

ABSTRACT
The ubiquitination of mitochondrial proteins labels damaged mitochondria for degradation through mitophagy. We recently developed an in vivo system in which mitophagy is slowed by inhibiting mitochondrial division through knockout of Dnm1l/Drp1, a dynamin related GTPase that mediates mitochondrial division. Using this system, we revealed that the ubiquitination of mitochondrial proteins required SQSTM1/p62, but not the ubiquitin E3 ligase PRKN/parkin, during mitophagy. Here, we tested the role of PINK1, a mitochondrial protein kinase that activates mitophagy by phosphorylating ubiquitin, in mitochondrial ubiquitination by knocking out Pink1 in dnm1l-knockout liver. We found mitochondrial ubiquitination did not decrease in the absence of PINK1; instead, PINK1 was required for the degradation of MFN1 (mitofusin 1) and MFN2, two homologous outer membrane proteins that mediate mitochondrial fusion in dnm1l-knockout hepatocytes. These data suggest that mitochondrial ubiquitination is promoted by SQSTM1 independently of PINK1 and PRKN during mitophagy. PINK1 and PRKN appear to control the balance between mitochondrial division and fusion in vivo.
Abbreviations: DNM1L/DRP1: dynamin 1-like; KEAP1: kelch-like ECH-associated protein 1; KO: knockout; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MFN1/2: mitofusin 1/2; OPA1: OPA1, mitochondrial dynamin like GTPase; PDH: pyruvate dehydrogenase E1; PINK1: PTEN induced putative kinase 1; PRKN/parkin: parkin RBR E3 ubiquitin protein ligase.
KEYWORDS: Dnm1l/Drp1, mitochondria, mitochondrial division, mitophagy, PINK1, PRKN/parkin
Introduction
Mitochondria are essential organelles that function in a variety of bioenergetic, metabolic, and developmental processes; therefore, cells employ multiple quality control mechanisms to maintain the functional integrity of mitochondria. For example, mitochondria contain many proteases that degrade misfolded proteins inside mitochondria, such as ATP-dependent AAA proteases [1,2]. Misfolded proteins inside mitochondria can induce the mitochondrial unfolded protein response to import more chaperone proteins that increase the folding capacity or induce the degradation of these misfolded proteins [3,4]. Mitochondrial unfolded protein responses also remodel cellular metabolism through changes in gene expression. There are several ubiquitination enzymes that target surface-exposed outer membrane proteins for proteasomal degradation [5]. In addition, a portion of the mitochondrion that is damaged can be pinched off by generating small vesicles that bud out from the mitochondrial membrane, called mitochondrial-derived vesicles, which are transported to lysosomes [6,7].
Damaged mitochondria are also eliminated as whole structures through selective autophagic degradation, which is termed mitophagy [8,9]. To mark mitochondria for mitophagy, mitochondrial proteins undergo ubiquitination and autophagosomes are recruited to the ubiquitinated mitochondria through receptor proteins that interact with both polyubiquitin chains and autophagy-related proteins, such as MAP1LC3/LC3. It has been reported, in response to the loss of the mitochondrial membrane potential, that the protein kinase PINK1, which is usually imported into healthy mitochondria, becomes trapped at the outer membrane and phosphorylates ubiquitin and the ubiquitin E3 ligase PRKN on the surface of mitochondria [8,9]. These phosphorylation events activate PRKN and further induce ubiquitination of mitochondrial proteins. However, our current understanding of the role of PRKN and PINK1 in mitophagy in vivo is limited since most of the studies have been done in cell culture systems.
We have developed in vivo systems in which mitophagy can be blocked by increasing the size of mitochondria by knocking out Dnm1l/Drp1, a dynamin-related GTPase that mediates mitochondrial division [10–12]. Using the floxed alleles of Dnm1l and tissue-specific CRE recombinase, we deleted Dnm1l individually in multiple organs, such as the brain, heart, and liver [13–17]. In all organs we tested, the loss of DNM1L enlarged mitochondria and decreased mitophagy. Consequently, halted mitophagy intermediates accumulated, such as mitochondria that piled up ubiquitinated proteins and LC3 on their surfaces. By analyzing these mitophagy intermediates, we found that the ubiquitination of enlarged mitochondria is independent of PRKN in neurons in the brain, cardiomyocytes in the heart, and hepatocytes in the liver [13–17]. Additional knockout (KO) of Prkn in dnm1l KO hearts, brains, or livers did not decrease mitochondrial ubiquitination. Supporting these findings, recent studies have shown that mitophagy occurs in vivo in the absence of PRKN or PINK1 [18–21]. The molecular mechanisms that ubiquitinate mitochondrial proteins and drive mitophagy independently of PRKN and PINK1 are largely unknown.
The mitophagy intermediates that were captured in the absence of DNM1L accumulated SQSTM1 on their surfaces [13,14,16]. SQSTM1 is thought to function as an autophagy receptor that connects autophagy substrates and autophagosomes by binding both ubiquitin and LC3 [22,23]. Surprisingly, in contrast to this current view, we have recently reported that the additional KO of Sqstm1 dramatically decreased mitochondrial ubiquitination in dnm1l KO hepatocytes [13]. At mitochondria, SQSTM1 associates with KEAP1 (kelch-like ECH-associated protein 1), a protein that forms a cullin-RING ubiquitin ligase with the E3 ligase RBX1. Both KEAP1 and RBX1 were recruited to the mitophagy intermediates by SQSTM1 in dnm1l KO hepatocytes [13]. Knockdown of Rbx1 also decreased mitochondrial ubiquitination. These data suggest a new mechanism by which the SQSTM1-KEAP1-RBX1 complex ubiquitinates mitochondrial proteins in mitophagy. Although association of SQSTM1 blocks the function of KEAP1 in the suppression of the transcription factor NFE2L2/NRF2 in autophagy-defective atg7 KO livers [24], we found that sqstm1 KO livers maintain the low level of Nfe2l2 target genes [13]. Therefore, the role of KEAP1 in SQSTM1-driven mitophagy is independent of the NFE2L2 pathway. We also showed that general macroautophagy is not activated in dnm1l KO livers [13].
SQSTM1-dependent mitochondrial ubiquitination takes place independently of PRKN [13]; however, it is possible that SQSTM1 and PRKN have overlapping functions in mitochondrial ubiquitination, i.e. in the presence of SQSTM1, the role of PRKN may be masked. It also remains to be determined whether this SQSTM1-independent ubiquitination involves PINK1. PINK1 has been reported to promote mitochondrial ubiquitination independently of PRKN, likely through other ubiquitin E3 ligases [25]. In the current study, to address these critical mechanistic questions, we simultaneously knocked out Sqstm1 and Prkn in dnm1l KO livers and tested a potential functional overlap between SQSTM1 and PRKN. We also generated double KO livers for dnm1l and pink1 to examine the role of PINK1 in mitochondrial ubiquitination.
Results
PRKN is dispensable for mitochondrial ubiquitination in the absence of SQSTM1
We have previously shown that mitochondrial ubiquitination requires SQSTM1, but not PRKN, in mitophagy in the liver, heart, and brain [13,16,17]. In the absence of SQSTM1, mitochondrial ubiquitination is decreased by 80% in dnm1l::sqstm1 KO livers [13]. To test whether the residual level of ubiquitination is mediated by PRKN, we combined the KO of Sqstm1 and Prkn in Alb-dnm1l KO mice (Alb-dnm1l::prkn::sqstm1 KO mice). At 3 months of age, the mice were chemically fixed by cardiac perfusion with paraformaldehyde and frozen liver sections were analyzed by immunofluorescence microscopy with antibodies against ubiquitin, SQSTM1, and a mitochondrial protein, pyruvate dehydrogenase (PDH). Consistent with our previous findings [13], mitochondria were highly ubiquitinated in Alb-dnm1l KO livers (Figure 1A and B). Biochemical fractionation of mitochondria confirmed the association of SQSTM1 with mitochondria in Alb-dnm1l KO livers (Figure 1C-F). The additional loss of SQSTM1 decreased mitochondrial ubiquitination by 80% in Alb-dnm1l::sqstm1 KO livers, whereas the loss of PRKN did not decrease ubiquitination in Alb-dnm1l::prkn KO livers (Figure 1A and B). Compared to Alb-dnm1l::sqstm1 KO livers, Alb-dnm1l::prkn::sqstm1 KO livers did not show decreased mitochondrial ubiquitination (Figure 1A and B). Therefore, it appears that PRKN does not account for the remaining mitochondrial ubiquitination activity in Alb-dnm1l::sqstm1 KO livers. These data exclude the possibility that SQSTM1 and PRKN have partially overlapping functions in mitochondrial ubiquitination.
Figure 1.
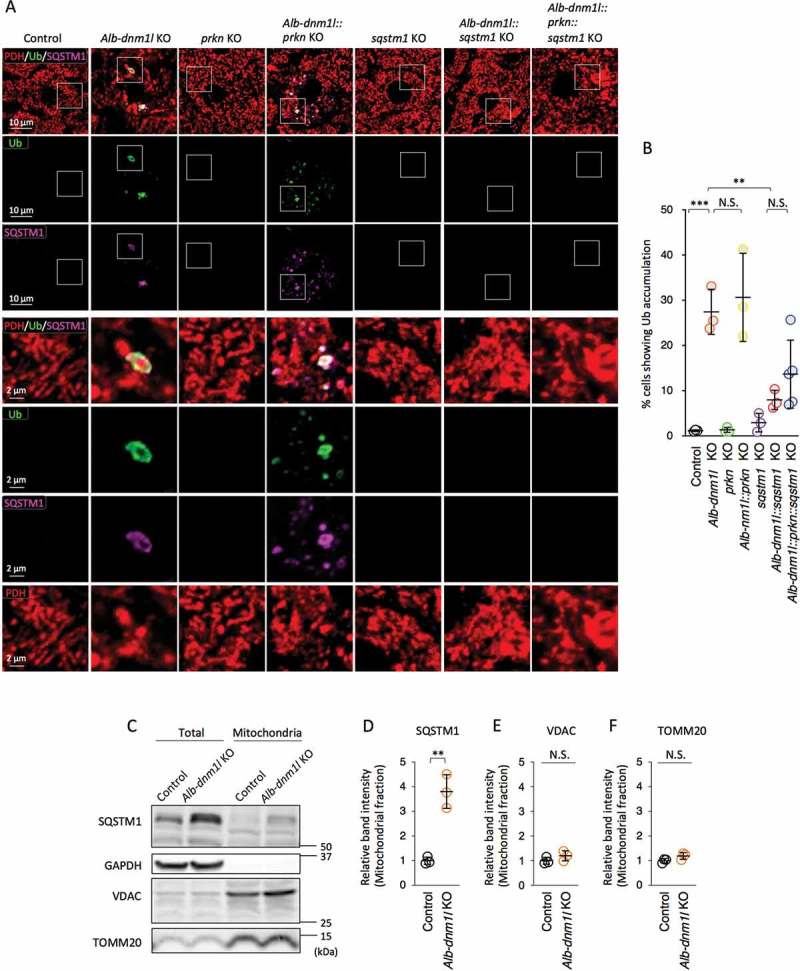
Mitochondrial ubiquitination requires SQSTM1, but not PRKN. (A) Frozen liver sections in the indicated mice were subjected to immunofluorescence microscopy with ubiquitin, SQSTM1, and a mitochondrial protein, pyruvate dehydrogenase (PDH). Lower panels are magnified images of boxed regions. Bars in higher panels: 10 µm. Bars in lower panels: 2 µm. (B) Quantification of cells showing the accumulation of ubiquitin on mitochondria. Values are average ± SD (n = 3–5 mice). (C) Mitochondria were isolated from control and Alb-dnm1l KO livers and analyzed by western blotting with antibodies to SQSTM1, GAPDH, VDAC and TOMM20. 20 µg of protein was loaded in each lane. (D-F) Band intensity was quantified, and values are presented as average ± SD (n = 3 mice). Statistical analysis was performed using one-way ANOVA followed by the post-hoc Tukey test in (B) and Student’s t-test in (D-F): *p < 0.05, **p < 0.01, ***p < 0.001.
The loss of PINK1 does not decrease mitochondrial ubiquitination
PINK1 stimulates ubiquitination of mitochondrial proteins by phosphorylating and activating the ubiquitin E3 ligase PRKN [26,27], but also promotes mitochondrial ubiquitination independently of PRKN [25]. To ask if mitochondrial ubiquitination in Alb-dnm1l KO livers depends on PINK1, we generated Alb-dnm1l::pink1 KO mice by crossing Alb-dnm1l KO mice with pink1 KO mice [28]. At 3 months of age, frozen liver sections were analyzed by immunofluorescence microscopy with antibodies against ubiquitin, SQSTM1, LC3, and PDH. We found no significant decrease in mitochondrial ubiquitination in Alb-dnm1l::pink1 KO livers, compared to Alb-dnm1l KO livers (Figure 2A and C). Similarly, the accumulation of SQSTM1 on mitochondria was not decreased in Alb-dnm1l::pink1 KO livers (Figure 2A, B and D). SQSTM1 was colocalized with both ubiquitin and LC3 on mitochondria in Alb-dnm1l and Alb-dnm1l::pink1 KO mice. These data suggest that recruitment of SQSTM1 to mitochondria and the ubiquitination of mitochondrial proteins were independent of PINK1.
Figure 2.
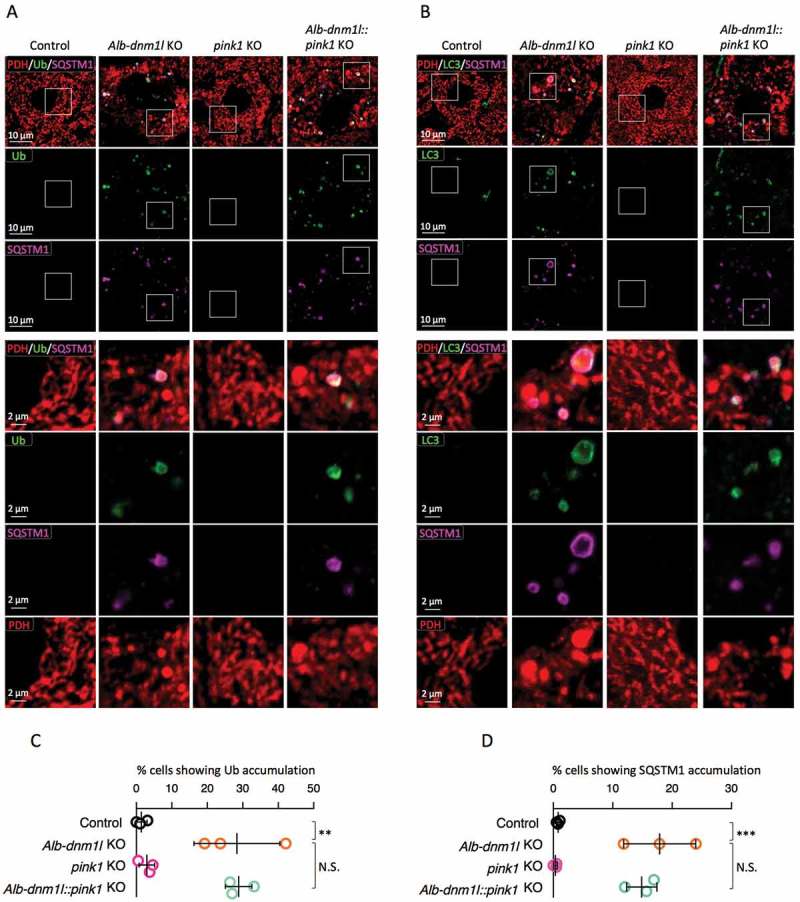
Mitochondria undergo ubiquitination in the absence of PINK1. (A and B) Frozen liver sections were examined by immunofluorescence microscopy with antibodies against ubiquitin, SQSTM1, and PDH (A) and LC3, SQSTM1, and PDH (B). Lower panels are magnified images of boxed regions. Bars in higher panels: 10 µm. Bars in lower panels: 2 µm. (C and D) Quantification of cells showing the accumulation of ubiquitin (C) and SQSTM1 (D) on mitochondria. Values are average ± SD (n = 3 mice). Statistical analysis was performed using one-way ANOVA followed by the post-hoc Tukey test: *p < 0.05, **p < 0.01, ***p < 0.001.
The loss of PINK1 restores normal levels of MFN1 and 2
The loss of mitochondrial division leads to the inactivation of three dynamin related GTPases that mediate mitochondrial fusion: the outer membrane proteins MFN1 and MFN2 and the inner membrane protein OPA1. In the absence of DNM1L, the levels of MFN1 and 2 are decreased by proteasomal degradation, whereas OPA1 is subjected to inactivating site-specific proteolytic cleavage [29,30]. These changes in these three mitochondrial fusion GTPases are thought to counterbalance decreased mitochondrial division by inhibiting mitochondrial fusion. We have previously shown that this balancing mechanism recognizes the size of mitochondria rather than the loss of DNM1L or the loss of mitochondrial division since restoring mitochondrial size in the absence of DNM1L can restore normal levels of MFN1 and 2 [13]. As a mechanism that specifically controls the abundance of MFN1 and MFN2, we have found that PRKN is necessary for the degradation of MFN1 and MFN2, but not the proteolytic cleavage of OPA1, in Alb-dnm1l KO livers [13].
To determine whether the degradation of MFN1 and 2 requires PINK1, we analyzed Alb-dnm1l::pink1 KO livers using Immunoblotting. We found that the amounts of both MFN1 and 2 were significantly increased in Alb-dnm1l::pink1 KO livers, compared to Alb-dnm1l KO livers (Figure 3A, B, and C). Conversely, OPA1 processing did not change in Alb-dnm1l::pink1 KO livers, compared to Alb-dnm1l KO livers. We found similar levels of the cleaved forms of OPA1 (S3 and S5) in Alb-dnm1l KO and Alb-dnm1l::pink1 KO mice (Figure 3D and E). As a control, we found similar levels of GAPDH and a mitochondrial outer membrane protein, TOMM20 (Figure 3F and G). These results suggest that PINK1 selectively regulates MFN1 and 2, but not OPA1, in response to increases in mitochondrial size.
Figure 3.
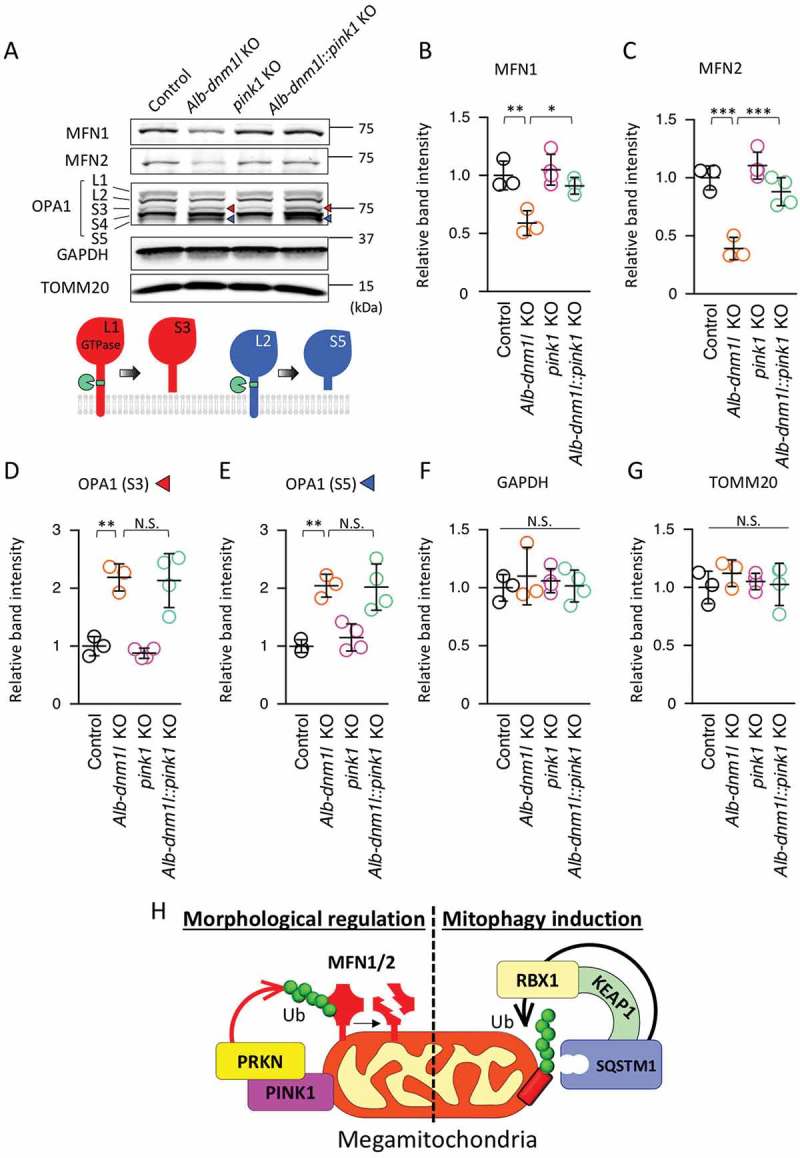
Additional loss of PINK1 restores the amounts of MFN1 and 2 that are decreased in Alb-dnm1l KO hepatocytes. (A) Immunoblotting of livers isolated from the indicated mice at 3 months using antibodies against MFN1 and 2, OPA1, GAPDH, and TOMM20. Lower image represents a model for OPA1 processing. (B–G) Quantification of band intensity of MFN1 (B), MFN 2 (C), OPA1-S3 (D), OPA1-S5 (E), GAPDH (F), and TOMM20 (G). Values are average ± SD (n = 3–4). (H) Model for mitochondrial ubiquitination mediated by the SQSTM1-KEAP1-RBX1 complex in mitophagy and by the PINK1-PRKN pathway in morphological regulation. Statistical analysis was performed using ANOVA followed by the post-hoc Tukey test: *p < 0.05, **p < 0.01.
Discussion
In our previous studies, we found that mitochondrial proteins are ubiquitinated by a mechanism that requires SQSTM1, but not PRKN [13,16,17]. Since PINK1 can work together with PRKN, as well as other ubiquitin E3 ligases, in mitochondrial ubiquitination, it was critical to determine whether PINK1 functions in this newly discovered mitochondrial ubiquitination pathway that is mediated by SQSTM1. In the present study, we addressed this question and found that SQSTM1-dependent ubiquitination occurs in the absence of PINK1. Therefore, it appears that there are multiple distinct mechanisms that ubiquitinate mitochondrial proteins to label damaged mitochondria for mitophagy (Figure 3H). We speculate that different mechanisms label damaged or dysfunctional mitochondria depending on the types and extent of mitochondrial damage.
We found that, rather than globally controlling mitochondrial ubiquitination, PINK1, along with PRKN, controls the specific degradation of MFN1 and 2 when mitochondrial size is increased due to the loss of DNM1L-mediated mitochondrial division (Figure 3H). Since the loss of DNM1L does not cause gross decreases in the mitochondrial membrane potential, PINK1 and PRKN may be activated independently of the membrane potential, e.g., when oxidative stress levels increase. It is also possible that PINK1 and PRKN respond to increases in the size of mitochondria. Targeted degradation of MFNs could enable the cell to reversibly readjust the balance between mitochondrial division and fusion without grossly removing mitochondria. In future studies, it would be important to further decipher how the balance of mitochondrial dynamics is maintained.
Materials and methods
Animals
All experiments with animals were conducted according to the guidelines established by the Johns Hopkins University Committee on Animal Care and Use. Dnm1lflox/flox [29], prkn-/- [16], sqstm1-/- [24] and pink1-/- [28] mice have been described previously. Alb-Cre mice [31] were obtained from the Jackson Laboratory. By breeding, we generated control (Dnm1lflox/flox and Alb-Cre+/-), Alb-dnm1l KO (Alb-Cre+/-::Dnm1lflox/flox), prkn KO (Dnm1lflox/flox::prkn-/-), Alb-dnm1l::prkn KO (Alb-Cre+/-::Dnm1lflox/flox::prkn-/-), sqstm1 KO (Dnm1lflox/flox::sqstm1-/-), Alb-dnm1l::sqstm1 KO (Alb-Cre+/-::Dnm1lflox/flox::sqstm1-/-), Alb-dnm1l::prkn::sqstm1 KO (Alb-Cre+/-::Dnm1lflox/flox::prkn-/-::sqstm1-/-), pink1 KO (Dnm1lflox/flox::pink1-/-), and Alb-dnm1l::pink1 KO (Alb-Cre+/-::Dnm1lflox/flox::pink1-/-) mice.
Immunoblotting
Mouse livers were dissected, flash-frozen in liquid nitrogen, and homogenized in RIPA buffer (Cell Signaling Technology, 9806S) containing complete mini protease inhibitor (Roche, 11,836,170,001). Lysates were centrifuged at 14,000 × g for 10 min and the supernatants were collected. Protein concentrations were determined by the Bradford method using a Bio-Rad protein assay (Bio-Rad, 5,000,006). Proteins were separated by SDS-PAGE and transferred onto Immobilon-FL membranes (Millipore, IPFL00010). After blocking in 3% BSA (Sigma-Aldrich, A9647) in PBS (137mM NaCl, 2.7mM KCl, 4.3 mM Na2HPO4pH, 1.1mM KH2PO4, pH 7.4)-Tween-20 (Sigma-Aldrich, P7949) for 1 h at room temperature, the blots were incubated with primary antibodies. Immunocomplexes were visualized using appropriate fluorophore-conjugated secondary antibodies and a PharosFX Plus Molecular Imager (Bio-Rad, CA, USA). Band intensity was determined using Fiji image analysis software.
Antibodies
The following primary antibodies were used: OPA1 (BD Biosciences, 612,607), PDH (Abcam, ab110333), GAPDH (Thermo, MA5-15,738), TOMM20/TOM20 (Santa Cruz Biotechnology, sc-11,415), MFN1 (Abcam, ab57602), MFN2 (Abcam, ab56889), SQSTM1 (Progen, GP-62C), ubiquitin (DAKO, z0458), and LC3 (MBL, PM036). The following secondary antibodies were purchased from Invitrogen: Alexa Fluor 488 anti-rabbit IgG (A21206), Alexa Fluor 488 anti-mouse IgG (A21202), Alexa Fluor 568 anti-mouse IgG (A10037), Alexa Fluor 647 anti-mouse IgG (A31571), and Alexa Fluor 647 anti-guinea pig IgG (A21450).
Immunofluorescence microscopy
Mice were anesthetized by intraperitoneal injection of Avertin solution (2,2,2-Tribromoethanol (Sigma-Aldrich, T48402) is dissolved in 2-Methhyl-2-butanol (Sigma-Aldrich, 240,486)) and fixed by cardiac perfusion of ice-cold 4% paraformaldehyde (Sigma-Aldrich, 441,244) in PBS (Quality Biological, 119–069-131) as previously described [15,16]. Perfused livers were dissected and further fixed in 4% paraformaldehyde in PBS for 2 h at 4°C. The samples were further incubated in PBS containing 30% sucrose overnight and frozen in OCT compound (Fisher Healthcare, 23–730-571). The frozen sections were cut, washed in PBS, and blocked in 10% chicken serum (Sigma-Aldrich, C5405). The sections were then incubated with primary antibodies followed by fluorescently labeled secondary antibodies. Samples were visualized using Zeiss (Oberkochen, Germany) LSM510-Meta, LSM780-FCS, and LSM800 GaAsP laser scanning confocal microscopes equipped with a 63× objective, as described previously [15,16]. Image analysis was performed using Fiji image analysis software.
Mitochondrial isolation
Mitochondria were isolated from the liver tissues as described [32] with some modifications. Livers dissected from control and Alb-dnm1l KO mice were rinsed in isolation buffer (i.e., 1 mM EGTA-Tris, 0.2 M sucrose [Fisher chemicals, S5-3], and 10 mM Tris-MOPS, pH of 7.4) to remove blood. Rinsed livers were minced and homogenized using a glass potter homogenizer with a Teflon pestle. Homogenates were centrifuged at 600 x g for 10 min at 4°C. The supernatants, which was used as the total fraction, were centrifuged at 7,000 x g for 10 min at 4°C. The pellets were washed twice in isolation buffer and used as the mitochondrial fraction.
Statistical analysis
ANOVA followed by the post-hoc Turkey test was used for the analysis of immunofluorescence and band intensity of MFN1 and 2, OPA1 S3 and S5, GAPDH and TOMM20. Student t-test was used for the analysis of band intensity of SQSTM1, VDAC and TOMM20 in the mitochondrial fraction. Statistical analysis was done with GraphPad Prism 8.0 software. * P < 0.05, ** P < 0.01, *** P < 0.001.
Funding Statement
This work was supported by the Japan Society for the Promotion of Science [Postdoctoral Fellowship for Research Abroad]; the Japan Society for the Promotion of Science [KAKENHI, JP19H03846]; National Institute of General Medical Sciences [GM123266]; National Institute of General Medical Sciences [GM131768].
Acknowledgments
We thank past and present members of the Iijima and Sesaki labs for helpful discussions and technical assistance. This work was supported by grants to T. Yamada (JSPS, Postdoctoral Fellowship for Research Abroad), T. Yanagawa (JSPS KAKENHI, JP19H03846), M.I. (NIH, GM084015) and H.S. (NIH, GM123266).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Quiros PM, Langer T, Lopez-Otin C.. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015. June;16(6):345–359. .PubMed PMID: 25970558 [DOI] [PubMed] [Google Scholar]
- [2].Goard CA, Schimmer AD. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene. 2014. May 22;33(21):2690–2699. . PubMed PMID: 23770858. [DOI] [PubMed] [Google Scholar]
- [3].Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2018. February;19(2):109–120. .PubMed PMID: 29165426 [DOI] [PubMed] [Google Scholar]
- [4].Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016. April;17(4):213–226. .PubMed PMID: 26956194 [DOI] [PubMed] [Google Scholar]
- [5].Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011. August;23(4):476–482. .PubMed PMID: 21705204; PubMed Central PMCID: PMC3155757. eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sugiura A, McLelland GL, Fon EA, et al. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. Embo J. 2014. October 1;33(19):2142–2156. PubMed PMID: 25107473; PubMed Central PMCID: PMC4282503. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Misgeld T, Schwarz TL. Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron. 2017. November 1;96(3):651–666. . PubMed PMID: 29096078; PubMed Central PMCID: PMCPMC5687842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018. February 19;28(4):R170–R185. . PubMed PMID: 29462587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016. March;17(3):300–316. .PubMed PMID: 26882551; PubMed Central PMCID: PMCPMC4772979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tamura Y, Itoh K, Sesaki H. SnapShot: mitochondrial dynamics. Cell. 2011. June 24;145(7):1158–1158.e1. . PubMed PMID: 21703455; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kameoka S, Adachi Y, Okamoto K, et al. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 2018. January;28(1):67–76. .PubMed PMID: 28911913; PubMed Central PMCID: PMCPMC5742555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Roy M, Reddy PH, Iijima M, et al. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol. 2015. February 18;33:111–118. . PubMed PMID: 25703628; Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yamada T, Murata D, Adachi Y, et al. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018. October 2;28(4):588–604. . PubMed PMID: 30017357; PubMed Central PMCID: PMCPMC6170673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yamada T, Adachi Y, Yanagawa T, et al. p62/sequestosome-1 knockout delays neurodegeneration induced by Drp1 loss. Neurochem Int. 2017. May 18 DOI: 10.1016/j.neuint.2017.05.012 [PubMed PMID: 28527629]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yamada T, Adachi Y, Fukaya M, et al. Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration. Am J Pathol. 2016. September 15;186(11):2798–2802. . PubMed PMID: 27640145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kageyama Y, Hoshijima M, Seo K, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. 2014. December 1;33(23):2798–2813. . PubMed PMID: 25349190; PubMed Central PMCID: PMC4282557. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kageyama Y, Zhang Z, Roda R, et al. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012. May 14;197(4):535–551. . PubMed PMID: 22564413; PubMed Central PMCID: PMC3352955. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McWilliams TG, Prescott AR, Montava-Garriga L, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018. February 6;27(2):439–449 e5. . PubMed PMID: 29337137; PubMed Central PMCID: PMCPMC5807059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Villa E, Proics E, Rubio-Patino C, et al. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 2017. September 19;20(12):2846–2859. . PubMed PMID: 28930681. [DOI] [PubMed] [Google Scholar]
- [20].Allen GF, Toth R, James J, et al. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013. December;14(12):1127–1135. .PubMed PMID: 24176932; PubMed Central PMCID: PMCPMC3981094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee JJ, Sanchez-Martinez A, Zarate AM, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018. May 7;217(5):1613–1622. . PubMed PMID: 29500189; PubMed Central PMCID: PMCPMC5940313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009. June 12;137(6):1001–1004. . PubMed PMID: 19524504; PubMed Central PMCID: PMCPMC3971861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. Febs J. 2015. December;282(24):4672–4678. .PubMed PMID: 26432171 [DOI] [PubMed] [Google Scholar]
- [24].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007. December 14;131(6):1149–1163. PubMed PMID: 18083104. [DOI] [PubMed] [Google Scholar]
- [25].Rojansky R, Cha MY, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife. 2016. November 17;5. doi: 10.7554/eLife.17896 PubMed PMID: 27852436; PubMed Central PMCID: PMCPMC5127638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014. June 5;510(7503):162–166. . PubMed PMID: 24784582; eng. [DOI] [PubMed] [Google Scholar]
- [27].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. August 20;524(7565):309–314. . PubMed PMID: 26266977; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kitada T, Pisani A, Porter DR, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007. July 3;104(27):11441–11446. . PubMed PMID: 17563363; PubMed Central PMCID: PMCPMC1890561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wakabayashi J, Zhang Z, Wakabayashi N, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009. September 21;186(6):805–816. PubMed PMID: 19752021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Saita S, Ishihara T, Maeda M, et al. Distinct types of protease systems are involved in homeostasis regulation of mitochondrial morphology via balanced fusion and fission. Genes Cells. 2016. May;21(5):408–424. .PubMed PMID: 26935475 [DOI] [PubMed] [Google Scholar]
- [31].Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999. January 1;274(1):305–315. PubMed PMID: 9867845 [DOI] [PubMed] [Google Scholar]
- [32].Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–295. PubMed PMID: 17406588 [DOI] [PubMed] [Google Scholar]