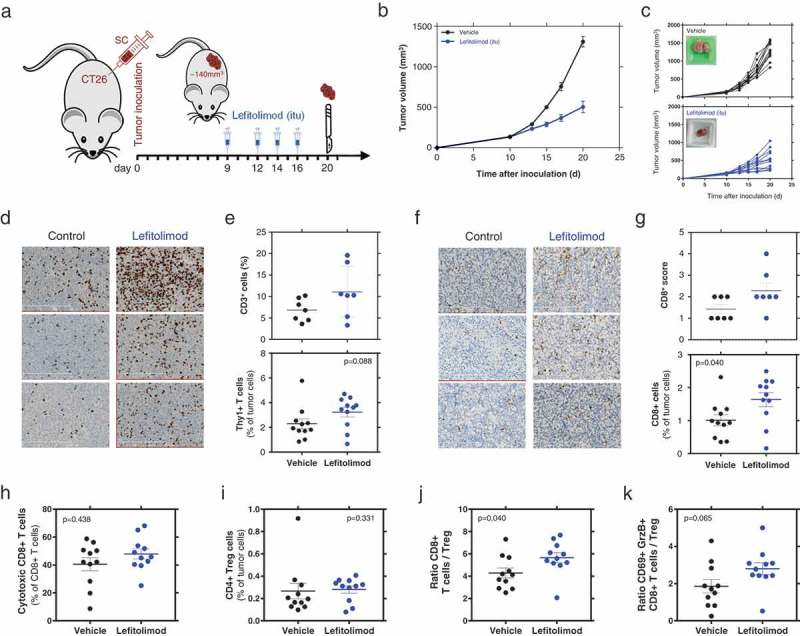
Beneficial modulation of the TME by lefitolimod in vivo. (a), Balb/c mice (N = 14) were inoculated sc with 5 × 104 CT26 tumor cells in 50% matrigel. Established tumors (app. 140 mm3) were injected with 200/250 µg lefitolimod (itu) or vehicle (control) at day 10, 13, 15 and 17. Mice were sacrificed at day 20 for tumor preparation. Tumor tissue was embedded to perform FFPE sections or frozen sections or used to prepare single-cell suspensions which are analyzed by flow cytometry. (b), mean tumor growth (± SEM), p ≤ 0.0001 at days 15, 17, 21 (Sidak`s multiple comparison test). (c), tumor growth from individual mice (top: vehicle, bottom: lefitolimod) – inlay: example of tumor at sacrifice. (d), examples from three representative mice each treated with vehicle or lefitolimod of CD3+ IHC staining (FFPE sections), length of scale bar 300 µm. (e), determination of CD3+ cells in the tumor using the Tissue studioTM software (Definiens®) (p = .097, N = 7, top) or flow cytometry (p = .088, N = 11, bottom). (f), examples from three representative mice each treated with vehicle or lefitolimod of CD8+ IHC staining (frozen sections), length of scale bar 300 µm. (g), determination of CD8+ cells in the tumor via IHC scoring (p = .122, N = 7, top) or flow cytometry (*p = .04, N = 11, bottom). (h-k), Flow cytometric assessment of T cell subpopulations: activated cytotoxic CD8+ T cells (CD8+CD69+GranzymeB+) (p = .438), (h), CD4+ regulatory T cells (p = .331), (i), ratio CD8+ T cells/CD4+ regulatory cells (*p = .04), (j), ratio cytotoxic T cell/CD4+ regulatory T cells (p = .065)(k), Mann–Whitney test was used.
Figure 2.
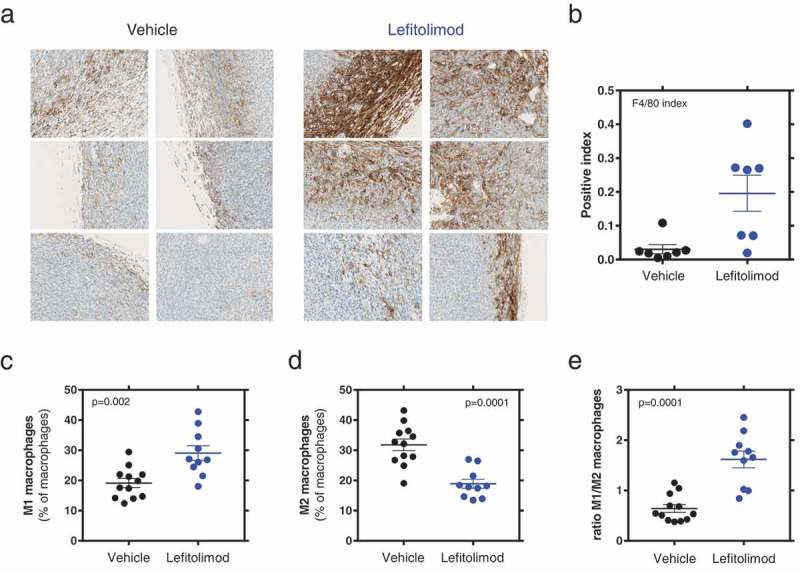
Beneficial modulation of macrophage population in the TME by lefitolimod. Experiments were performed as described in Figure 1. (a), examples from three representative mice each treated with vehicle or lefitolimod of F4/80+ macrophage IHC staining (FFPE sections), display windows are the same as in Figure 1(d). (b), determination of F4/80+ macrophages in the tumor border via IHC scoring (*p = .011, N = 7). (c-d), Flow cytometric assessment of macrophage subpopulations: M1 macrophages (MHC-II/CD86 high) (**p = .002), (c), M2 macrophages (MHC-II/CD86 low) (**p = .0001), (d), ratio M1/M2 macrophages (***p = .0001), (e). Mann–Whitney test was used.
Generation of tumor-specific CD8 + T cells
Itu treatment with lefitolimod in the syngeneic colon carcinoma model CT26 was used to evaluate lefitolimod’s ability to induce a systemic tumor-specific immune response. Splenocytes from lefitolimod-treated mice were re-stimulated with CT26 cells or the MHC-I restricted AH1 peptide derived from the immunodominant antigen gp-70 of CT26 cells. A significant increase of IFN-gamma secreting cells was detected in the spleens of lefitolimod-treated mice after re-stimulation with CT26 cells in comparison to vehicle-treated mice (Figure 3(a)). Moreover, an increase of IFN-gamma secreting cells within the spleens of lefitolimod-treated mice was detected after re-stimulation with the tumor-specific AH1 peptide antigen (Figure 3(b)), indicating the generation of a systemic tumor-specific CD8 + T cell response. The magnitude of response correlated inversely with the measured tumor volume in the lefitolimod-treated group (Figure 3(c,d)).
Figure 3.
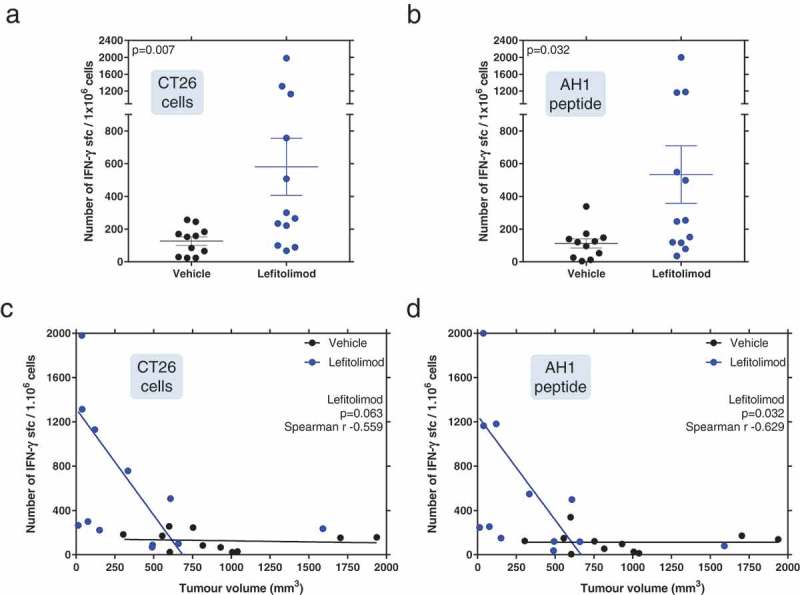
Detection of systemic tumor-specific immune responses. Mice were inoculated with CT26 tumor cells and treated with lefitolimod as described in Figure 1. 20 days after tumor inoculation spleens were harvested and re-stimulated with CT26 cells (a) or AH1 peptide (b) for 20 h. IFN-gamma ELISpot was performed. Number of IFN-gamma secreting cells per 1 × 106 splenocytes are shown. Mann–Whitney test was used. Correlation of tumor volume with the number of IFN-gamma secreting cells after stimulation with CT26 cells (c) or AH1 peptide (d), was calculated with the Spearman r test.
Anti-tumor effects of lefitolimod in low-immunogenic B16 melanoma model
Following the data from CT26 colon carcinoma model, we investigated the effect of lefitolimod in the B16 melanoma model known as low-immunogenic.22 Subcutaneous treatment with lefitolimod led not only to reduced tumor growth but this also translated well into augmented survival (Figure 4(a–d)). The results suggest that lefitolimod is able to reactivate the immune surveillance against another tumor of low immunogenicity.
Figure 4.
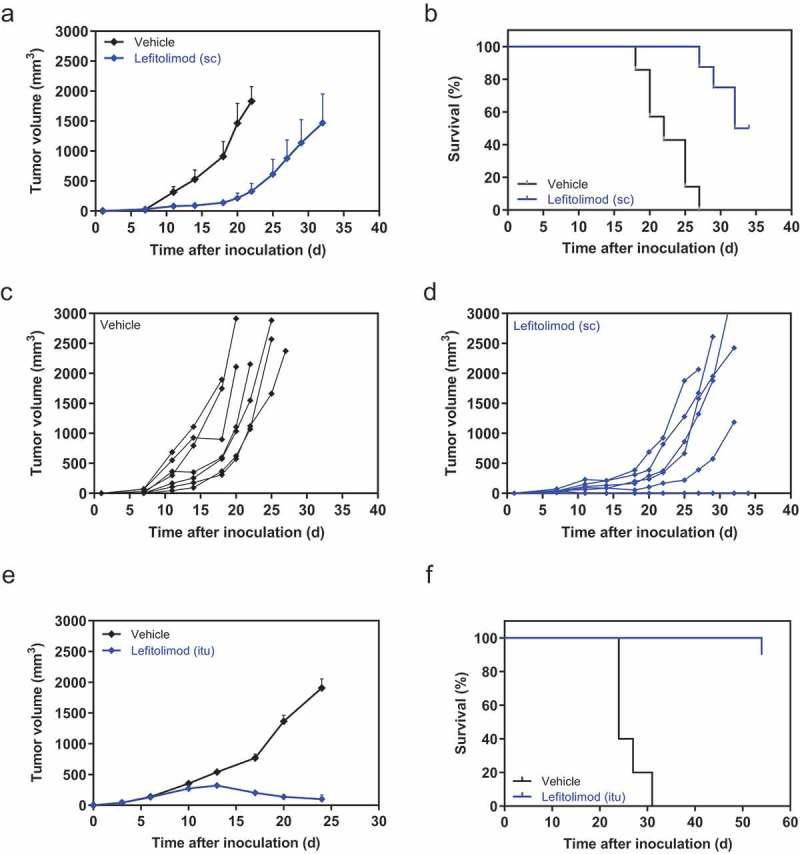
Anti-tumor effect of single-agent lefitolimod in syngeneic B16 and EMT-6 tumor models. (a-d), peritumoral sc injection in B16 model: 5 × 105 B16 tumor cells were inoculated sc into the flank of the mice (day 0). Seven to eight mice each were assigned per group via body weight stratification. 200 µg lefitolimod was injected sc (9x, starting day 3) with vehicle as control. The tumor size at day 7 was 37 mm3 (lefitolimod) and 32 mm3 (vehicle), respectively. Mean tumor growth (±SEM), p = .02 for days 11/14, p = .006 at days 18/20 (multiple t-tests), analyzed until day 20 (a), survival, p = .0001 (log-rank) (b), individual tumor volumes of vehicle (c, black) and lefitolimod group (d, blue) are shown. (e-h), anti-tumor effect and induction of long-lasting immunity in the EMT-6 model after itu treatment: Balb/c mice were inoculated sc with 5 × 105 EMT-6 tumor cells. Established tumors (app. 40 mm3, 3 days after tumor inoculation) were injected with 250 µg lefitolimod (9x, starting day 3). Mean tumor growth (±SEM), p = .04 at day 13, p ≤ 0.0001 at days 17/20/24 (Sidak`s multiple comparison test) (e) and survival, p ≤ 0.0001 (log-rank) (f) are shown. Tumor growth inhibition by lefitolimod was 86% (day 17–24). (g), surviving mice from (f) were inoculated with 5 × 105 EMT-6 tumor cells at day 54 (1st re-challenge) and subsequently with 5 × 105 CT26 cells at day 115 (2nd re-challenge) without further treatment. Age-matched naïve mice were used as controls. Individual tumor volumes are shown. (h), detection of systemic anti-tumor immunity in mice surviving the two re-challenges (EMT-6 and CT26 cells). Spleens were collected and splenocytes were re-stimulated with mitomycin-treated EMT-6, CT26, Renca, A20, 4T1 or WEHI164 cells for 24 h. IFN-gamma ELISpot was performed. Spleen cells of age-matched naïve mice were used as controls. Shown are numbers of spots per 8 × 105 splenocytes corrected by subtraction of numbers of spots obtained for the respective “splenocyte only” control.
Induction of tumor regression and long-lasting immune memory by lefitolimod in EMT-6 breast cancer model
The EMT-6 breast cancer model, described as more immunogenic in comparison to CT26 and B16 but with a comparable degree of low immune cell infiltration,23 is non-responsive to immunotherapy, i.e. with anti-PD-L1.24 However, treatment of mice with single-agent lefitolimod in the syngeneic EMT-6 breast cancer model showed substantial tumor growth inhibition and a highly significant increase of survival (Figure 4(e,f)). Notably, the tumors of 9/10 mice completely disappeared. All nine mice survived a second inoculation of EMT-6 cells as well, in contrast to age-matched naïve mice, indicating a sustained anti-tumor immune memory against EMT-6 tumor cells. Furthermore, all nine mice survived a subsequent inoculation with a distinct CT26 colorectal cancer cell line (Figure 4(g)). This indicates that lefitolimod was not only able to induce a sustained anti-tumor immune memory against EMT-6 tumor cells, but also generated a comprehensive immune response against other cell type, such as CT26 tumor cells, likely via shared antigens of both tumor types. Higher numbers of IFN-gamma secreting cells were detected after stimulation of splenocytes from the double-surviving mice with EMT-6 and CT26, but also with Renca (renal cancer) and 4T1 (breast cancer) cells in comparison to spleen cells of age-matched controls (Figure 4(h)). This indicates the generation of a broad systemic immune memory.
Figure 4.
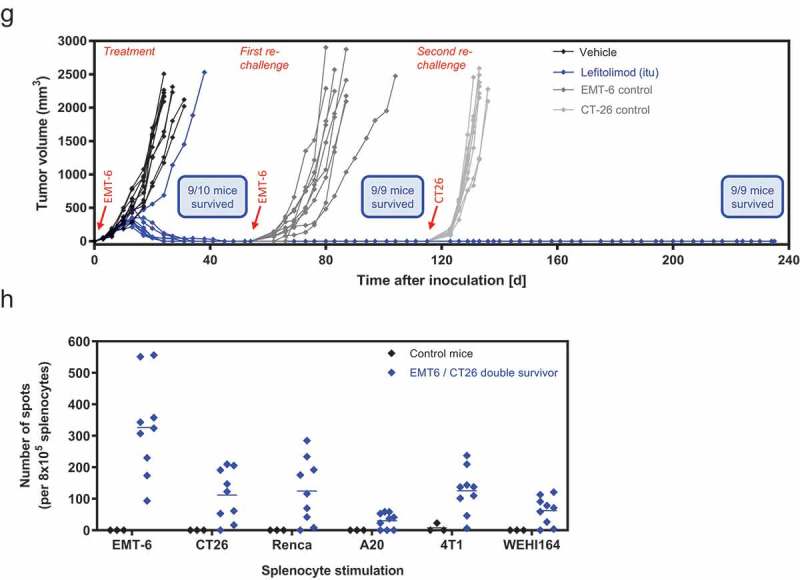
(Continued).
Indirect anti-tumor effect of lefitolimod
To evaluate if a direct effect of lefitolimod on tumor cells contributed to the observed anti-tumor effect, lefitolimod’s influence on the viability of selected tumor cell lines was studied – i.e. cell lines used for tumor inoculation in previously employed tumor models (EMT-6, B16F10, CT26, A20). Neither lefitolimod nor a lefitolimod variant without functional CG-motifs showed relevant induction of cytotoxicity in a relevant concentration range (even at higher concentrations than necessary for optimal immune activation in vitro) (Figure S2). The results were confirmed in an apoptosis-assay, where no increase of caspase 3/7 activity was detected after treatment of the relevant tumor cell lines with lefitolimod (data not shown). This confirmed that lefitolimod does not directly reduce the viability of the tumor cells. Reduced growth of tumor cells required further components of the immune system, such as pDC and NK cells.8,10
Combination of lefitolimod with checkpoint inhibitors in syngeneic tumor models
It was speculated that the mode-of-action of lefitolimod starts upstream of the initiation points of immune checkpoint inhibitors (CPI) like anti-PD-1/anti-PD-L1 so that a treatment strategy combining both agents, lefitolimod and CPI, may result in an enhanced anti-tumor effect. Using the previously employed colon carcinoma CT26 model the combined treatment with lefitolimod and anti-PD-L1 led to a further reduction in tumor growth and also to a prolonged survival of the mice (Figure 5(a,b)). The synergistic anti-tumor effect of lefitolimod with a checkpoint inhibitor was even more pronounced in the A20 lymphoma model, which was selected as model of hematologic origin. The combined treatment with lefitolimod and anti-PD-1 resulted in complete disappearance of the tumors and tumor-free survival of all mice (Figure 5(c,d)). To immunologically explain these anti-tumor data, the impact of lefitolimod on T cell responses was analyzed employing an in vitro assay using human PBMC which were treated with peptides selected from HLA class I-restricted T-cell epitopes of recall-antigens (CMV, EBV, Flu = CEF), lefitolimod and anti-PD-1 as checkpoint inhibitor. The combination of lefitolimod and anti-PD-1 augmented IFN-gamma secretion of PBMC by about 5–6-fold (single agent lefitolimod: 3-fold, single-agent anti-PD-1: 2-fold) (Figure 5(e)). These data suggest that lefitolimod alone can enhance the activation of CD8 + T cells, but also has the potential to synergistically improve the effect of the checkpoint inhibitor anti-PD-1 (Figure 5(f)).
Figure 5.
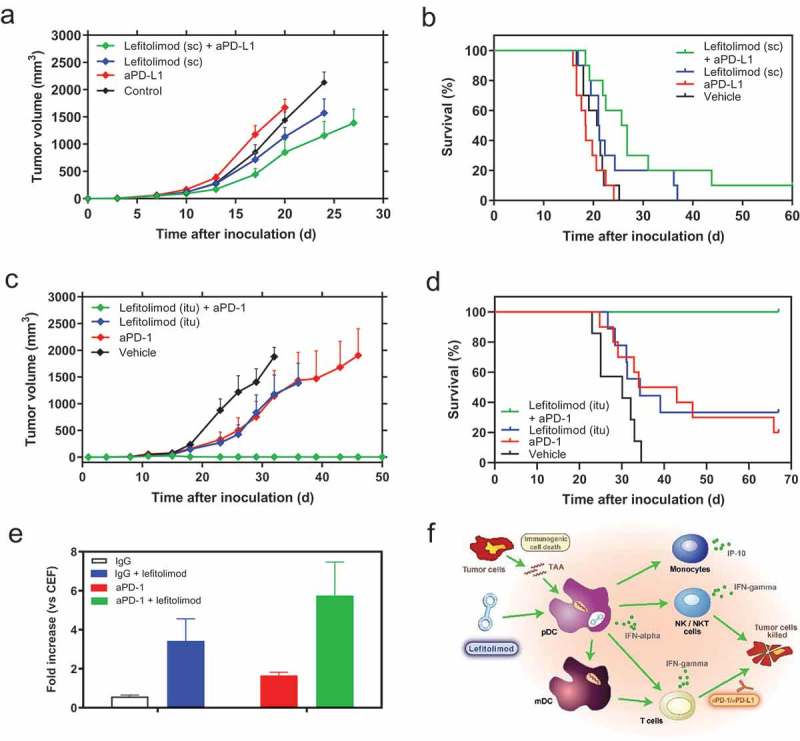
Combination of lefitolimod with immune checkpoint inhibitors (CPI). (a&b), anti-tumor activity of the combination of sc lefitolimod (11x 250 µg, starting at day 3) with aPD-L1 in the CT26 model: mean tumor growth (+SEM) p = .001 for lefitolimod+anti-PD-L1 at day 17, p = .02 for lefitolimod, and p ≤ 0.0001 for lefitolimod+anti-PD-L1 at day 20, all vs vehicle (Dunnett`s multiple comparison test), analyzed until day 20 (a) and survival, p = .0024 (log-rank) (b). (c&d), anti-tumor activity of the combination of itu lefitolimod (11x 250 µg, starting at day 13 at a mean tumor volume of at least 40 mm3) with aPD-1 in the A20 model: mean tumor growth (+SEM), significant for all comparisons vs vehicle from day 23 to 32 ranging from: p = .04 to ≤ 0.0001 (Dunnett`s multiple comparison test), analyzed until day 32 (c) and survival, p = .05 for lefitolimod, p = .03 for anti-PD-1, p ≤ 0.0001 for lefitolimod and anti-PD-1 (log-rank), all vs vehicle (d). (e), activation of human PBMC with peptides selected from HLA class I-restricted T-cell epitopes of recall-antigens, lefitolimod and anti-PD-1 (mean+SEM, n = 4). PBMC were activated with a peptide pool consisting of peptides each corresponding to HLA class I-restricted T-cell epitope from cytomegalo virus, Epstein-Barr virus and Influenza virus (CEF-peptides) in the presence of lefitolimod and anti-PD-1 for 48 h. IFN-gamma was measured in the cell culture supernatants. IFN-gamma values were normalized on the values obtained after stimulation with CEF-peptides. (f), rationale for combination of lefitolimod with CPI based on its mode-of-action: Lefitolimod activates TLR9-positive pDC. IFN-alpha, secreted by pDC, initiates broad activation of the innate and adaptive immune system: NK cells and NKT cells are activated for cytotoxicity against tumor cells. Monocytes secrete IP-10 which is chemotactic for NK cells and CD8 + T cells and has angiostatic properties. Activated pDC/mDC take up and present tumor-associated antigens (TAA) to T cells. T cells proliferate and differentiate into effector and/or memory cells. This provides the basis for a combination with CPI, who take advantage of the immune activation provided by lefitolimod.
Discussion
Lefitolimod is a TLR9 agonist and triggers the secretion of IFN-alpha by TLR9-expressing pDC.8–10 IFN-alpha stimulates several key regulatory immune cells and thereby initiates innate and also adaptive immune responses,16 the latter specially by activating CD8-alpha+ dendritic cells able to cross-present antigens to cytotoxic T cells.25,26 Activated cytotoxic T cells express the CXC-chemokine receptor 3 (CXCR3), and can migrate into tumors in response to the TH1-type chemokines CXC-chemokine ligand 9 (CXCL9) and IP-10 (CXCL10).27–29 Lefitolimod induces the secretion of the chemokine IP-10 from human PBMC. Secretion of IP-10 depends on the presence of pDC and IFN-alpha.8,18
The presence of a T cell-inflamed TME in so-called “hot tumors” is linked with improved responses to cancer immunotherapies including checkpoint inhibitors.30,31 However, MSS cancers are characterized by a lower infiltration of T cells and are known to be poorly responsive to CPI32,33 rendering it as an ideal model for immunotherapeutic approaches to modulate the TME and enhance the effect of CPI.
Indeed, lefitolimod has shown to activate cytotoxic T cells and initiating their recruitment into the TME in the MSS colon carcinoma CT26 model. These data indicate that the mode-of action of lefitolimod via TLR9 starts upstream of the targets of checkpoint inhibitors like anti-PD-1/anti-PD-L1.
The combination of lefitolimod with the CPI anti-PD-1/anti-PD-L1 resulted in synergistic anti-tumor effects in mouse tumor models. Similar data have recently been obtained by PTO-modified TLR9 agonists in mice34,35 and also in a clinical trial in advanced melanoma.36 However, it should be considered that PTO-modified TLR9 agonists induce a different cytokine pattern in vitro.8,18
The described immunomodulatory effects of lefitolimod, the potent synergistic anti-tumor responses observed in combination with CPI in murine models, during which no signs of toxicological effects have been observed, and the absence of toxic effects in more than 450 patients in clinical trials10-13,37 renders lefitolimod as an ideal combination partner for immunotherapeutic approaches in humans. In fact, a clinical trial in melanoma patients with a combination of lefitolimod and ipilimumab is ongoing and has shown encouraging first data on increase of tumor-infiltrating lymphocytes and a favorable safety profile, and no dose-limiting toxicities have been encountered at any dose level of lefitolimod together with ipilimumab.38
Lefitolimod induced an increase of immune cells within the tumors, shown for T cells as well as for macrophages. Moreover, the ratio of anti-tumoral M1 vs pro-tumoral M2 macrophages39,40 was increased after treatment with lefitolimod. The complexity of the population of tumor-associated macrophages is high and they play a dual role in tumor growth. They have anti-tumor features in the early stages of tumors, whereas with tumor progression, tumor-associated macrophages (TAM) adopt a tumor-promoting M2-like phenotype characterized by activation of Th2 signaling in the TME.41,42 There is also growing evidence that specific populations of monocytes/macrophages are correlated with improved responses to CPI43,44 in humans. Therefore, a promising therapeutic strategy would be the specific targeting of pro-tumoral M2 macrophages or repolarizing M2-like TAM to the tumor-suppressive phenotype.41,42
In our murine models, the strongest anti-tumor effect was observed in the EMT-6 breast cancer model after treatment with lefitolimod showing a complete tumor disappearance in 9/10 mice. The complete regression of established tumors in the immune-excluded EMT-6 model is especially remarkable. In the hands of others, therapeutic blockade of PD-L1 in the EMT-6 model resulted in an anti-tumor effect only in combination with anti-TGF-beta.24 Moreover, all surviving mice rejected not only the initially used EMT-6 tumor cells in a re-challenge study, but also the distinct CT26 colorectal cancer cells. The detection of IFN-gamma secreting cells with reactivity not only against EMT-6 and CT26, but also Renca and 4T1 cells in the surviving mice indicate a sustained and broad immune memory after potent initial anti-tumor responses, potentially against shared antigens between different tumor types.
Reduction of tumor growth was shown in the colon carcinoma CT26 model after itu injection of lefitolimod into established tumors. In the clinical setting, tumor burden at the start of treatment have an influence on the benefit from the treatment, with early treatment and smaller tumors being advantageous.45,46 In keeping with this, we observed the expected augmented anti-tumor effect in a tumor model when inoculating a lower dose of CT26 cells for tumor growth (Figure S3(a–c)). Furthermore, an earlier treatment – starting as early as 8 days before tumor inoculation – led to clearly better tumor inhibition (Figure S3).
It is also known that itu injection of immunotherapeutic compounds induces potent anti-tumoral immune responses.47 This is line with our data (Figure 1, Figure 4(e–h)). The underlying MoA may be a local priming of immune cells like APC present in the tumor which in turn take up, process and present available TAA.47,48 However, the itu approach has several clinical limitations: (i) itu administration may be associated with adverse events such as local inflammation, pain, and bleeding,49,50 (ii) not all tumor lesions are easily assessable and for the majority of patients with solid cancer, and (iii) additional clinical/surgical/radiological or other imaging methods are required to achieve accurate injection of the target lesion.50 Interestingly, we observed anti-tumor effects also after peritumoral and even distant sc injection of lefitolimod (Figure 5(a) and S4). Supposedly, systemically applied lefitolimod stimulates pDC patrolling within the skin or draining lymph nodes and – after direct activation – leading to their migration to the secondary lymphoid organs triggering immune activation and cell differentiation followed by their migration into the tumor to initiate anti-tumor immune responses. This was recently shown in HIV patients who developed augmented type-I IFN responses in gut biopsies after sc lefitolimod administration.51 This data supports a clinical strategy of sc lefitolimod administration.
To support the anti-tumor effects obtained from syngeneic models, we employed a skin painting model which mimics the de novo development of tumors mimicking more closely the cancer progression in patients52 and investigated the tumor prophylactic activity of sc or ip injection of lefitolimod (Figure S5(a)). A reduction in tumor incidence and multiplicity as well as increase of tumor-free survival was observed for animals treated with lefitolimod for both injection routes (Figure S5(b–d)) confirming the previously described anti-tumor effect of lefitolimod in a de novo tumor model.
In summary, we have shown that single-agent lefitolimod has the potential to beneficially modulate the TME, increase tumor-specific T cell responses and anti-tumor memory, resulting in pronounced anti-tumor effects. These properties of a potent immune surveillance reactivator improve the effect of CPI and are the basis for combination approaches of lefitolimod with anti-PD-1 and anti-PD-L1.
Material and methods
TLR9 agonist
Lefitolimod was synthesized as described previously17 by Mologen. In short, two identical 5ʹ phosphorylated, hairpin-shaped 58-mer ODN were ligated and the resulting dumbbell-shaped covalently closed molecules were purified by HPLC. Lefitolimod consists of a double-stranded stem and two single-stranded loops of 30 nucleotides each with three non-methylated CG motifs in each loop. Lefitolimod was dissolved in phosphate-buffered saline (15 mg/ml). Endotoxins were below 10 EU/mg.
Mouse tumor models
Selection of tumor models
The MoA of lefitolimod includes a strong activation of cells of the innate and adaptive immune system, necessary for an anti-tumor response, which should be applicable for different tumor types. To confirm this hypothesis, lefitolimod was tested in diverse tumor models covering a range of different tumor indications with diverse immunological background (including low-immunogeneic tumors). A summary table of the tumor models used is provided in the Supplement (Table S1).
Syngeneic tumor models
Female C57BL/6 and Balb/c mice (age 6–8 weeks) were housed and treated in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Tumors were engrafted by subcutaneous (sc) injection of 100–200 µl tumor cell suspension in the flank. Tumor length and width were determined using calipers and volume was calculated as (width2 × length)/2). Animals were sacrificed when tumor size exceeded a pre-determined endpoint.
CT26 model for TME evaluation
Sc tumor inoculation was done with 5 × 104 CT26 cells (+50% matrigel). Begin of treatment with 200 or 250 µg lefitolimod (in PBS) or vehicle was started at day 10, when the mean tumor volume was about 140 mm3. In total, four intratumoral (itu) applications were performed, at day 10, 13, 15, and 17. At day 20 the tumors were sampled and subjected to immunohistochemistry (IHC) or tumor-infiltrating lymphocytes (TIL) were isolated and analyzed by FACS.
B16 model:
Tumor inoculation was done with 5 × 105 B16 cells. Begin of treatment with 200 µg lefitolimod (in PBS) or vehicle was started at day 3. In total, nine sc applications were performed every other day. Tumor growth and survival were assessed.
EMT-6 model
Sc tumor inoculation was done with 5 × 105 EMT-6 cells. Mice were randomized prior to treatment when tumors were well established (about 40 mm3). 250 µg lefitolimod (in PBS) or vehicle was given itu three times per week for 3 weeks. For re-challenge experiments surviving mice were re-inoculated with 5 × 105 EMT-6 cells at day 54 and 5 × 105 CT26 cells at day 115 – distant from the primary tumor inoculation site, but without further treatment. Naïve mice were inoculated with tumor cells without any treatment as re-challenge controls.
CT26 model, combination with aPD-L1
Sc tumor inoculation was done with 1 × 106 CT26 cells. Mice were randomized prior to tumor inoculation according to their body weight. 250 µg lefitolimod (in PBS) or vehicle was given peritumorally three times per week (11 applications in total), starting at day 3. 10 mg/kg aPD-L1 (clone 10F.9G2 manufacturer: Bio X Cell) was given intraperitoneally (ip), two times per week for 4 weeks, starting at day 3. Tumor growth and survival (time to reach tumor volume of 1500 mm3) were assessed.
A20 model, combination with aPD-1
Sc tumor inoculation was done with 1 × 106 A20 cells. Mice were randomized prior to tumor inoculation according to their bodyweight. 250 µg lefitolimod (in PBS) or vehicle was given itu three times per week for 3 weeks with 11 applications in total, starting at day 14. 100 µg (per mouse) aPD-1 (clone RMP1-14, company Bio X Cell) was given ip, two times per week for 2 weeks, starting at day 8. Tumor growth and survival (time to reach tumor volume of 1800 mm3) were assessed.
De novo tumor model
Skin painting studies are frequently used as an experimental carcinogenesis model based on OCDE Guideline 451 “Carcinogenicity studies” and NTP Guideline “Specifications for the conduct of studies to evaluate the toxic and carcinogenic potential of chemical, biological and physical agents in laboratory animals for the National Toxicology Program”. The two-step skin carcinogenesis model is defined by an irreversible tumor initiation process that often involves the activation of the c-H-ras oncogene, and tumor promotion caused by topical application of a promoting test substance. One hundred and fifty outbreed female (hairless) SKH1 mice were grouped according to NTP-guidelines into three groups (50 mice each). A single dose of DMBA (7,12-Dimethylbenzanthracene) dissolved in acetone was applied to the back of each mouse (40 µg/mouse/application). As a promoter substance, TPA (12-O-tetradecanoylphorbol-13-acetate) was applied three times per week starting day 7. Lefitolimod (20 µg once-weekly) application was started in parallel and administered sc or ip in the respective group. Clinical observations, body weight, and skin tumors were recorded once a week. All surviving mice were sacrificed after 18 weeks. All mice sacrificed at that time and those that died during the application period were examined for skin tumors.
Evaluation of tumor microenvironment (TME)
The syngeneic colon carcinoma model CT26 was used to evaluate lefitolimod’s modulation of the TME. The localization of immune cells within the tumor was investigated by immunohistochemistry. The phenotype of T cells and macrophages present within tumor-infiltrating immune cells were analyzed by flow cytometry.
Immunohistochemical (IHC) analysis of tumors
Tumors were embedded in Tissue-TEK OCT (Sakura Finetek Inc.) and stored at −20°C or formalin-fixed paraffin-embedded (FFPE) sections were prepared. For the detection of CD8 + T cells 5 µm cryosections were prepared, fixed in acetone and stained using the Ventana Discovery XT system (Roche). Sections were incubated with Rat anti-mouse CD8alpha (clone 53–6.7, eBioscience) at a dilution 1:500 (v/v) for 60 min. For the detection of CD3 + T cells in FFPE sections after pre-treatment of slides for dewaxing and heat antigen retrieval with the Ventana Cell Conditioning Solution 1 (Roche) for 40 min, a rat anti-mouse CD3 antibody (clone CD3-12, Abcam,) was added for 60 min at a dilution 1/200. For the detection of F4/80+ macrophages in FFPE sections after a pre-treatment of slides for dewaxing and heat antigen retrieval with the Ventana Protease 3 (Roche) for 8 min, a rat anti-mouse F4/80 antibody (clone BM8, eBiosciences) was added for 60 min at a dilution 1/50. Sections for CD3- and CD8- and F4/80-staining were incubated with Rabbit anti-Rat IgG Fc (clone R18-2, abcam) at a dilution 1/500 for 32 min. Thereafter, sections were incubated with omniMap anti-RabbitHRP (Roche) for 16 min. The chromoMap DAB kit (Roche) was used as substrate for visualization. Counterstaining was performed with hematoxylin. Slides were scanned (NanoZoomer, Hamamatsu) and for CD8-staining tumor center and margin were manually scored by two experienced operators in a blinded fashion (1: no labeling, 2: few labeling, 3: intermediate labeling, 4: intense labeling). Quantification of the CD3+ cells within the tumor was performed using the Tissue studioTM software from Definiens®.
Flow cytometry of tumor cells
Single-cell suspensions of tumors from mice inoculated with CT26 tumor cells and treated itu with 250 μg lefitolimod (in PBS) or vehicle were prepared at day 20 after inoculation. Cells were surface-stained with monoclonal antibodies in PBS containing 3% serum on ice. The following antibodies were used: anti-CD4 (clone REA604), anti-CD8a (clone 53–6.7), anti-CD45 (clone 30-F11), anti-Thy1 (clone 5E10), anti-FOXP3 (clone FJK-16s), anti-Granzyme B (clone GB11), anti-CD69 (clone H1.2F3), anti-F4/80 (clone T45-234), anti-Ly6C (clone AL-21), anti-CD11b (clone M1/70), anti-Gr1 (clone M1/70), anti-MHC II (clone M5/114.15.2) and anti-CD86 (clone GL1). All flow cytometry parameters of cells were acquired on a FACS Canto II (BD Biosciences). Percentages of cell subsets were related to the indicated parent populations; geometric means of fluorescence for markers were indicated as mean fluorescence intensity (MFI). Data were analyzed with the BD FACSDiva software.
Elispot assay
Splenocytes from mice sc inoculated with CT26 tumor cells and itu treated with 250 μg lefitolimod or vehicle were prepared at day 20 after inoculation. 9 × 105 splenocytes were ex vivo stimulated with 1 × 105 CT26 cells or 1 μg/ml MHC I-restricted AH1 peptide (Eurogentec) for 20 h at 37°C. As a positive control for polyclonal T-cell responsiveness, splenocytes were stimulated with 0.5 µg/ml of anti-mouse CD3 antibody (clone 145-2C11, BD Biosciences). The frequency of IFN-gamma secreting cells was determined in triplicates using an IFN-gamma ELISPOT assay (Mabtech), according to the manufacturer’s protocol. Spot-forming cells were counted using an automated ELISpot reader system (Biosys).
Spleen cells of nine mice surviving double EMT-6 tumor inoculation as well as CT26 tumor inoculation and spleen cells from three naïve mice were prepared. For ELISpot assay (Mabtech, No. 3321-4HPW-2) 8 × 105 spleen cells were co-cultured with 8 × 104 mitomycin C-treated (100 µg/ml) tumor cells (EMT-6, CT26, Renca, A20, 4T-1, WEHI164) for 24 h in triplicates. Detection of IFN-gamma secreting cells was done according to the instructions of the manufacturer. For positive controls spleen cells were incubated with 500 ng/ml PMA plus 1 µg/ml Ionomycin; for negative controls, spleen cells were cultured without any additives. Number of spots was analyzed in an ELISpot reader (AID iSpot). For analysis number of spots in the “splenocytes only” approach was subtracted from the respective approaches with tumor cells.
In vitro PBMC stimulation assay
Buffy coats from anonymized healthy donors were obtained from the “DRK-Blutspendedienst – Ost”. Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation using Ficoll (Biochrom). Cells were cultured in complete medium (RPMI1640 [Lonza] with 2 mM UltraGlutamine [Lonza] supplemented with 10% [v/v] fetal calf serum [Linaris], 100 U/ml Penicillin and 100 µg/ml Streptomycin [Lonza]) in flat-bottom plates (PBMC, 6 million cells/ml). Cells were stimulated with 1 µg/ml extended CEF (cytomegalo virus, Epstein-Barr virus, influenza virus) peptide pool (JPT Peptide Technologies GmbH), 3 µM lefitolimod and 10 µg/ml anti-PD-1 (Miltenyi Biotec) for 2 days. IFN-gamma in the culture supernatant was determined by ELISA (OptEIA Human IFN gamma ELISA Set, BD Biosciences) and was performed in duplicates according to the manufacturer’s instructions. Optical density was measured at 450 nm; the data were analyzed with the MicroWin software (Berthold Technologies).
Statistical analyses
Data were analyzed with GraphPad Prism 7 (GraphPad Software Inc.). P values <.05 were considered significant. The statistical analyses are specified in the figure legends.
Acknowledgments
We are grateful to Jaqueline Schneider and Lisa Schneider for their expert technical assistance.
Disclosure of interest
KK, BV, DO, MB and MS are employees at Mologen AG.
BW holds shares of Mologen AG and receives compensation for consulting services.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Connolly DJ, O’Neill LA.. New developments in Toll-like receptor targeted therapeutics. Curr Opin Pharmacol. 2012;12:1–11. doi: 10.1016/j.coph.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial watch: experimental toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012;1:699–716. doi: 10.4161/onci.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanzler H, Barrat FJ, Hessel EM, Coffman RL.. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 4.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 6.Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4:249–258. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 7.Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–1194. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapp K, Kleuss C, Schroff M, Wittig B. Genuine immunomodulation with dSLIM. Mol Ther Nucleic Acids. 2014;3:e170. doi: 10.1038/mtna.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt M, Hagner N, Marco A, Konig-Merediz SA, Schroff M, Wittig B.. Design and structural requirements of the potent and safe TLR-9 agonistic immunomodulator MGN1703. Nucleic Acid Ther. 2015;25:130–140. doi: 10.1089/nat.2015.0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wittig B, Schmidt M, Scheithauer W, Schmoll HJ. MGN1703, an immunomodulator and toll-like receptor 9 (TLR-9) agonist: from bench to bedside. Crit Rev Oncol Hematol. 2015;94:31–44. doi: 10.1016/j.critrevonc.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham D, Van Cutsem E, Salazar R, Ducreux M, Scheithauer W, Sclafani F, Wiegert E, Schmidt M, Tournigand C, Sobrero A, et al. Patient characteristics after completion of recruitment from the phase 3 IMPALA study with lefitolimod in metastatic colorectal carcinoma. Ann Oncol TiP 2017;28:93. doi: 10.1093/annonc/mdx711.074. [DOI] [Google Scholar]
- 12.Schmoll HJ, Wittig B, Arnold D, Riera-Knorrenschild J, Nitsche D, Kroening H, Mayer F, Andel J, Ziebermayr R, Scheithauer W. Maintenance treatment with the immunomodulator MGN1703, a Toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: a randomised, double-blind, placebo-controlled trial. J Cancer Res Clin Oncol. 2014;140:1615–1624. doi: 10.1007/s00432-014-1682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas M, Ponce-Aix S, Navarro A, Riera-Knorrenschild J, Schmidt M, Wiegert E, Kapp K, Wittig B, Mauri C, Domine Gomez M, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase 2 IMPULSE study. Ann Oncol. 2018. doi: 10.1093/annonc/mdy326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guiducci C, Coffman RL, Barrat FJ. Signalling pathways leading to IFN-alpha production in human plasmacytoid dendritic cell and the possible use of agonists or antagonists of TLR7 and TLR9 in clinical indications. J Intern Med. 2009;265:43–57. doi: 10.1111/j.1365-2796.2008.02050.x. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt M, Anton K, Nordhaus C, Junghans C, Wittig B, Worm M.. Cytokine and Ig-production by CG-containing sequences with phosphorodiester backbone and dumbbell-shape. Allergy. 2006;61:56–63. doi: 10.1111/j.1398-9995.2005.00908.x. [DOI] [PubMed] [Google Scholar]
- 18.Kapp K, Schneider J, Schneider L, Gollinge N, Jansch S, Schroff M, Wittig B, Kleuss C. Distinct immunological activation profiles of dSLIM® and ProMune® depend on their different structural context. Immun Inflamm Dis. 2016;4:446–462. doi: 10.1002/iid3.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 20.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, Boisguerin V, Bukur T, Sorn P, Paret C, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. doi: 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, Hermann-Kleiter N, Lower M, Baier G, Krogsdam A, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. 2018;9:32. doi: 10.1038/s41467-017-02424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu JW, Bhattacharya S, Yanamandra N, Kilian D, Shi H, Yadavilli S, Katlinskaya Y, Kaczynski H, Conner M, Benson W, et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS One. 2018;13:e0206223. doi: 10.1371/journal.pone.0206223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer. 2017;3:797–808. doi: 10.1016/j.trecan.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol. 2017;1036:19–31. doi: 10.1007/978-3-319-67577-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28:383–391. doi: 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164:1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, Varner JA, Pu M, Messer KS, Guiducci C, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2. doi: 10.1172/jci.insight.93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Campos J, Gallotta M, Gong M, Crain C, Naik E, Coffman RL, Guiducci C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A. 2016;113:E7240–E9. doi: 10.1073/pnas.1608555113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8:1250–1257. doi: 10.1158/2159-8290.CD-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weihrauch MR, Richly H, von Bergwelt-Baildon MS, Becker HJ, Schmidt M, Hacker UT, Shimabukuro-Vornhagen A, Holtick U, Nokay B, Schroff M, et al. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur J Cancer. 2015;51:146–156. doi: 10.1016/j.ejca.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Reilley M, Tsimberidou A, Piha-Paul S, Yap T, Fu S, Naing A, Rodon J, Nguyen L, Ager C, Meng M, et al. Phase 1 trial of TLR9 agonist lefitolimod in combination with CTLA-4 checkpoint inhibitor ipilimumab in advanced tumors. J Clin Oncol. 2019;37 abstractTPS2669. doi: 10.1200/JCO.2019.37.15_suppl.TPS2669. [DOI] [Google Scholar]
- 39.Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435–445. doi: 10.1084/jem.20150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. 2013;35:585–600. doi: 10.1007/s00281-013-0367-7. [DOI] [PubMed] [Google Scholar]
- 41.Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904. doi: 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- 42.Zheng X, Turkowski K, Mora J, Brune B, Seeger W, Weigert A, Savai R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017;8:48436–48452. doi: 10.18632/oncotarget.17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, Becher B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018. doi: 10.1038/nm.4466. [DOI] [PubMed] [Google Scholar]
- 44.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112:6140–6145. doi: 10.1073/pnas.1417320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuczynski EA, Krueger J, Chow A, Xu P, Man S, Sundaravadanam Y, Miller JK, Krzyzanowski PM, Kerbel RS. Impact of chemical-induced mutational load increase on immune checkpoint therapy in poorly responsive murine tumors. Mol Cancer Ther. 2018;17:869–882. doi: 10.1158/1535-7163.MCT-17-1091. [DOI] [PubMed] [Google Scholar]
- 46.Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Totterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J ImmunoTher. 2010;33:225–235. doi: 10.1097/CJI.0b013e3181c01fcb. [DOI] [PubMed] [Google Scholar]
- 47.Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res. 2014;20:1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28:xii33–xii43. doi: 10.1093/annonc/mdx683. [DOI] [PubMed] [Google Scholar]
- 49.Ellmark P, Mangsbo SM, Furebring C, Norlen P, Totterman TH. Tumor-directed immunotherapy can generate tumor-specific T cell responses through localized co-stimulation. Cancer Immunol Immunother. 2017;66:1–7. doi: 10.1007/s00262-016-1909-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marabelle A, Andtbacka R, Harrington K, Melero I, Leidner R, de Baere T, Robert C, Ascierto PA, Baurain JF, Imperiale M, et al. Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann Oncol. 2018;29:2163–2174. doi: 10.1093/annonc/mdy423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krarup AR, Abdel-Mohsen M, Schleimann MH, Vibholm L, Engen PA, Dige A, Wittig B, Schmidt M, Green SJ, Naqib A, et al. The TLR9 agonist MGN1703 triggers a potent type I interferon response in the sigmoid colon. Mucosal Immunol. 2017. doi: 10.1038/mi.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16:759–773. doi: 10.1038/nrc.2016.91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.