

Abstract
Hereditary colorectal cancer accounts for less than 5% of all colorectal cancer cases. Some of the unique characteristics that are commonly encountered in cases of hereditary colorectal cancer include early age at onset, synchronous/metachronous occurrence of the cancer, and association with multiple cancers in other organs, necessitating different management from sporadic colorectal cancer. While the diagnosis of familial adenomatous polyposis might be easy because usually 100 or more adenomas that develop in the colonic mucosa are in this condition, Lynch syndrome, which is the most commonly associated disease with hereditary colorectal cancer, is often missed in daily medical practice because of its relatively poorly defined clinical characteristics. In addition, the disease concept and diagnostic criteria for Lynch syndrome, which was once called hereditary non‐polyposis colorectal cancer, have changed over time with continual research, thereby possibly creating confusion in clinical practice. Under these circumstances, the JSCCR Guideline Committee has developed the “JSCCR Guidelines 2016 for the Clinical Practice of Hereditary Colorectal Cancer (HCRC)," to allow delivery of appropriate medical care in daily practice to patients with familial adenomatous polyposis, Lynch syndrome, or other related diseases. The JSCCR Guidelines 2016 for HCRC were prepared by consensus reached among members of the JSCCR Guideline Committee, based on a careful review of the evidence retrieved from literature searches, and considering the medical health insurance system and actual clinical practice settings in Japan. Herein, we present the English version of the JSCCR Guidelines 2016 for HCRC.
Keywords: hereditary colorectal cancer, guideline, familial adenomatous polyposis, Lynch syndrome
Introduction
1. Guideline objectives
In Japan, the incidence of colorectal cancer has been steadily increasing; it is presently one of the most frequently encountered cancers in clinical practice and a cause for great concern. Most colorectal cancers are thought to be caused by the accumulation of gene mutations in the colonic mucosa or adenomas, the occurrence of which is thought to be influenced by lifestyle, environmental factors, advanced age, etc. (sporadic colorectal cancer). Another type of colorectal cancer, called familial colorectal cancer, which accounts for 20-30% of all colorectal cancer cases, is commonly found among relatives (familial clustering). In less than approximately 5% of colorectal cancer cases, the causative genes have been identified, irrespective of the presence or absence of familial clustering, and these cases are collectively referred to as cases of hereditary colorectal cancer. Early age at onset, synchronous/metachronous occurrence, association with multiple cancers in other organs, etc., are usually seen in cases of hereditary colorectal cancer; therefore, this type of cancer should be managed differently from sporadic colorectal cancer. However, hereditary colorectal cancer is still not well-recognized by general clinicians.
Familial adenomatous polyposis and Lynch syndrome have high incidence rates in cases of hereditary colorectal cancer. While detection of familial adenomatous polyposis might be facilitated by its common occurrence in 100 or more adenomas of the colonic mucosa, Lynch syndrome, which is the most commonly associated disease with hereditary colorectal cancer, is often missed in daily medical practice owing to its relatively poorly defined clinical characteristics. In addition, Lynch syndrome was once called hereditary non-polyposis colorectal cancer (HNPCC), and its disease concept and diagnostic criteria have changed with continuing research, thereby possibly creating confusion in clinical practice.
Under these circumstances, the “JSCCR Guidelines 2016 for the Clinical Practice of Hereditary Colorectal Cancer” (hereafter referred to as the JSCCR Guidelines 2016 for HCRC), intended for doctors and medical personnel engaged in providing medical care to patients with familial adenomatous polyposis, Lynch syndrome, and other related diseases, have been developed for the following purposes:
(1) To deepen the understanding of the concept of hereditary colorectal cancer
(2) To provide guidance on management strategies, including diagnosis and surveillance, for hereditary colorectal cancer
(3) To emphasize the importance of the need to consider the psychosocial burden caused by hereditary diseases in patients and their families (relatives) as well as their need for support
(4) To enhance mutual understanding between healthcare professionals and patients by making these guidelines available to the public.
2. How to use the JSCCR Guidelines 2016 for HCRC
The JSCCR Guidelines 2016 for HCRC can be used as a tool for the treatment of hereditary colorectal cancer under actual clinical practice settings. More specifically, they can be referred to for the diagnosis/treatment/surveillance of individual patients or for obtaining informed consent for genetic testing and optimal treatment selection etc. from patients and their families. The JSCCR is responsible for the statements in the JSCCR Guidelines 2016 for HCRC. However, the personnel directly in charge of treatment, and not the JSCCR or the Guideline Committee, must take responsibility for treatment outcomes.
3. Method of development for the JSCCR Guidelines 2016
1) Circumstances of the development of JSCCR Guidelines 2016 for HCRC
The JSCCR planned to develop “the JSCCR Guidelines for the Clinical Practice of Hereditary Colorectal Cancer” as a project of the Familial Colorectal Cancer Committee, and published the “JSCCR Guidelines 2012 for HCRC” in July 2012. Subsequently, several new findings and clinical practice guidelines, particularly regarding Lynch syndrome, were published from overseas. In addition, the Familial Colorectal Cancer Committee itself analyzed data from “A Retrospective Multicenter Study of Familial Adenomatous Polyposis” and “Registration and Genetic Analysis of HNPCC -Secondary Study-,” which were studies conducted by the JSCCR, and obtained new findings. Under these circumstances, the clinical genetics departments have been established, mainly in specialized institutions, and hereditary tumors have increasingly become an issue of social concern in Japan. Based on the above, revision of the guidelines published in 2012 was initiated in 2015. A draft revision was prepared after many discussions and submitted to the Guideline Evaluation Committee in May 2016. In addition, a public hearing was held in the 85th annual meeting of the JSCCR in July 2016, and subsequently, the revised points were published on the website of the JSCCR to collect opinions from the public. Further revisions were made by reference to these opinions, and finally the “JSCCR Guidelines 2016 for HCRC” was published in November 2016.
We attempted to develop the JSCCR Guidelines 2016 for HCRC in accordance with the concept of evidence-based medicine. However, the incidence of hereditary colorectal cancer is relatively low, and it is not easy to design high-evidence-level studies. In view of this difficulty in obtaining sufficient evidence, the guidelines have been developed by consensus reached among members of the JSCCR, based on information obtained from literature searches, and considering the medical health insurance system and actual clinical practice situation in Japan. In addition, members of the Japanese Society for Familial Tumors also participated in the Guideline Development Committee.
2) Principles behind Guideline development
The JSCCR Guidelines 2016 for HCRC presents evidence for each management strategy to enable clearer understanding of the management strategies, including the diagnosis, treatment, and surveillance of hereditary colorectal cancer; however, the technical aspects of each treatment method have not been discussed.
3) Extraction and evaluation of evidence
The method adopted for guideline development was in accordance with the concept of EBM. However, because hereditary colorectal cancer is a relatively rare disease and it is difficult to conduct randomized controlled trials, the evidence levels have not been shown.
4. Description method
Familial adenomatous polyposis and Lynch syndrome, which have relatively high incidence rates among cases of hereditary colorectal cancer, were selected, and (1) the disease concept, (2) diagnosis, (3) treatment, (4) postoperative surveillance, (5) management of patients and their families, etc., were briefly described for each disease. Next, contents suitable for inclusion in clinical questions (CQs) were selected and discussed by the Guideline Development Committee.
5. Method for describing the recommendations
Each recommendation in response to a CQ is accompanied, as much as possible, by classifications of the evidence and recommendation categories, based on consensus reached among members of the Guideline Development Committee. In determining the recommendation categories, in addition to an evaluation of the validity of the source of evidence for each recommendation, a comprehensive investigation of the validity and clinical applicability of each recommendation was performed, by ascertaining that the diagnosis and treatment methods are based on clear scientific evidence, are the best and safest available, are minimally invasive, and are in line with those used in actual clinical practice in Japan.
Classification of the recommendation categories are as follows:
●Category A: unanimous recommendation by the Guideline Development Committee based on high-level evidence
●Category B: unanimous recommendation by the Guideline Development Committee based on low-level evidence
●Category C: recommendation that was not agreed upon completely by all the members of the Guideline Development Committee, irrespective of the level of evidence
●Category D: recommendation that was not agreed upon by the members of the Guideline Development Committee
6. Method of literature search
The PubMed and Ichushi-Web databases were selected for the literature search, and the English and Japanese literature was systematically searched in both databases for the period from the earliest possible date to August 2015. The exhaustive literature search was performed for the broad category, namely, “familial adenomatous polyposis,” to obtain articles on familial adenomatous polyposis, and for the broad categories, “Lynch syndrome,” “hereditary non-polyposis colorectal cancer,” “microsatellite instability,” and “mismatch repair,” to obtain articles on Lynch syndrome; manual searching was added as required. The full texts of the articles selected from 25,941 extracted documents with abstracts (familial adenomatous polyposis: 1,049 Japanese articles and 7,897 English articles; Lynch syndrome: 1,050 Japanese articles and 16,045 English articles) were critically examined. In addition, important articles published from September 2015 onward were also adopted after full examination.
7. Revision
In cooperation with the Japanese Society for Familial Tumors, the JSCCR Guideline Committee and Familial Colorectal Cancer Committee, as the central organizations, shall aim to revise the JSCCR Guidelines 2016 for HCRC in 4 years, in principle.
8. Publication
The JSCCR Guidelines 2016 for HCRC will be published as a pamphlet and will be made available to the public on the website of the JSCCR, etc., so that the guidelines can be widely used under clinical settings throughout Japan.
Chapter 1: Familial Adenomatous Polyposis
1. Outline
●Familial adenomatous polyposis (FAP) is a hereditary autosomal dominant disease caused by germline mutations in the Adenomatosis Polyposis Coli (APC) gene, and it is characterized by the development of multiple colorectal adenomas (Side Memo 1: Method for describing genomic alterations, germline and somatic mutations; Appendix: II. Method for describing genomic variants).
●If not treated, almost all FAP patients develop colorectal cancer.
●FAP patients can develop not only colorectal cancer, but also various other associated tumorous and non-tumorous lesions in the gastrointestinal tract and other organs.
[Clinical features]
●Some FAP patients have been reported to develop colorectal cancer while still in their teenage years while approximately 50% of the patients develop colorectal cancer by their 40s. If left untreated, almost all patients develop colorectal cancer by around 60 years of age1). (Attachment I).
●The most common cause of death in FAP patients is colorectal cancer2), which accounted for approximately 80% of all causes of death in FAP patients until the 1980’s; however, the proportion has been decreasing toward approximately 60% from the 1990’s (Table 1).
Table 1.
Changes in the Mortality Rates of Various Cancers and Other Conditions Over Time in Patients with Familial Adenomatous Polyposis.
| Cause of death | Up to 1980 (n=268) |
1981-1990 (n=166) |
1990-2003 (n=71) |
|---|---|---|---|
| Colorectal cancer | 80.2% | 77.7% | 60.6% |
| Desmoid tumor | 3.0% | 4.8% | 9.9% |
| Gastric cancer | 3.0% | 2.4% | 2.8% |
| Duodenal/periampullary cancer | 1.8% | 2.4% | 5.6% |
| Pancreatic cancer | 0 | 0 | 1.4% |
| Small bowel cancer | 1.2% | 1.2% | 1.4% |
| Myocardial infarction/heat failure | 1.8% | 2.4% | 2.8% |
| Stroke | 1.4% | 1.2% | 2.8% |
| Lung cancer | 0.9% | 2.4% | 5.6% |
| Hepatocellular carcinoma | 0.7% | 0.6% | 0 |
| Uterine cancer | 0.5% | 0.6% | 1.4% |
| Gastric ulcer | 0.2% | 0 | 0 |
| Esophageal cancer | 0.2% | 0 | 1.4% |
| Gallbladder cancer | 0.2% | 0.6% | 0 |
| Sarcoma | 0.2% | 0 | 0 |
| Ovarian cancer | 0.2% | 0 | 0 |
| Thyroid cancer | 0 | 0 | 1.4% |
| Accident | 2.1% | 2.4% | 0 |
| Other diseases | 1.6% | 1.2% | 2.8% |
| Unknown | 0.2% | 0 | 0 |
| Suicide | 0.2% | 0 | 0 |
| All cases | 100% | 100% | 100% |
| Age at the time of death (average ± SD) | 41.9 ± 11.9 | 44.0 ± 13.9 | 46.0 ± 15.6 |
Modifications with ref. 2)
●Among the main extracolonic manifestations (Table 2), duodenal cancer and desmoid tumor are major causes of death in addition to colorectal cancer in FAP patients.
Table 2.
Major Neoplastic Lesions Associated with Familial Adenomatous Polyposis.
| ・Fundic gland polyposis* |
| ・Gastric adenoma* |
| ・Duodenal adenoma* |
| ・Periampullary adenoma* |
| ・Jejunal/ileal adenoma* |
| ・Desmoid tumor |
| ・Skull osteoma/jaw osteoma/unerupted teeth/extra teeth (supernumerary teeth) |
| ・Epidermoid cyst |
| ・Thyroid cancer |
| ・Congenital hypertrophy of the retinal pigment epithelium |
| ・Hepatoblastoma |
| ・Adrenal tumor* |
| ・Brain tumor |
*: possibility of malignant transformation
[Causative gene]
●APC gene on chromosome 5 (5q22.2)
[Mode of inheritance]
●Autosomal dominant inheritance
[Mechanisms of tumorigenesis] (Figure 1A, 1B)
Figure 1.
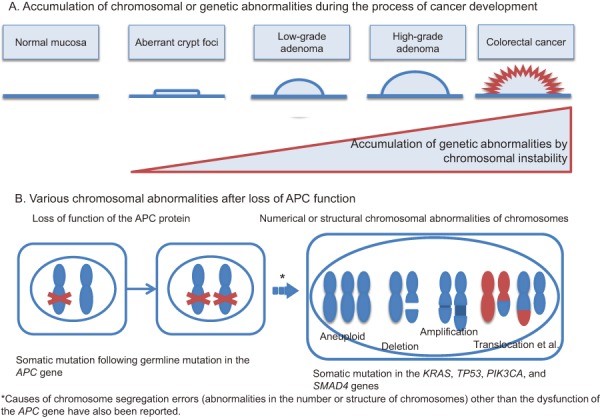
Predicted mechanisms underlying colorectal cancer development in patients with FAP.
●In addition to a germline mutation in one of the two alleles of the APC gene, an acquired second-hit somatic alteration, such as a deletion in the other allele of the APC gene in the epithelial cells of the large intestine (loss of heterozygosity [LOH]), is thought to be the cause of aberrant crypt foci (ACF) (Side Memo 1: chromosomal instability, loss of heterozygosity). (Side Memo 1: aberrant crypt foci)3).
●Dysfunction of the APC protein causes accumulation and nuclear translocation of cytoplasmic β-catenin; then, nuclear β-catenin forms a complex with TCF4, which promotes transcription.
●The mechanism via which APC protein malfunction causes chromosomal instability (CIN) remains unknown; however, in the presence of APC protein malfunction, somatic alterations such as LOH tend to occur in oncogenesis-related genes. In the development of colorectal cancer from ACF via the adenoma-carcinoma sequence, additional mutations are thought to occur in carcinogenesis-related genes such as KRAS and TP53 genes (multi-hit theory or multi-stage model)4).
[Incidence]
●The estimated incidence of FAP in the overall population is 1:20,000 to 1:10,000 in Western countries, and 1:17,400 in Japan5). Less than 1% of all patients with colorectal cancer are estimated to have FAP6). According to a JSCCR multicenter study, 0.24% of all colorectal cancer patients have FAP.
Side Memo 1
■Method for describing genomic alterations, germline and somatic mutations
The description method validated by the Human Genome Variation Society (http://www.hgvs.org/mutnomen/) is generally used. So far, changes in genomic sequences have often been represented by terms such as “mutation” and “polymorphism.” However, because the use of these terms may be perceived differently by different individuals, thus causing confusion, the use of terms such as “sequence variant,” “alteration,” and “allelic variant” is recommended. These terms represent the presence of changes compared to reference sequences, but do not indicate any causal relationship with diseases. In addition, the expression “pathogenic” may be used, only after carefully considering what situations it can be used in, and other expressions such as “affect function” may be used in the future.
■Germline and somatic mutations
Gene mutations transmitted through the sperm or ovum are called germline mutations. Since these mutations are present in the fertilized ovum, all cells of the body have these mutations. On the other hand, new gene mutations in non-germ cells constituting the body, or non-germline mutations, are called somatic mutations.
■APC gene mutations in FAP
In the tumors of patients with FAP, germline and somatic mutations of the APC gene result in the production of a truncated APC protein, which is thought to be an inactive form of the protein.
■Chromosomal instability (CIN)
CIN represents abnormalities in the number or structure (deletion, duplication, translocation, etc.) of chromosomes seen in cancer and other cells, and it is thought to cause tumorigenesis.
■Loss of heterozygosity (LOH)
Heterozygosity indicates the presence of different base sequences in a homologous region of a pair of genetic information inherited from the parents. In the case of FAP, pathogenic mutations are present only in one of the two alleles of the APC gene, and the other allele is normal (wild type) in normal cells. This state is called heterozygosity. However, loss of the wild-type APC allele by deletion, referred to as LOH, occurs during the process of oncogenesis.
■Aberrant crypt foci (ACF)
ACF cannot be distinguished from normal mucosa by normal endoscopic observation, and can only be confirmed by magnifying endoscopy as clusters of abnormal crypts showing strong staining with methylene blue. Some ACF are thought to be precursor lesions of adenomas and/or carcinomas.
2. Diagnosis
1) Flow of diagnosis (Figure 2)
Figure 2.
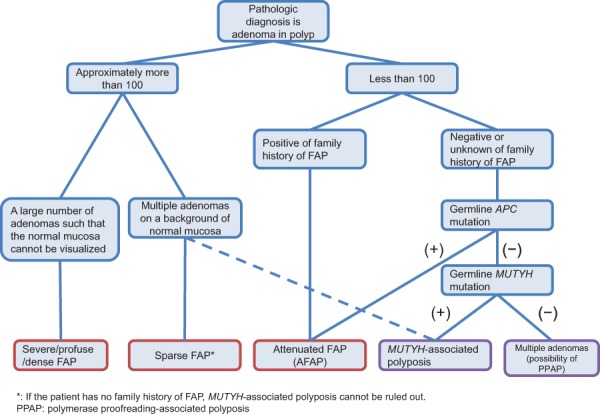
Flow-chart for the diagnosis of FAP.
●FAP may be diagnosed clinically and/or genetically7).
[Clinical diagnosis]
If either of the following criteria (1) or (2) is satisfied, a diagnosis of FAP is made.
(1) Detection of approximately 100 or more adenomas in the large intestine, irrespective of the presence/absence of a family history of FAP.
(2) Detection of less than 100 adenomas in the presence of a family history of FAP.
[Genetic diagnosis]
If a pathogenic germline mutation is present in the APC gene, a diagnosis of FAP is made.
●There are exceptional pathologies other than FAP that are characterized by the presence of approximately 100 or more adenomas in the large intestine (MUTYH-associated polyposis, an autosomal recessive disease). Therefore, a family history consistent with autosomal dominant inheritance is a useful clue for the diagnosis of FAP.
●Irrespective of the number of colorectal adenomas, the presence of characteristic extracolonic manifestations is a useful clue for the diagnosis of FAP. In 20-40% of patients clinically diagnosed with FAP, no mutations are detected in the APC gene8,9). (CQ1)
●If a patient wishes to undergo genetic testing for his/her own treatment or for the diagnosis in his/her relatives, or if attenuated FAP (AFAP) has to be differentiated from MUTYH-associated polyposis and polymerase proofreading-associated polyposis (PPAP), genetic testing of the APC gene is considered. This testing can be performed in testing companies (not covered by the national health insurance program) (Side Memo 2: genetic testing) (CQ1).
2) Classification according to the density of the colorectal adenomas
●FAP is sometimes classified as profuse FAP, sparse FAP, and attenuated FAP, according to the density of the adenomas. Profuse FAP and sparse FAP are sometimes collectively called typical (classical) FAP.
●Density of adenomas has been reported to be associated with the site of the germline mutation in the APC gene and with the risk of development of colorectal cancer.
●Profuse FAP: Normal mucosa cannot be visualized macroscopically because of the profusion of adenomas (Figure 3) However, often, adenoma density is found to differ even among regions of the large intestine.
Figure 3.
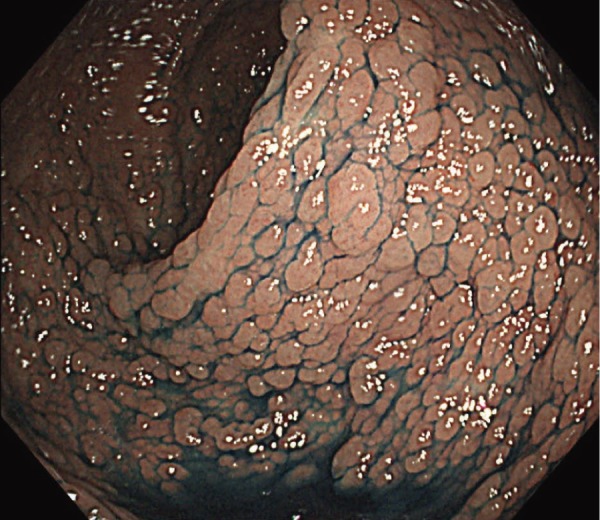
Severe/profuse/dense FAP.
●Sparse FAP: Multiple adenomas can be observed on a background of normal mucosa. The number of adenomas is approximately ≥100 (Figure 4).
Figure 4.
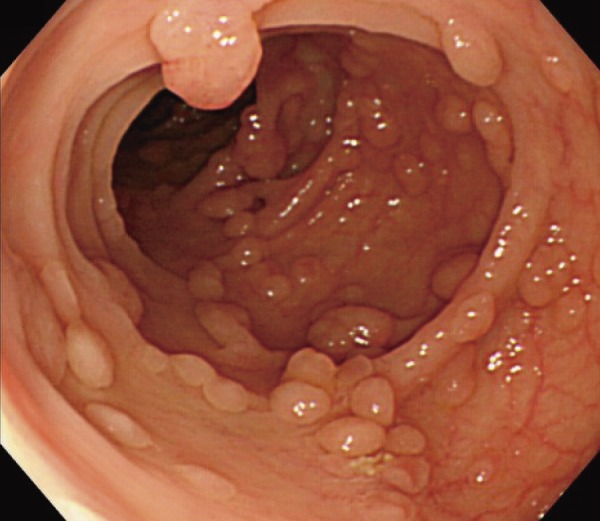
Sparse FAP.
●Attenuated FAP (AFAP)Note 1: The number of adenomas is approximately ≥10 and <100 (CQ2).
●In cases of profuse FAP, a germline mutation is often seen between codons 1250 to 1464 (in particular, codon 1309) in the APC gene10,11). In AFAP, the germline mutation is often seen in the alternative splicing region (in which an exon is skipped during transcription because of the mutation), or in the 5’ or 3’ region of the APC gene12).
According to the JSCCR multicenter study, the age at diagnosis of adenomas and the age at diagnosis of cancer in the colorectum are often lower among patients with profuse FAP than in those with other types of FAP. It has been reported that approximately half of the patients with profuse, sparse, and attenuated types develop colorectal cancer by age 40, 47, and 55 years, respectively.
Side Memo 2
■Difference between the profuse and sparse types
FAP is sometimes classified according to the number of colorectal adenomas into the profuse (>1,000 or 2,000 adenomas) and sparse (100-1,000 or 2,000 adenomas) types. Many reports classify these types of FAP as typical FAP, and FAP associated with a smaller number (10-99) of adenomas as AFAP. Strict differentiation between the profuse and sparse types is of little clinical significance.
3) FAP-associated lesions
●FAP is often associated with extracolonic tumorous and/or non-tumorous lesions.
●Presence of tumorous lesions, such as fundic gland polyposis (Figure 5), gastric adenoma (Figure 6) (CQ10), duodenal adenoma (Figure 7) (CQ11), ampullary adenoma (CQ12), desmoid tumor (Figure 8) (CQ14), subcutaneous soft tissue tumor/osteoma, and dental abnormalities (Figure 9) serve as useful clues for the diagnosis of FAP (Side Memo 3: Gardner syndrome).
Figure 5.
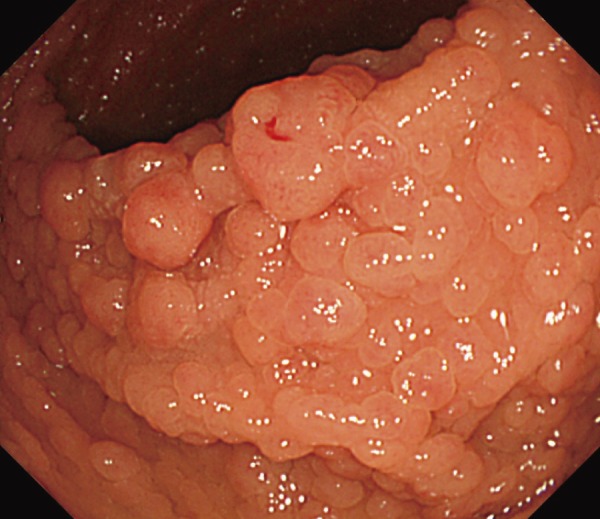
Fundic gland polyposis.
Figure 6.

Gastric adenoma (left, depressive type; right, elevated type).
Figure 7.
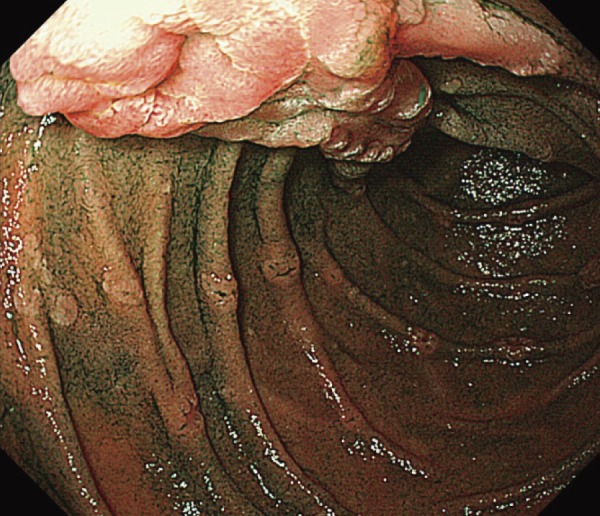
Duodenal adenoma and periampullary adenoma.
Figure 8.
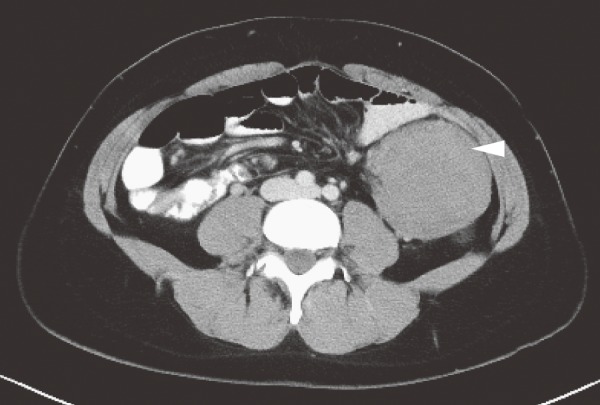
Intra-abdominal desmoid tumor (arrowhead).
Figure 9.
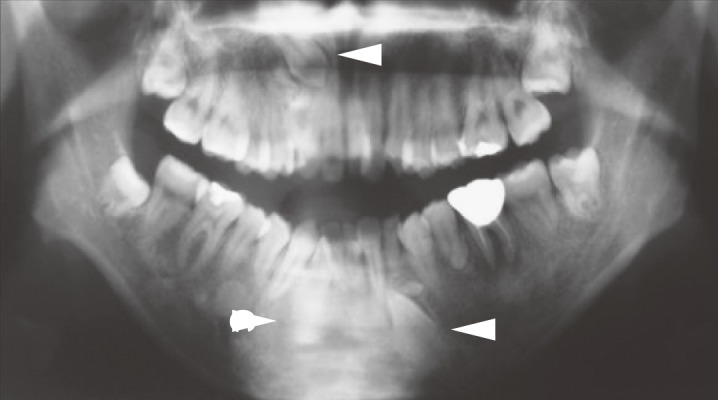
Dental abnormalities (unerupted teeth, arrowheads).
●FAP patients without Helicobacter pylori (H. pylori) infection often tend to have fundic gland polyposis, as compared to those with H. pylori infection13). Surveillance for fundic gland polyposis is required in FAP patients, because of the risk of malignant transformation of fundic gland polyps in these patients.
●FAP patients often develop depressed-type or protruded-type gastric adenomas (Figure 6).
●Congenital hypertrophy of the retinal pigment epithelium (Figure 10), a non-tumorous lesion, is detectable before the development of colorectal adenomas in FAP patients, and it is a helpful clue for diagnosis (Side Memo 3: Congenital hypertrophy of the retinal pigment epithelium).
Figure 10.
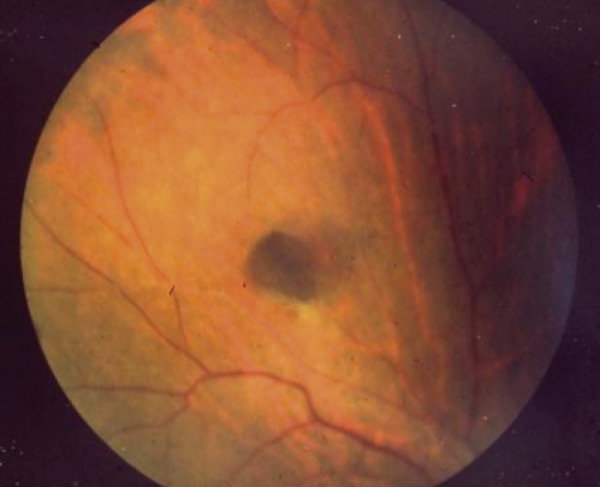
Congenital hypertrophy of the retinal pigment epithelium.
FAP patients may also develop other tumorous lesions, including desmoid tumor, thyroid cancer, adrenal tumor, hepatoblastoma, and brain tumor (Side Memo 3: Turcot syndrome).
Side Memo 3
■Gardner syndrome
Colorectal polyposis associated with subcutaneous soft tissue tumors, osteomas, dental abnormalities, desmoid tumors, etc., was once called Gardner syndrome, and was regarded as different from FAP, but subsequently, like FAP, this syndrome was also found to be caused by germline mutations in the APC gene. At present, the term Gardner syndrome is usually not used.
■Congenital hypertrophy of the retinal pigment epithelium (CHRPE)
CHRPE is a discontinuous flat pigmented lesion of the retina without clinical symptoms, which does not require any treatment. It does not affect visual acuity and does not show malignant transformation. Because approximately 80% of FAP patients have CHRPE, and because it can be detected at birth, it is a helpful clue for the diagnosis of FAP in children.
■Turcot syndrome (type 2)
Colorectal polyposis associated with brain tumor (mainly cerebellar medulloblastoma), and with a germline mutation in the APC gene is called Turcot syndrome, type 2 (see Lynch syndrome for Turcot syndrome, type 1).
4) Diseases and pathological conditions that should be differentiated from FAP
APC mosaicism:
If somatic mutations in the APC gene were to occur during the process of tumorigenesis, a mosaic of cells with and without the mutations in the APC gene would result. If this abnormality were to occur in cells that differentiate into mucosal cells of the large intestine, multiple colorectal adenomas would develop, like in FAP. It has been reported that APC mosaicism occurs in 1.6-4% of FAP patients with identified mutations in the APC gene and 11-20% of FAP patients with a negative family history14,15). Clinically, this condition is managed as FAP. In addition, mutations in the APC gene, if present in some germ cells (sex mosaicism), may be passed on to the next generation.
MUTYH-associated polyposis (MAP):
MAP is a hereditary autosomal recessive disease caused by biallelic germline mutation of the MUTYH gene, which is one of the base excision repair genes16). MAP is characterized by the presence of about 10-100 adenomas in the large intestine, although some patients could have as many as 100-1,000 adenomas17). The incidence of germline mutations in the MUTYH gene is unknown among Japanese colorectal cancer patients. The penetrance of colorectal cancer (proportion of individuals who develop colorectal cancer among those with gene mutations) is 43-100% in individuals aged up to 60 years18). Some MAP patients have been reported to develop a variety of lesions like those found in FAP. In Japan, there are few case reports of MAP, and this disease remains poorly understood. Treatment for MAP is like that for AFAP.
Polymerase proofreading-associated polyposis (PPAP):
PPAP is a hereditary autosomal dominant disease caused by pathogenic germline mutations in the POLE or POLD1 gene, both of which repair errors in DNA replication (proofreading function)19). Many patients have a few dozen colorectal adenomas, while some patients have been reported to have no adenomas. As extracolonic manifestations, duodenal adenomas/cancers and brain tumors have been reported to develop in patients with PPAP carrying mutation of the POLE gene20) and endometrial cancers, breast cancers, and brain tumors have been reported to develop in patients with PPAP carrying mutations of the POLD1 gene21). Tumors of the large intestine (colorectal adenomas and cancers) in PPAP are histologically indistinguishable from these tumors in sporadic cases. Therefore, genetic testing is necessary for a definitive diagnosis.
3. Treatment
1) Treatment of colorectal adenomas
●Proctocolectomy or colectomy prior to the development of colorectal cancer is a reliable prophylactic treatment.
The main surgical procedures adopted are as follows (Figure 11, Table 3):
Figure 11.
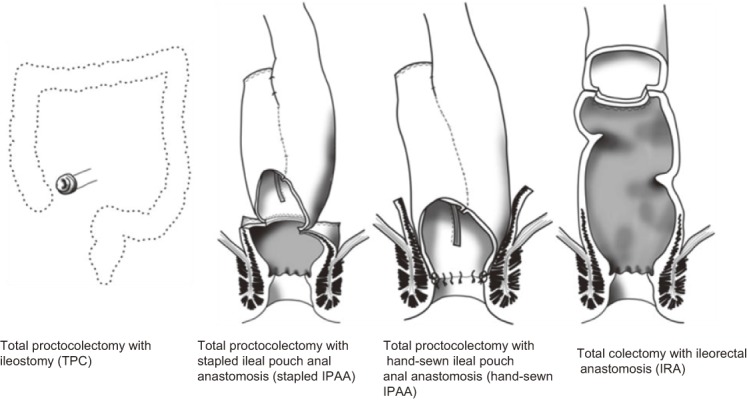
Surgical procedures for FAP (Side Memo 4: Nomenclature of the surgical procedures).
Table 3.
Characteristics of Surgical Procedures.
| Surgical procedures | Total proctocolectomy with ileostomy | Total proctocolectomy with ileal pouch anal anastomosis | Total colectomy with ileorectal anastomosis |
| Advantages | ・Complete prevention of colorectal cancer
・Less complications |
・Near-complete prevention of most colorectal cancer
・Preservation of anal function |
・Good anal function
・Easy operation ・Less complications |
| Disadvantages | ・Deteriorated body image and inconvenient stool management by stoma creation | ・Complex surgery
・Unstable anal function ・Possibility of cancer development at remaining rectal mucosa near the anus ・Possibility of pouchitis |
・Possibility of development of rectal cancer (depending on the number of adenomas, location of the germline mutation in the APC gene, or length of the residual rectum) |
(1) Total proctocolectomy + permanent ileostomy (TPC)
(2) Restorative proctocolectomy + ileal pouch-anal anastomosis (IPAA)
(3) Total colectomy + ileorectal anastomosis (IRA)
●At present, IPAA is thought to be the standard surgical procedure and it is commonly performed in FAP patients22-25). (CQ3,CQ4)
●In general, it is recommended that patients undergo prophylactic surgery when they are in their 20’s. (CQ5)
Side Memo 4
■Nomenclature of the surgical procedures
In Western countries, ileal pouch anal anastomosis with mucosectomy (IAA) and ileal pouch anal canal anastomosis (IACA) are often collectively called ileal pouch-anal anastomosis (IPAA), without discrimination between them. In addition, IAA is sometimes called hand-sewn IPAA, and IACA is sometimes called stapled IPAA. The height of the anastomosis (length of the residual rectum) is not clearly defined for ileorectal anastomosis (IRA). Total proctocolectomy + permanent ileostomy is often called total proctocolectomy (TPC).
●In recent years, laparoscopic surgery has been increasingly used for prophylactic proctocolectomy (colectomy). (CQ6)
●In cases where desmoid tumors are found in the mesentery during prophylactic surgery, IPAA is generally not recommended, owing to the risk of recurrence or enlargement of desmoid tumors and technical problems, but it may be acceptable under certain conditions. (CQ3)
●Total proctocolectomy for FAP may reduce fertility in females. (CQ7)
●Drug therapy with non-steroidal anti-inflammatory drugs (NSAIDs) has been attempted; however, its usefulness is unclear. (CQ8)
2) Treatment of colorectal cancer
●In FAP patients with locally advanced colorectal cancer, standard treatment for locally advanced colorectal cancer should be undertaken. If curative resection of the colorectal cancer can be expected, the surgical procedure should be selected according to the condition of the FAP.
●In FAP patients with locally advanced colorectal cancer, the surgical procedure should be determined after a comprehensive consideration of the stage and site of the colorectal cancer. If curative resection of the colorectal cancer can be expected, total proctocolectomy or total colectomy with dissection of the regional lymph nodes is an option; on the other hand, if the colorectal cancer cannot be expected to be curatively resected, a surgical procedure like that for sporadic colorectal cancer should be selected.
●Chemotherapy similar to that used for patients with sporadic colorectal cancer should be used for colorectal cancer associated with FAP.
●Even after total proctocolectomy or total colectomy, chemotherapy selection can be guided by the recommendations in the “JSCCR Guidelines 2016 for the Treatment of Colorectal Cancer.”
●If metastatic lesions can be expected to be curatively resected, treatment similar to that for metastases from sporadic colorectal cancer should be used.
3) Examinations for extracolonic manifestations before proctocolectomy (colectomy)
●It is desirable to carry out extensive examinations to check for extracolonic manifestations prior to colorectal resection, irrespective of the presence or absence of associated locally advanced colorectal cancer, although there is little evidence of its usefulness.
●It is recommended to check for the presence of gastroduodenal lesions, including ampullary and desmoid tumors prior to colectomy.
●Examinations for other tumorous lesions can be performed during the surveillance after proctocolectomy (colectomy).
●Presence/absence of adenomas and cancers of the stomach and duodenum, including of the ampulla of Vater, should be checked by upper gastrointestinal endoscopy.
●The presence/absence of desmoid tumors should be checked for by palpation, CT, and/or MRI.
●Ultrasonography to check for thyroid cancer need not necessarily be performed before colectomy, but it must be incorporated into the postoperative surveillance plan, especially in female patients.
●In general, small-bowel follow-through and small-bowel endoscopy (capsule endoscopy) are not performed before proctocolectomy (colectomy) except when there are symptoms/findings (including preoperative diagnostic imaging findings) raising the suspicion of intestinal lesions.
●Because adrenal tumors develop at a low frequency, and hepatoblastomas and brain tumors develop commonly only until 2 to 3 years of age and up to adolescence, respectively, preoperative examinations for these tumorous lesions are, in general, not required.
4. Postoperative surveillance
1) Surveillance specific to proctocolectomy (colectomy)
●If there is any residual colorectal mucosa after prophylactic proctocolectomy (colectomy), regular colonoscopic examination is required, in view of the possibility of new colorectal cancer development.
●In FAP patients undergoing surgery for colorectal cancer, postoperative surveillance similar to that in sporadic colorectal cancer patients should be planned/performed.
●Long-term surveillance to monitor the development of cancer in the remaining rectum is required after IRA (CQ9).
●Usually 2 to 3 cm of rectal mucosa is left behind after stapled IPAA, and a small amount of rectal mucosa may also be left behind after hand-sewn IPAA. Therefore, long-term surveillance of the remaining rectum is required after stapled IPAA and after hand-sewn IPAA.
●Adenomas in the ileal pouch have been reported to develop in 6.7-74% of patients after IPAA26-29), and cancer has also been reported to develop30,31). Therefore, long-term surveillance is necessary.
●Pouchitis occurs in approximately 5% of patients undergoing IPAA for FAP, but the incidence is lower than that after surgery for ulcerative colitis32). The condition usually manifests with fever, diarrhea, and anemia, and if these symptoms are noted, colonoscopic examination should be performed immediately.
●In FAP patients with advanced colorectal cancer treated by curative resection, surveillance for recurrence should be performed as in patients with sporadic colorectal cancer.
2) Surveillance for extracolonic manifestations
●Surveillance should be conducted bearing in mind the possible development of desmoid tumors, which tend to develop within 2 to 3 years after colectomy, and the possible development of malignancies such as duodenal cancers.
●Extracolonic manifestations requiring treatment often develop after proctocolectomy (colectomy). A method for surveillance of the remaining rectum and for extracolonic manifestations after proctocolectomy (colectomy) is proposed, as shown in Table 433).
Table 4.
Surveillance for the Remaining Rectum after Surgery and the Major Associated Lesions in Familial Adenomatous Polyposis.
| Associated lesions | Initiation age and screening procedures |
| Remaining rectal adenomas | · Annual colonoscopy with polypectomy or ablation after IPAA
· Colonoscopy with polypectomy or ablation for the patients after IRA every 6 months (depending on age or density of adenoma) |
| Duodenal adenoma/cancer (including ampullary lesions) | Baseline upper gastrointestinal endoscopy starting at the time of colectomy or at 20-25 years old, whichever is earlier. Thereafter, upper gastrointestinal endoscopy repeated regularly depending on the severity. |
| Gastric adenoma/cancer | Upper gastrointestinal endoscopy every year (or simultaneously with examination for duodenal lesions) |
| Thyroid cancer (for females) | Thyroid ultrasound and palpation every year starting in late teenage |
| Intra-abdominal desmoid tumor | · Abdominal palpation every year
· After colectomy, abdominal and pelvic CT or MRI every 3 years for patients with a family history of desmoid tumors |
| Brain tumor | Radiologic examination every year |
| Jejunal/ileal adenoma or cancer | Data to support any recommendation are lacking. Simultaneously with radiological examinations (CT/MRI) for desmoid tumors |
Modification with ref. 33)
[Gastrointestinal tract]
●Polyps in fundic gland polyposis usually show the histological features of hyperplastic polyps, and therefore, do not constitute an indication for surgery. Gastric adenomas develop mainly in the antrum. In Japan, FAP patients are at higher risk of developing gastric cancer than the general population. Gastric surveillance should be conducted simultaneously with duodenal surveillance. (CQ10)
●The incidence of cancer in the duodenum (including ampullary tumors) is high, necessitating regular endoscopic follow-up and treatment of adenomas. (CQ11, CQ12)
●No recommended method for surveillance of the ileum/jejunum has been established yet. Jejunal/ileal cancer develops rarely. (CQ13)
[Desmoid tumors]
●Desmoid tumors often develop in the abdominal wall, mesentery, or retroperitoneum within 2 to 3 years after proctocolectomy (colectomy)26,34). Palpation and diagnostic imaging should be performed carefully and careful attention should be paid to the clinical symptoms (abdominal pain, abdominal distension, mass, gastrointestinal obstruction, etc.). (CQ14)
[Others]
●Among malignancies, attention should be paid to the development of thyroid cancer (especially in females). Neck palpation and ultrasonography should be performed once a year. (CQ15)
3) Management of families (relatives)
●It is desirable to provide genetic counseling not only to patients, but also to their relatives. (CQ16)
●Surveillance of the gastrointestinal tract, mainly of the large intestine, should be performed in first-degree relatives (parents, children, and siblings) after obtaining informed consent.
●It is indispensable to take a family history in patients with hereditary tumors, including FAP, and it is desirable to accurately describe/record the family history using a pedigree chart35,36). (Figure 12)
Figure 12.
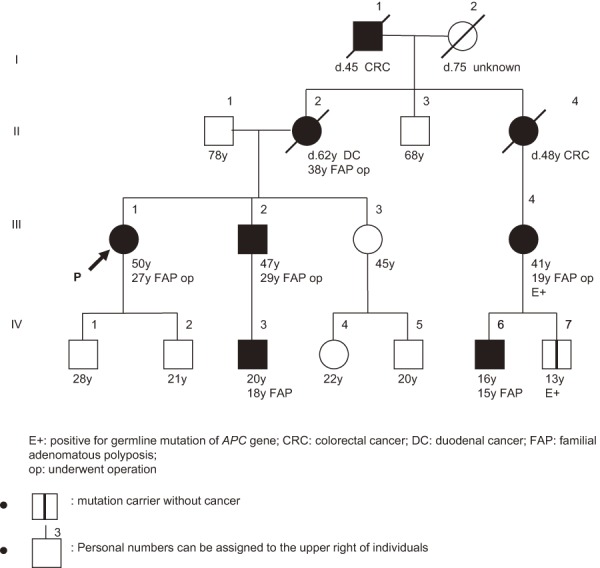
Example of description of family tree for FAP.
●If any relatives have colorectal adenomas (particularly 2 or more colorectal adenomas), the FAP diagnostic chart (Figure 2) should be followed.
●If no adenomas are detected by colonoscopy, colonic examination should be performed approximately every 3 years.
●If no adenomas are detected by multiple colonic examinations up to 35 years of age, FAP can almost definitely be ruled out.
●If genetic testing is performed, genetic counseling needs to be provided by a physician and/or specialist before and after the testing. (CQ15)
●If a patient has been diagnosed with FAP by genetic testing, his/her relatives can be diagnosed with FAP by genetic testing of a blood sample.
Clinical Questions
CQ1: Under what circumstances is genetic testing required for the diagnosis and treatment of FAP?
Recommendation category: B
Genetic testing of the APC gene is required in the following cases.
(1) When the results of genetic testing are planned to be used as reference for treatment selection or surveillance in patients clinically diagnosed with FAP.
(2) When, in a pedigree in which a pathogenic germline mutation of the APC gene has been detected, relatives of the patient wish to undergo genetic testing.
(3) When the results of genetic testing are to be used for the diagnosis of AFAP or in the differential diagnosis of MAP and PPAP.
1. Genetic testing in patients clinically diagnosed as having FAP
FAP is often diagnosed clinically, even in the absence of a family history. However, a relationship between the site of the pathogenic germline mutation in the APC gene and the number of colorectal adenomas, other associated lesions, etc., has been reported, and genetic testing is sometimes useful for treatment selection or surveillance37).
2. Genetic testing in the relatives of a patient with a known pathogenic germline mutation in the APC gene
Genetic testing allows FAP to be diagnosed in the relatives (e.g., children) of a patient with a known pathogenic germline mutation of the APC gene.
3. Diagnosis of AFAP or differential diagnosis of MUTYH-associated polyposis
Although AFAP can often be clinically diagnosed based on the number of polyps in the colorectum (less than 100) and a family history consistent with autosomal dominant inheritance, extracolonic manifestations, etc., identification of a pathogenic germline mutation in the APC gene is useful for a definitive diagnosis. If only the patient or only the sibling(s) of the patient among the family members have less than 100 colorectal adenomas, the patient or the siblings may have MUTYH-associated polyposis, and genetic testing of the APC gene, followed by, or simultaneously with, genetic testing of the MUTYH gene is useful in the differential diagnosis between the two conditions. MUTYH-associated polyposis is inherited in an autosomal recessive manner, and it is important to clarify which gene is mutated, considering risk assessment, surveillance, etc., in relatives.
No pathogenic germline mutations of the APC gene are identified in some patients who have been clinically diagnosed with FAP. According to a report from Western countries9), pathogenic germline mutations of the APC gene are identified by usual testing methods in approximately 60% of patients with classical (typical) FAP, and pathogenic APC germline mutations and biallelic mutations of the MUTYH gene are identified in 10% and 7% of patients respectively, with 20 to 99 colorectal adenomas, and in 5% and 4%, of patients respectively, with 10 to 19 colorectal adenomas. Possible reasons for the failure to detect pathogenic germline mutations of the APC gene include: (1) difficulty in the detection of APC gene alterations by the analysis method used, (2) presence of unknown causative genes for adenomatous polyposis, (3) APC mosaicism, (4) MAP, and (5) PPAP.
The “Guidelines for Genetic Tests and Diagnosis in Medical Practice” of the Japanese Association of Medical Sciences38) and the guidelines of genetics-related societies should be observed, in principle, in genetic testing. Genetic testing under these circumstances is not covered by the national health insurance program in Japan; however, genetic testing of the APC gene can be outsourced to testing companies. Approximately 2 to 3 mL of whole blood is required for the genetic testing.
CQ2: What are the points that should be kept in mind while treating attenuated FAP (AFAP)?
Recommendation category: C
Total colectomy + ileorectal anastomosis (IRA) and long-term surveillance by colonoscopy should be considered in patients with AFAP.
It is difficult to confidently diagnose AFAP based only on the number of colorectal adenomas (less than 100); however, a family history of FAP and fundic gland polyposis, duodenal adenoma, exostosis, desmoid tumor, congenital hypertrophy of the retinal epithelium (CHRPE), etc., that may be associated with FAP or AFAP serve as helpful clues for diagnosis39,40).
If the above-mentioned characteristics are unclear, differentiation from MUTYH-associated polyposis and polymerase-proofreading-associated polyposis is difficult, necessitating genetic testing for definitive diagnosis.
In AFAP, pathogenic germline mutations of the APC gene are often found in the 5’ or 3’ region, alternative splicing region (in which an exon is skipped during transcription due to the mutation), etc. of the APC gene12), while no mutations may be identified in many cases.
The mean age of colorectal cancer development is higher in AFAP patients than in typical FAP patients. Burt et al.41) reported that the mean age at the diagnosis of AFAP was 41 years and that the number of colorectal adenomas was variable, with a mean of 25 (0-470) in 120 individuals from 2 families surveyed. The mean age at onset of colorectal cancer was 58 (21-81) years, and 75% of the patients had cancer of the right colon. The cumulative incidence of colorectal cancer up to 80 years of age (69%) was lower than that in typical FAP patients (almost 100%). According to the JSCCR multicenter study, the mean age at onset of colorectal cancer was 50 years in AFAP patients, and half of the patients developed colorectal cancer by 55 years of age, which was later in life than that in typical FAP patients. Therefore, in AFAP patients without rectal cancer, IRA12) and long-term follow-up by colonoscopy41) are valid options.
CQ3: What are the points that should be considered when selecting a surgical procedure for FAP patients?
Recommendation category: B
Total proctocolectomy + ileal pouch-anal anastomosis (IPAA) is the standard surgical procedure. Total colectomy + ileorectal anastomosis (IRA) is also a valid option in patients with sparse FAP and those with a small number of rectal adenomas.
IPAA is the standard surgical procedure for typical FAP23) (Figure 13). An ileal pouch is generally constructed in a J-shape42). IPAA is largely divided into hand-sewn IPAA, in which the rectal mucosa is dissected from the dentate line and an ileal pouch is anastomosed manually to the dentate line, and stapled IPAA, in which stapling anastomosis of the surgical anal canal and ileal pouch is performed. The former procedure leaves only a small amount of rectal mucosa, but requires a greater level of skill in the operator. The JSCCR multicenter study revealed that recently, in Japan, laparoscopic surgery has been used in more than 70% of cases and that hand-sewn IPAA has been selected in an increasing proportion of cases24). (CQ6)
Figure 13.
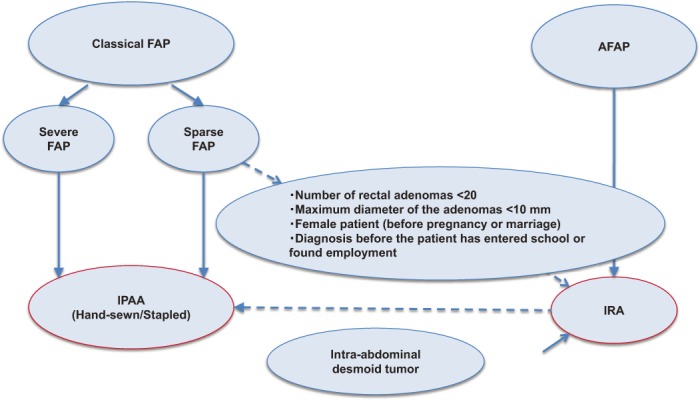
Flow-chart for choice of surgical method of prophylactic colectomy in FAP.
IRA is recommended for AFAP cases. IRA is considered not only in AFAP patients, but also in sparse-type FAP patients who have less than 20 rectal adenomas with a maximum diameter of less than 10 mm, young females who wish to become pregnant, and children/adolescents before school age/employment, etc23,43,44). A metaanalysis45) comparing IPAA and IRA revealed that IRA was better in the improvement of the stool frequency, defecation at night and pad use than IPAA; however, IPAA was better than IRA in the improvement of fecal urgency. The incidence of postoperative complications (within 30 days) was significantly higher after IPAA (23.4% vs. 11.6%). There were no differences in the postoperative sexual function, dietary restriction, long-term complications, or incidence of developing desmoid tumors between IPAA and IRA. It has been reported that the incidence of postoperative complications after IPAA is reduced with increased surgical skill level of the surgical team46).
In patients with mesenteric desmoid tumors, it is often difficult to perform IPAA, and IRA is usually selected. However, there is an opinion that if the bottom of the ileal pouch reaches the pelvic floor, IPAA can also be performed.
Total proctocolectomy + permanent ileostomy, which was used before the spread of anus-preserving surgery, is rarely performed as prophylactic surgical treatment. According to the JSCCR multicenter study, this procedure was performed in approximately only 3% of all cases including colorectal cancer24). Total proctocolectomy + permanent ileostomy should be reserved for patients with locally advanced lower rectal cancer, those with poor anal function, and those in whom the ileal pouch does not reach the pelvic floor, etc.
In patients with colorectal cancer, the choice of surgical procedure should be based on a comprehensive consideration of the degree of progression and site of the cancer (See Chapter I; 3. Treatment; 2) Treatment of colorectal cancer).
CQ4: Is temporary ileostomy required in cases undergoing IPAA for FAP?
Recommendation category: None
Temporary ileostomy is not required in all cases, but its need should be determined on a case-by-case basis considering its advantages and disadvantages.
A recent meta-analysis of patients treated by IPAA revealed that patients in whom temporary ileostomy was performed had a lower incidence of anastomotic leakage, but higher incidence of anastomotic stricture and bowel obstruction47). It has been reported that temporary ileostomy can be avoided under the following circumstances: in patients with (1) stapling anastomosis, (2) no anastomotic tension, (3) complete anastomosis, (4) sufficient hemostasis, (5) no anastomotic air leak, and (6) no evidence of malnutrition, infection, anemia or regular steroid use48). Anastomotic leakage after IPAA may cause an important long-term complication, namely, pouch failure. Anal dysfunction and poor expansion of the ileal pouch have been reported as possible causes of pouch failure. From these points of view, it is considered that temporary ileostomy may be useful in the prevention of anastomotic leakage and pelvic abscess, or suppress the degree of these adverse events as much as possible after IPAA. However, it should be kept in mind that the above studies included both patients with ulcerative colitis and those with FAP, the latter accounting only for a small proportion of the subjects.
Studies of IPAA conducted on only FAP patients have reported that temporary ileostomy is performed in most patients, except some of those undergoing stapled IPAA49,50). In a study on the usefulness of temporary ileostomy in FAP patients aged less than 20 years old, patients in whom temporary ileostomy was not performed showed favorable long-term defecation control, but had significantly higher incidence of anastomotic leakage within 30 days of surgery (17.2% vs. 0%, P = 0.002) and a higher reoperation rate (20.7% vs. 4.6%, P = 0.02)51). However, most subjects included in this study underwent stapled IPAA, and further studies of temporary ileostomy in patients undergoing hand-sewn IPAA are required.
The JSCCR multicenter study showed that temporary ileostomy was performed in 55% of patients who had undergone IPAA25).
A systematic review of the closure of temporary ileostomy52) revealed that closure was safe, but that 16.5% of all subjects had postoperative complications, including bowel obstruction in 7.6% (reoperation in 2.9% of all cases), anastomotic leakage in 2.0%, wound infection in 4.0%, and late complications such as incisional hernia in 1.9% and bowel obstruction in 9.4%.
Considering the above, temporary ileostomy can be avoided in selected FAP patients undergoing IPAA, but it is not easy to clearly determine its indications. Therefore, it is practical to determine the need for temporary ileostomy on a case-by-case basis, taking into consideration its advantages and disadvantages.
CQ5: At what age is prophylactic proctocolectomy (colectomy) recommended to prevent colorectal cancer in FAP patients?
Recommendation category: B
In general, the surgery is performed when the patients are in their 20’s; however, the decision should be made after comprehensively considering the patient sex, density of colorectal adenomas, presence/absence of malignant transformation of the adenomas, associated lesions, the patient’s social background, etc.
The most important considerations in deciding the timing for prophylactic (procto)colectomy in FAP patients are: (1) cumulative prevalence of colorectal cancer2); (2) density of the adenomas53); (3) size and morphology of the adenomas; (4) age at death, age at cancer onset, and presence/absence of desmoid tumors in members of the pedigree54); (5) germline mutation site in the APC gene55); (6) educational, work, and other environments of the patient56); (7) fertility57) and presence/absence of male sexual dysfunction58) after IPAA; (8) presence/absence of gastrointestinal symptoms, such as diarrhea, abdominal pain and melena; and (9) the histopathological findings of the tumor. Considering the prevalence of colorectal cancer, it is recommended that some classic FAP patients should undergo surgery while in their teens, and that most FAP patients should undergo surgery while they are in their 20’s59,60).
According to the JSCCR multicenter study, the cumulative incidence of developing colorectal cancer (excluding intramucosal cancer) was approximately 1% in patients aged 20 years and increased to 9.6% and 21.4% in AFAP and classic FAP patients aged 30 years, respectively. Thus, the incidence is higher in classic FAP patients (Attachments: I. Familial adenomatous polyposis; Attachment Table 3: Cumulative incidence of developing colorectal cancer and duodenal adenoma).
CQ6: Is laparoscopic surgery useful for FAP?
Recommendation category: C
The decision to undertake laparoscopic surgery for FAP should be made after a full informed consent is obtained from the patient, according to the skill of the operator at the institution.
Recently, laparoscopic surgery has been used in an increasing proportion of patients undergoing IPAA or IRA (IPAA: 23-53%; IRA: 58-62%)61-64). According to previously published reports, laparoscopic surgery takes a longer time, but there are no differences between laparoscopic and open surgery in the incidence of postoperative complications, mortality, reoperation rate or readmission rate63); furthermore, the laparoscopic approach yields better esthetic outcomes with less intraoperative bleeding. In addition, laparoscopic surgery was also reported to be associated with a lower incidence of postoperative bowel obstruction, due to lower risk of occurrence of intra-abdominal adhesions and a lower incidence of postoperative fertility impairment in females61). According to the JSCCR multicenter study conducted recently, laparoscopic surgery has been used in more than 70% of cases24), and among the subjects of this study, the laparoscopic approach had been used in 74 out of 171 (43%) patients undergoing IPAA and 52 out of 85 (61%) patients undergoing IRA25).
Concerning the short-term outcomes of laparoscopic surgery, the operation time is long; however, safety is secured. The decision to undertake laparoscopic surgery for FAP should be made after a full informed consent is obtained from the patient, according to the skill of the operating surgeon at the institution.
CQ7: Does IPAA have adverse effects on fertility, pregnancy, and delivery in female patients with FAP?
Recommendation category: None
IPAA may be associated with postoperative reduction in fertility, but has little adverse effect on pregnancy or delivery.
A study involving 58 female Danish patients with FAP65) showed that their fertility rate was 90%, equivalent to that in the general population. A study involving 162 female European patients with FAP demonstrated that the fertility rate in FAP patients who had not undergone any surgery was equivalent to that in the general population. In addition, while the fertility rate of FAP patients who had undergone IRA was also equivalent to that in the general population, the fertility ratio was reduced to 0.46 in FAP patients who had undergone IPAA57). On the other hand, a study involving 138 Dutch patients with FAP reported that fertility was not related to the surgical procedure, but to the age at first surgery66).
The reduction in fertility after IPAA is thought to be caused by postoperative adhesions. Oresland et al.67) reported that hysterosalpingography after total proctocolectomy revealed adhesion of the fallopian tube to the pelvic wall in 48% of the cases, unilateral obstruction of the fallopian tube in 43% and bilateral obstruction of the fallopian tubes in 10% of the cases.
A study in which both patients with FAP and those with ulcerative colitis were included, reported that the fertility was significantly higher after laparoscopic IPAA than after IPAA via open surgery68). However, there have been no prospective studies including only FAP patients.
Studies including both patients with FAP and those with ulcerative colitis have reported that pregnancy and transvaginal delivery are safe after IPAA69,70). However, the possibility of anal sphincter muscle damage and nerve damage of the pelvic floor muscles after perineal incision must be considered during transvaginal delivery after IPAA.
CQ8: Is there any effective pharmacotherapy for the adenomas in FAP patients?
Recommendation category: C
NSAIDs have been attempted for colorectal adenomas and duodenal adenomas. Although many studies have reported that NSAIDs reduced polyp number, it is unclear whether they are useful in suppressing the development of new adenomas.
Many studies have evaluated the efficacy of sulindac, one of the NSAIDs, for the control of colorectal adenomas in FAP patients. Treatment with sulindac (150-300 mg/day) for 6 weeks to 98 months reduced the number of colorectal adenomas, or the number of rectal adenomas after total colectomy, by 50% or more, whereas 2-year treatment with 150-300 mg/day of sulindac failed to suppress the development of new adenomas71).
High-dose (800 mg/day) treatment with celecoxib, one of the selective cyclooxygenase-2 (Cox-2) inhibitors, for 6 months reduced the number of colorectal adenomas by 28% in FAP patients72). Celecoxib should be administered at a high dose for a long period of time for suppressing the development of colorectal adenomas in FAP patients. Rofecoxib, another of the selective Cox-2 inhibitors, was also reported to reduce the number of rectal adenomas by approximately 7% after total colectomy73). However, because long-term treatment with rofecoxib resulted in a high incidence of cardiovascular adverse events74), its use for the prevention or treatment of adenomas is not recommended.
So far, no useful pharmacotherapy to suppress the development of new colorectal or duodenal adenomas has been reported.
CQ9: How should the risk of rectal cancer development be managed after total colectomy + ileorectal anastomosis (IRA)?
Recommendation category: C
Long-term surveillance for the development of cancer in the remaining rectum is necessary.
Long-term follow-up after IRA has revealed that 24-43% of patients develop cancer in the remaining rectum75,76). During a 20-year period after IRA, the rectum had to be resected in 10% of patients with AFAP, 39% of patients with sparse FAP, and 61% of patients with profuse FAP37).
With advances in surgical techniques, IPAA has been used in an increasing proportion of cases22-24), and the use of IPAA in patients with a greater number of risk factors for rectal cancer has reduced the proportion of patients undergoing proctectomy after IRA from 40 to 13%, and has also reduced the cumulative incidence of cancer development in the remaining rectum after IRA61,77,78).
CQ10: How should gastric lesions be managed in FAP patients?
Recommendation category: C
FAP patients in East Asia have higher risk of gastric cancer than the general population, necessitating long-term endoscopic surveillance.
Approximately 50% of FAP patients develop multiple protrusive polyps in the fundus to the body (fundic gland polyposis). Foveolar-type adenomas (according to the WHO classification) and pyloric gland adenomas are known to develop in the background of fundic gland polyposis, and although rare, development of invasive cancer has also been reported. Particularly, large polyp clusters, with some showing dysplastic or malignant changes, indicate endoscopic resection13,79). Gastrectomy should not be performed for fundic gland polyposis. Solitary or sporadic, depressed- or elevated-type adenomas develop in the antrum80). From the above, adenomas measuring 1 cm or more in diameter, as well as sporadic adenomas not associated with FAP, are relative indicators of endoscopic resection, considering the risk of malignant transformation. While the incidence of gastric cancer in FAP patients has been reported to be equivalent to that in the general population in Western countries81), it has been reported to be 3-7 times higher in FAP patients than in the general population in East Asia82,83). It is desirable to perform upper gastrointestinal endoscopy once a year (or simultaneously with surveillance for duodenal adenoma surveillance).
CQ11: How should duodenal adenomas (excluding those of the ampulla) be managed in FAP patients?
Recommendation category: C
No consensus has been reached on the treatment or surveillance for duodenal adenomas, but the Spigelman classification can be referred to for optimal treatment and surveillance.
After excluding colorectal cancer, which accounts for death in the majority (61-69%) of FAP patients, duodenal cancer (including ampullary cancer) ranks as the second most common cause of death after desmoid tumors, and accounts for death in approximately 3% of FAP patients2,84). The relative risk of duodenal cancer in FAP patients as compared with that in the general population is 250-330.881,82). The cumulative incidence of duodenal cancer by 57 years of age is estimated to be approximately 4.5%85). Duodenal adenomas are seen in 30-90% of FAP patients86-88), and the prevalence of adenomas increases after 40 years of age, eventually reaching 90%87,88). Duodenal adenomas grow extremely slowly87,89); however, regular endoscopic surveillance/treatment is necessary. The JSCCR multicenter study found that the cumulative incidence of duodenal adenomas by the age of 50 years was 39.2%, and significantly higher in classic FAP patients than in AFAP patients (42.5% vs. 23.5%)90). There exists a clinicopathological classification of duodenal adenomas, called the Spigelman classification91). In the Spigelman classification, the number and maximum diameter of duodenal adenomas are assessed by endoscopy, and biopsy (Figure 14) is used to evaluate the histology and severity of dysplasia. Over time, some modifications have been introduced to this classification (modified Spigelman classification)91) (Figure 15). (Side Memo 5: Changes in the evaluation methods for Spigelman classification)
Figure 14.
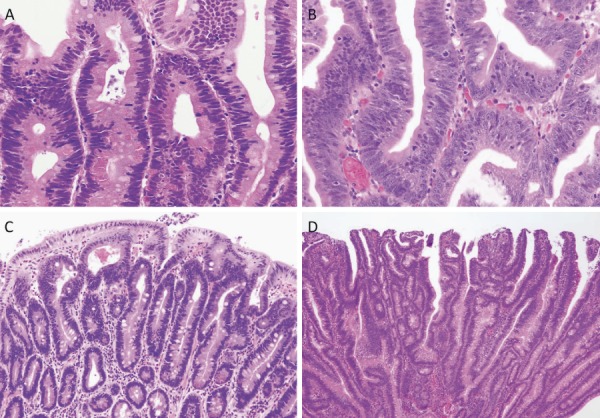
Histology of FAP-associated duodenal adenomas. A: Low-grade adenoma: The tumor glands are rather uniform and the adenomatous epithelial cells show basally oriented, elongated nuclei. B: Intramucosal carcinoma: Tumor glands show significant irregularity, nuclear stratification, and occasional prominent nucleoli. Note that high-grade dysplasia in the Spigelman classification includes non-invasive intramucosal carcinoma in the Japanese classification. C: Tubular adenoma: This lesion shows a relatively regular tubular architecture. D: Tubulo-villous adenoma: This lesion partially exhibits villous architecture, composed of fibrovascular cores lined by dysplastic epithelium.
Figure 15.
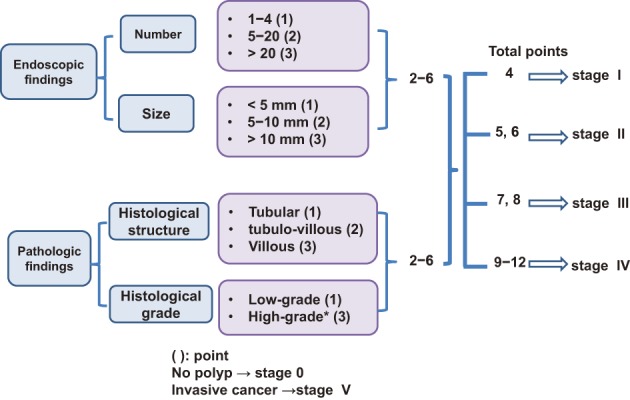
Classification of duodenal adenomas by the modified Spigelman classification.
Direct-view endoscopy and side-view endoscopy are used to diagnose duodenal adenomas. A study of 37 Dutch patients with FAP showed that the use of narrow-band imaging increased the number of duodenal adenomas detected, but did not affect the results of classification according to Spigelman classification93).
Endoscopic treatments of duodenal adenomas include snare resection, electrocautery, and argon plasma coagulation. Endoscopic electrocautery should be used for adenomas classified as Spigelman stage I/II. Endoscopic or transduodenal resection is not sufficient for patients with many adenomas85). It was reported that endoscopic complete resection of adenomas classified as Spigelman stage II/III was associated with a high incidence of complications and a recurrence rate of 50-100%86). To date, no clinical trials have been conducted for comparing endoscopic treatment and follow-up of duodenal lesions in FAP patients.
No consensus has been reached on the interval of testing, but it is recommended that testing be performed every 4 to 5 years for cases with stage 0, every 2 to 5 years for cases with stage I, every 2 to 3 years for cases with stage II, and every 6 months to 2 years for cases with stage III duodenal adenomas85,88). Assessment of the indication for surgery or half-yearly to yearly surveillance by a specialist is recommended for patients with stage IV high-grade adenomas, severe adenomatosis, etc. Pancreaticoduodenectomy (PD), pylorus-preserving pancreaticoduodenectomy (PPPD), or pancreas-sparing duodenectomy (PSD) should be considered for patients with stage IV adenomas, because malignant transformation occurs in 7-36% of cases85,94).
Among surgical procedures, PD or PPPD is generally selected, and it was reported that PSD was performed in 13 Danish patients with FAP between 1999 and 2010. Six (46%) of these patients developed postoperative complications, and of these, 3 had anastomotic leakage, but recovered with conservative treatment95). According to a report from the Netherlands, 43 out of 1,066 FAP patients underwent duodenectomy (PSD was performed in 22 of these), and PSD has been the first-line surgical procedure for “prophylactic duodenectomy” since 199996). However, in Japan, PSD is performed in only some hospitals for FAP patients. Management of duodenal adenomas according to the modified Spigelman classification is shown in Figure 16.
Figure 16.
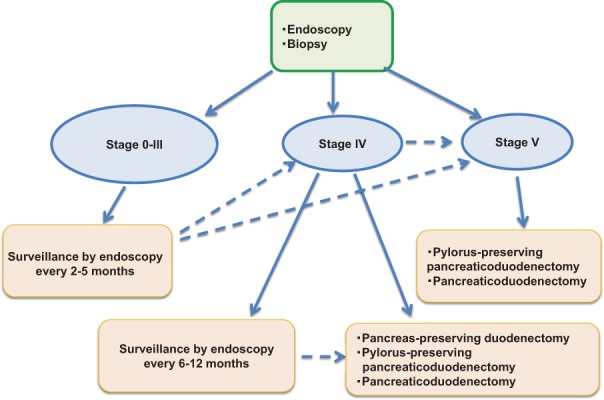
Surveillance of duodenal adenomas based on the modified Spigelman classification.
Side Memo 5
■Changes in the evaluation methods for the Spigelman classification
The Spigelman classification is a staging system for duodenal adenomas associated with FAP that was proposed in 198991). The polyp number, maximal diameter, histology and severity of dysplasia are assessed on a scale ranging from 1 to 3, and the total score is used to determine the disease stage. In the Vienna classification of 200097), the grading of the severity of dysplasia was changed from 3 levels, that is, mild, moderate, and severe, to 2 levels, namely, low-grade and high-grades, and a modified classification was proposed, in which 1 and 3 points are given to the low and high grades, respectively92). Recently, the National Comprehensive Cancer Network (NCCN) Guidelines (Genetic/Familial High-Risk Assessment: Colorectal V.2.2015) proposed a classification that was a simpler form of the Spigelman classification, or the modified Spigelman classification. This classification consists of stages 0 (no adenomas), I (1 to 4 tubular adenomas measuring 1-4 mm in diameter), II (5-19 tubular adenomas measuring 5-9 mm in diameter), III (20 or more adenomatous lesions measuring 1 cm or more in diameter), and IV (dense or high-grade adenomas). No prospective studies of the validity of surveillance or treatment based on these staging systems have been conducted, and this issue needs to be addressed in the future.
CQ12: How should ampullary tumors (adenomas/cancers) be managed in FAP patients?
Recommendation category: C
Endoscopic or surgical treatment should be selected for ampullary tumors according to the clinical condition and symptoms.
Approximately 50% of FAP patients develop ampullary tumors98,99). Some AFAP patients also develop ampullary tumors100). The relative risk of ampullary cancer in FAP patients as compared to that in the general population was reported to be 123.791). Endoscopic ampullectomy or transduodenal ampullectomy101,102) is indicated for tumors localized to the papilla. The former has often been adopted, with recent advances in colonoscopic treatment techniques.
Electrocautery of periampullary lesions (within 2 cm of the papilla), including those of the papilla, has been reported to be safe and effective103), while it has also been suggested that aggressive treatment is not recommended, because the lesions were found to remain benign over long-term observation for more than 10 years104). Ma et al.105) retrospectively investigated the data of 26 FAP patients who underwent endoscopic ampullectomy in the United States between 1990 and 2010. Complications in these patients included pancreatitis (19.2%), abdominal pain (7.6%), and bleeding (3.8%). Of the 24 patients who could be followed up, 14 (58.3%) had local recurrence, and the authors called attention to this problem. Gluck et al.106) reported that endoscopic follow-up of 80 FAP patients for an average of 7.2 years revealed ampullary tumors in 38 patients (47.5%), of whom 10 had advanced adenomas (tumor diameter 10 mm or more, villous type, high-grade dysplasia), and that endoscopic ultrasonography (EUS) is important for their diagnosis. In addition, 15 underwent endoscopic ampullectomy, of which 2 eventually underwent surgery for recurrent lesions. Regarding surgery, if there is a periampullary lesion that is difficult to treat endoscopically, pancreas-sparing duodenectomy (PSD)107) may be selected and if any evidence of malignant change is noted, pancreaticoduodenectomy (PD), pylorus-preserving pancreaticoduodenectomy (PPPD), etc., should be selected.
CQ13: How should jejunal/ileal tumors be managed in FAP patients?
Recommendation category: C
Small-bowel endoscopy and capsule endoscopy have been attempted, but no consensus has been reached on the examination or treatment of jejunal/ileal tumors in FAP patients.
Jejunal/ileal adenomas develop in 60-75% of FAP patients108-111). A study using capsule endoscopy showed that patients with duodenal adenomas also tend to have jejunal/ileal adenomas109,112). Most of these adenomas measure 10 mm or less in diameter111,113,114). Studies on relatively large numbers of patients have shown that the number of adenomas tends to be higher in the jejunum and lower in the ileum109,114). In principle, because jejunal/ileal cancer develops rarely115), endoscopic resection is not indicated for jejunal adenomas. However, how jejunal/ileal adenomas should be examined and treated remains to be established, and this issue needs to be addressed in the future116).
CQ14: What are the management strategies that should be used for desmoid tumors in FAP patients?
Recommendation category: C
No consensus has been reached concerning the treatment of desmoid tumors in FAP patients. Pharmacotherapy, surgery, conservative treatment (follow-up), etc., could be selected according to the site and severity of the tumors.
Management strategy for desmoid tumors should be selected taking into consideration the characteristics of the desmoid tumors, types of treatment available, tumor stage, etc.
1. Characteristics
Desmoid tumor is a type of fibroma, which does not metastasize, but tends to show invasive growth. Desmoid tumors are seen in 8-20% of FAP patients54,83,117,118), intra-abdominal desmoid tumors accounting for 70% of all cases119). They often develop in the abdominal wall, mesentery or retroperitoneum after (procto)colectomy (in particular, within 2 to 3 years)26,34), and when developing intra-abdominally (including in the retroperitoneum), they can cause bowel obstruction, perforation, abscess formation, ureteral obstruction, etc., often making treatment difficult. The mortality rate of FAP patients developing desmoid tumors is reported to be 0-14%26,83,118,120,121).
2. Types of treatment
Desmoid tumors should be treated taking into account their characteristics including: (1) spontaneous decrease of size or size stabilization120,122,123), and (2) recurrence that has been reported to occur in 10-68% of cases after resection117). Pharmacotherapy (including chemotherapy), surgical resection, radiation therapy, etc., have been used for the treatment of desmoid tumors. In FAP patients, desmoid tumors are often adjacent to the intestine, such as those in the mesentery, and radiation therapy is generally not recommended, because it can cause bowel injury and is poorly effective124).
Treatment with sulindac (300 mg/day), which is one of the NSAIDs, and tamoxifen (40 to 120 mg/day) or toremifene (180 mg/day), which are antiestrogens, could be selected for large and/or rapidly growing intra-abdominal or abdominal-wall desmoid tumors125,126).
Both sulindac and antiestrogens have been reported to have a limited effect in reducing the tumor size, but they suppress tumor growth127-129). Recently, the efficacy of a tyrosine kinase inhibitor, imatinib, has also been examined. Desurmont et al.128) reported that imatinib reduced the tumor size or stabilized the tumor size in 36% of treated cases. On the other hand, Chugh et al.130) reported a 1-year progression-free rate of 66% in inoperable desmoid tumor patients treated with imatinib, but reduction of the tumor size occurred in only 3% of the patients. Therefore, at present, the efficacy of imatinib remains to be clearly established.
Regarding cytotoxic chemotherapy, high response rates were reported with a combination regimen of doxorubicin (DOX) plus dacarbazine (DTIC)131). In Japan also, DOX + DTIC therapy has been found to be effective132). In addition to DOX + DTIC therapy, methotrexate (MTX) plus vinblastine (VBL) has also been reported to be effective133).
Desurmont et al.128) compared the response rates of intra-abdominal desmoid tumors to various pharmacotherapies. They found that the response rates were 77% to treatment with cytotoxic anticancer drugs, 50% to treatment with sulindac + tamoxifen, 40% to treatment with tamoxifen, 36% to treatment with imatinib, and 28% to treatment with sulindac. Thus, the response rate of intra-abdominal desmoid tumors was the highest to treatment with cytotoxic anticancer drugs, and they concluded that cytotoxic anticancer drugs could be the first-line treatment. However, it has not been clearly established in which type of intra-abdominal desmoid tumors cytotoxic anticancer drugs should be used as the first-line treatment.
Extra-abdominal desmoid tumors have been reported to show high recurrence rates after resection (20-25%), although the incidence of postoperative complications is low. Because recurrence after resection may not only be caused by incomplete resection, but also possibly by new tumor development at the site of incision, excessive peritumoral resection should be avoided134). Although surgery should be considered for bowel obstruction due to intra-abdominal desmoid tumors, it may not be successful due to the difficulty of resection or the necessity for massive intestinal resection129). Smith et al.135) reported the absence of any difference in survival between patients treated by complete resection and patients not treated by complete resection, including by-pass cases.
3. Treatment of intra-abdominal desmoid tumors based on the Church classification
A staging system for intra-abdominal desmoid tumors has been developed by reference to the classification of Church et al.121) (Table 5). Although no prospective studies have been conducted, options include follow-up or use of NSAIDs for stage I tumors, surgery and NSAIDs + tamoxifen, if possible, for stage II tumors, NSAIDs + tamoxifen + chemotherapy for stage III tumors, and chemotherapy or by-pass surgery for stage IV tumors (Figure 17). According to one report, mortality was 0 in stage I/II patients and 15% and 44% in stage III and IV patients, respectively. Stent placement is recommended for ureteral obstruction.
Table 5.
Staging System for Intra-Abdominal Desmoid Tumors according to Church’s Classification ref. 121).
| I | II | III | IV | |
|---|---|---|---|---|
| Maximal size | <10 cm | 10-20 cm | >20 cm | |
| Growth speed | No growth within 6 months | Growth within 6 months | More than 50% increase in maximal diameter within 3 months | |
| Uretic obstruction | No | Yes | ||
| Bowel obstruction | No | Yes | ||
| Sensation of tumor | No | Yes | ||
| Pain | No | Yes | ||
| Restriction of daily life | No | Yes | ||
| Hospitalization | Unnecessary | Necessary | ||
Figure 17.
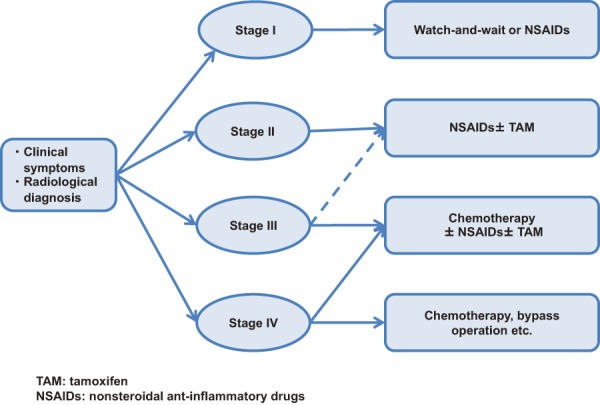
Treatment plan according to the staging system for intra-abdominal desmoid tumors.
CQ15: What malignancies other than those of the gastrointestinal tract should careful attention be paid to in FAP patients?
Recommendation category: None
Thyroid cancer, adrenal cancer, hepatoblastoma, brain tumor, etc., are known to develop in FAP patients, and particularly, many cases of thyroid cancer have been reported. The usefulness of screening tests and surveillance for these tumors has not yet been confirmed.
Thyroid cancer is reported to develop in 1-6.1% of FAP patients83,136,137), with papillary carcinoma accounting for most of the cases. Papillary thyroid cancer develops more commonly in females, with a female:male ratio of 44:1138). The relative risk of thyroid cancer in female FAP patients as compared to the general population is reported to be 23 to 16083,138,139). Thyroid cancer in FAP patients often shows a characteristic histology, i.e., the cribriform-morula variant140,141), and diagnosis of FAP is sometimes made during treatment of the thyroid cancer. Because reported incidences of multiple thyroid cancers and bilateral thyroid cancer are as high as 28.6-69%142-144) and 42-67%142,145), respectively, one report has recommended subtotal thyroidectomy140). However, papillary thyroid cancer associated with FAP has a favorable prognosis83,146), and the surgical procedure should be carefully selected. One report recommends ultrasonography in addition to palpation as a screening examination for thyroid cancer in FAP patients147).
Some FAP patients develop brain tumors (Turcot syndrome, type 2). The incidence is 2.4 times higher in females than in males. Medulloblastoma is the most common tumor (60%), while other tumors such as astrocytoma and ependymoma have also been reported148). The reported relative risks of brain tumors, overall, and of medulloblastoma in female FAP patients as compared to the general population are 7 and 92, respectively149). Medulloblastoma commonly develops in childhood to young adulthood.
Adrenal tumors develop in 7.4-13% of FAP patients8,150,151). The reported relative risk of adrenal tumors in FAP patients as compared to the general population is 2.3 to 12.58,150,151). These tumors are often accidentally detected by CT. In a study by Will et al. of 30 FAP patients145), 2 (6%) had bilateral adrenal tumors, the age at diagnosis ranged from 26 to 69 years, and there was no gender difference in the incidence. Surgery is indicated for patients with suspected hormone-producing tumors or malignant transformation of the tumor. The incidence of malignant adrenal tumors is unknown in FAP patients.
It is estimated that 0.42-0.75% of children with FAP develop hepatoblastoma152). The peak age at onset is approximately 3 years, and the relative risk in FAP patients as compared to the general population is reported to be 176 to more than 420153,154). Imaging examinations, including abdominal ultrasonography are used for the diagnosis, and 90% of the patients have high levels of α-fetoprotein (AFP)155). CHRPE is known to be frequently associated with hepatoblastoma, and FAP patients with a family history of hepatoblastoma are known to be at a higher risk156). Surgical resection or chemotherapy should be selected for treatment. Patients in whom the tumor is detected early have a favorable prognosis after complete resection157,158), and one report159) recommends surveillance at the peak age of onset.
CQ16: What are the points that should be kept in mind while providing genetic counseling to FAP patients/their families?
Recommendation category: B
It is necessary to provide information about FAP and psychosocial support while providing genetic counseling for FAP patients and their families (relatives), including at-risk individuals.
Genetic counseling should be provided to FAP patients, irrespective of whether genetic testing has been performed or not. Genetic testing should be performed mainly by a physician at a time and in a place where the patient can talk calmly, in accordance with the “Guidelines for Genetic Tests and Diagnosis in Medical Practice” of the Japanese Association of Medical Sciences (February 2011), guidelines of the Japanese Society for Familial Tumors, “Ethical Guidelines for Human Genome/Gene Analysis Research,” etc.
In genetic counseling for FAP, comprehensive information about the disease should be provided, including the fact that first-degree relatives of affected individuals have a 50% chance of inheriting the mutation, and that if left untreated, almost a 100% of APC mutation carriers develop colorectal cancer. Furthermore, the significance of genetic testing as one of the options, and its methods, limitations, costs, etc., should be explained, to help the patients and their families make an independent choice about genetic testing. Genetic counseling should be provided not only before and after genetic testing, but also continuously, as necessary.
Genetic or diagnostic testing (colonoscopic examination) of relatives of FAP patients frequently involves minors. When genetic or diagnostic testing is performed in minors, it is desirable to obtain not only the consent of the legal representatives of the subjects, but also informed consent from the subjects themselves after providing them with an explanation according to their level of understanding.
The results of genetic testing of relatives may differ among siblings (mutation carriers and unaffected individuals). Some individuals not carrying the pathogenic mutation have a guilty conscience (survivor guilt), e. g., a feeling that “I alone am safe and I am sorry,” and psychological support for family members not carrying the pathogenic mutation is also sometimes required as part of genetic counseling.
Various tumorous and non-tumorous lesions develop in FAP, necessitating collaboration with multiple departments. It is desirable to provide long-term social, economic, and psychological support as a medical team.
Chapter II: Lynch Syndrome
1. Outline
●Lynch syndrome is a hereditary autosomal dominant disease, mainly caused by germline mutations in one of the mismatch repair genes (Side Memo 6: Mismatch repair function; Appendix: II. Method for describing genomic variants).
●These patients and their families are at an elevated risk of developing various malignancies, including colorectal cancer and endometrial cancer.
[Clinical features]
●Colorectal cancers in Lynch syndrome are characterized by early age at onset and occurrence of multiple tumors (synchronous/metachronous), and preferentially develop in the right colon. The frequency of poorly differentiated adenocarcinoma is higher among Lynch syndrome-associated colorectal cancers than among sporadic colorectal cancers. The histological features of colorectal cancer associated with Lynch syndrome include the presence of tumor-infiltrating lymphocytes, a medullary growth pattern, mucinous/signet-ring cell differentiation, and Crohn’s-like lymphocytic reaction153,159-161). (CQ17, CQ18)
●A variety of other Lynch syndrome-associated (malignant) tumors can also develop besides colorectal cancer, including endometrial cancer, ovarian cancer (CQ19), gastric cancer, small-bowel cancer, bile duct cancer, pancreatic cancer, renal pelvic/ureteral cancer, brain tumor, and skin tumor (CQ17). Recently, breast cancer, bladder cancer162), and prostate cancer163) have also been reported to develop in association with Lynch syndrome.
●The risk of development of Lynch syndrome-associated tumors varies depending on the causative gene, the type of mutation, environmental factors, etc.153,164-170) (Table 6)
Table 6.
Cumulative Lifetime Risk of Lynch Syndrome-Associated Neoplasms (Up to the Age of 70 Years).
| Malignant tumors | Cumulative risk (%) | |
|---|---|---|
| Colorectal cancer | 54%-74% | (Male) |
| 30%-52% | (Female) | |
| Endometrial cancer | 28%-60% | |
| Stomach caner | 5.8%-13% | |
| Ovarian cancer | 6.1%-13.5% | |
| Small-bowel caner | 2.5%-4.3% | |
| Bile duct cancer | 1.4%-2.0% | |
| Pancreatic cancer | 0.4%-3.7% | |
| Renal pelvic/ureteral cancer | 3.2%-8.4% | |
| Brain tumor | 2.1%-3.7% | |
| Sebaceous gland tumor | Unknown | |
In addition, some individuals carrying the disease-causing genetic mutation remain totally asymptomatic throughout their lives.
[Major causative genes]
●Germline mutations in any of the following genes:
MLH1 gene on chromosome 3
MSH2 and MSH6 genes on chromosome 2
PMS2 gene on chromosome 7
[Mode of inheritance]
●Autosomal dominant inheritance
[Mechanism of malignant transformation] (Figure 18)
Figure 18.
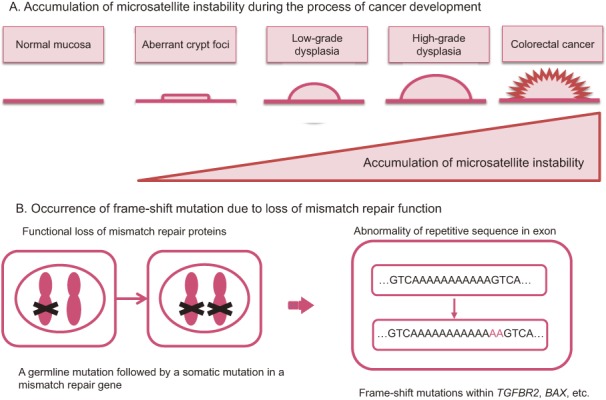
Possible mechanisms underlying colorectal cancer development in patients with Lynch syndrome.
●In Lynch syndrome, a pathogenic germline mutation is present in one allele of one of the mismatch repair genes, and an acquired mutation (or methylation of the promoter region) in the other (wild-type) allele impairs mismatch repair function. As a result, deviations in the number of tandem repeats (microsatellite instability) often occur in the tumors (microsatellites are simple repetitive sequences in the genome.). The genes involved in tumor suppression (such as TGFBR2), cell proliferation, DNA repair (such as MSH3 and MSH6), apoptosis (such as BAX), etc., contain repetitive sequences in the coding regions and mutations tend to develop in these regions.
●The adenoma-carcinoma sequence has been suggested to underlie the development of colorectal cancer in patients with Lynch syndrome, as in cases of sporadic colorectal cancer (Figure 18); however, the precise mechanisms of carcinogenesis in LS-associated colorectal cancer have not yet been fully clarified.
[Incidence]
●Lynch syndrome has been estimated to account for 2-4% of all colorectal cancer cases159,171).
●The incidence in the overall Japanese population is unknown.
Side Memo 6
■Changes in the nomenclature of Lynch syndrome
In 1966, Henry T. Lynch et al.172) reported families in which colorectal cancer and endometrial cancer were more frequently encountered than in the general population. In 1984, Boland et al.173) classified the conditions into two categories; Lynch syndrome I, characterized by an increased risk of development of only colorectal cancer, and Lynch syndrome II, characterized by increased risk of development of not only colorectal cancer, but also cancer of other organs in the family members. These two conditions have come to be collectively called Lynch syndrome or hereditary non-polyposis colorectal cancer (HNPCC). In 1990, the terms were unified as HNPCC, and in a workshop of the International Collaborative Group on HNPCC (ICG-HNPCC) held in Amsterdam, standardized Amsterdam criteria I174) were proposed to collect HNPCC pedigrees. Causative genes have been reported one after another since 1993. As a result, it had been found that there are many families that carry a causative gene mutation, but do not meet the Amsterdam criteria I, and many others that meet the Amsterdam criteria I, but in which no causative genes can be identified. Therefore, in 1998, the revised Amsterdam criteria (Amsterdam criteria II) (Table 7), developed taking into consideration the occurrence of malignant tumors other than colorectal cancer, such as endometrial cancer, were proposed, for collaborative research on HNPCC175). Thereafter, the appropriateness of the term HNPCC was repeatedly discussed, and it came to be thought that the term is inappropriate considering the characteristics of the disease, i.e., the occurrence also of various malignant neoplasms other than colorectal cancer. Currently, the term Lynch syndrome, named after - Dr. Lynch, is commonly used.
■Mismatch repair function
Cells are equipped with the function of detecting and repairing mismatches that occur during DNA replication. Mismatch repair dysfunction increases the frequency of mispairs and insertions/deletions of simple repeat sequences by 10- to 1,000-fold, which results in microsatellite instability (Side Memo 8: Method for MSI testing and evaluation of the results [p.58]).
■Recent research on the causative genes of Lynch syndrome
(1) Germline epimutation
Recently, it was found that epimutations are involved in tumorigenesis in some cases of Lynch syndrome. Epimutations refer to modifications of molecules involved in gene expression, such as aberrant DNA methylation, that can causes changes in gene expression without alterations of the DNA sequence. Although rare, aberrant germline methylation (hypermethylation) of the promoter region of the MLH1 gene has been reported as a cause of Lynch syndrome176).
(2) EPCAM deletion
The EPCAM (TACSTD1) gene is located upstream of the MSH2 gene, adjacent to it, and deletion of its 3’ region (sequences necessary for transcription termination) can cause Lynch syndrome. The deletion allows the EPCAM and MSH2 genes to be continuously transcribed, thereby inducing aberrant methylation of the promoter region of the MSH2 gene and loss of expression of the MSH2 protein. The risk of colorectal cancer development in individuals with EPCAM deletions is comparable to that in patients with Lynch syndrome caused by mutations in the MSH2 gene, although the risk of endometrial cancer development is lower in these individuals177). EPCAM deletions have been reported to account for 1-3% of Lynch syndrome cases178).
Table 7.
Amsterdam Criteria II ref. 175).
| At least three relatives must have a Lynch syndrome-associated cancer (colorectal, endometrium, small bowel, ureter, or renal pelvic cancer); all of the following criteria should be met: |
| • One must be a first-degree relative of the other two; |
| • At least two successive generations must be affected; |
| • At least one should have been diagnosed before the age 50 years; |
| • Familial adenomatous polyposis should be excluded; and |
| • Tumor diagnosis should be confirmed by histopathological examination. |
2. Diagnosis
1) Flow of diagnosis
●Definitive diagnosis should be made according to the following Steps 1 to 3 in patients with clinicopathological findings (including family history) suggestive of Lynch syndrome (Figure 19).
Figure 19.
Diagnostic process for Lynch syndrome. Microsatellite instability: MSI, high-frequency MSI: MSI-H.
Step 1: It should be checked whether the patient meets the Amsterdam criteria II175) (Table 7, Figure 20A, 20B) or the revised Bethesda guidelines179) (Table 8) (primary screening).
Figure 20.
Family tree fulfilling Amsterdam criteria II. (See appendix: Principles in drawing and reading pedigrees) A: Multiple family members with colorectal cancer B: Multiple members with Lynch syndrome-associated extra-colonic cancers.
Table 8.
The Revised Bethesda Guidelines (2004) ref. 179) for Testing Colorectal Tumors for High-frequency Microsatellite Instability (MSI-H) .
| Tumors from patients with colorectal cancer (CRC) should be tested for MSI in the following situations: |
| 1. CRC diagnosed in a patient less than 50 years old. |
| 2. Presence of synchronous, metachronous colorectal, or other Lynch syndrome (LS)-associated tumors,a) regardless of the age. |
| 3. CRC with MSI-H histologyb) diagnosed in a patient less than 60 years old. |
| 4. CRC diagnosed in a patient with one or more first-degree relatives with a LS-associated tumor, with one of the cancers being diagnosed under the age of 50 years. |
| 5. CRC diagnosed in two or more first- or second-degree relatives with LS-associated tumors, regardless of the age. |
a) LS-associated tumors include colorectal cancer, endometrial cancer, gastric cancer, small-intestinal cancer, ovarian cancer, pancreatic cancer, renal pelvic/ureteral cancer, biliary tract cancer, brain tumors, sebaceous gland adenomas, and keratoacanthomas.
b) Tumor infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern.
Step 2: Microsatellite instability (MSI) testing or immunohistochemistry for the causative gene products in the tumor tissue should be performed to confirm high-frequency MSI (MSI-H) or to confirm the absence of mismatch repair proteins.Note 2 (secondary screening) (CQ20, CQ21)
Step 3: Pathogenic germline mutations in the mismatch repair genes should be identified (not covered by the national health insurance program in Japan) for definitive diagnosis. (CQ22, CQ23, CQ24)
Recently, it was proposed that all patients (or patients aged 70 years or less) with colorectal cancer be screened for Lynch syndrome, irrespective of the clinicopathological findings or family history (Side Memo 7: universal tumor screening).
Note 2: If MSI testing shows MSI-H or immunohistochemistry shows loss of MLH1 expression, the tumor tissue should be tested for the BRAF V600E mutation. If the tumor is positive for this mutation, Lynch syndrome can almost certainly be ruled out. Thus, this allows patients who do not have to proceed to Step 3 to be selected. BRAF V600E mutation testing is also not covered by national health insurance in Japan, but can be outsourced to testing companies. It must be noted that the BRAF V600E mutation has been reportedly found in some colorectal cancers associated with Lynch syndrome caused by mutations in the PMS2 gene180).
Step 1. Criteria for primary screening (Table 7, Table 8)
●It has been reported that 27-41%172,181) of Lynch syndrome families meet the Amsterdam criteria II175) and that 68-89% meet the revised Bethesda guidelines179); thus, more patients with Lynch syndrome can be identified using the revised Bethesda guidelines181).
●Approximately one-fourth of all colorectal cancer patients fulfill the revised Bethesda guidelines182). Namely, a considerable proportion of non-Lynch syndrome patients with sporadic colorectal cancer also meet the revised Bethesda guidelines179).
●In the JSCCR project studies, 1.2% of all colorectal cancer patients were found to meet the Amsterdam criteria II183).
Step 2. Tests used for secondary screening
MSI testing:
In some tumor cells with impaired mismatch repair function, the number of repeats in microsatellites, which are repetitive sequences of one to several nucleotides in the genome, is different from that in normal cells. This phenomenon is called microsatellite instability (MSI). MSI-H (high-frequency MSI) is usually seen in the tumor tissues in Lynch syndrome. Thus, screening and auxiliary diagnosis of Lynch syndrome has been performed using this phenomenon. This testing is covered by national health insurance (CQ20, Side Memo 8: Method for MSI testing and evaluation of the results).
●In cases with clinical findings suggestive of Lynch syndrome where the results of MSI testing of the colorectal tumor (colorectal adenomas can also be examined, although the detection sensitivity is lower) show MSI-H, Lynch syndrome should be strongly suspected.
●In MSI testing, the microsatellite length is compared between normal and tumor tissues, usually using 5 markers (Bethesda markers) (Figure 21). If there are differences in the microsatellite length in the tumor tissue, the tumor tissue is judged as showing MSI. MSI detected with 2 or more markers is defined as MSI-H, MSI detected with a single marker is defined as low-frequency MSI (MSI-L), and MSI detected with none of the 5 markers is defined as microsatellite-stable (MSS).
Figure 21.

An example of MSI analysis using the Bethesda markers.
Immunohistochemistry:
Most Lynch syndrome-associated tumors have biallelic inactivation of one of the mismatch repair genes, namely, MLH1, MSH2, MSH6, and PMS2, and expression of the corresponding protein is lost in most cases. Because MSI-H is caused by mismatch repair deficiency, the results of MSI testing are highly consistent with those of immunohistochemistry for mismatch repair proteins. It has been reported that the results of MSI testing and immunohistochemistry are consistent in 90% of cases and that the false-negative rate of immunohistochemistry for Lynch syndrome is 5-10%159,184). The great advantage of immunohistochemistry, as compared to MSI testing, is that it allows the causative gene to be deduced. In addition, immunohistochemistry is simpler than MSI testing and has been used increasingly in institutions in Japan. (CQ20, CQ21)
Step 3. Tests for definitive diagnosis
Genetic testing for mutation in the mismatch repair genes:
The patients’ blood is used to directly determine the presence or absence of pathogenic germline mutations in the mismatch repair genes. If a pathogenic mutation is identified, the patient is definitively diagnosed as having Lynch syndrome. In Japan, this testing is not covered by the national health insurance program, and it is currently performed at the patient’s expense or as research (genetic testing can be outsourced to testing companies). Genetic counseling must be provided before and after the testing. (CQ22, CQ23, CQ24)
●Even if genetic testing for mutations of the mismatch repair genes is considered unnecessary in the screening process, or genetic testing does not reveal any pathogenic mutations in the causative genes, the patient may still have Lynch syndrome.
●For families with clinical features strongly suggestive of Lynch syndrome, genetic testing for mutations in the mismatch repair genes is sometimes performed directly without screening by MSI testing or immunohistochemistry.
●It is desirable to perform genetic testing for mutations in the mismatch repair genes in individuals whose family members show clinical features suggestive of Lynch syndrome (multiple cancers, including colorectal cancer, endometrial cancer, early-onset cancer, etc.)
Side Memo 7
■Muir-Torre syndrome
Muir-Torre syndrome is a disease characterized by synchronous/metachronous development of various Lynch syndrome-associated tumors, such as colorectal cancer with sebaceous tumors (sebaceous adenoma, sebaceous epithelioma or sebaceous carcinoma), and/or keratoacanthoma. Germline mutations are found mainly in the MSH2 gene185).
■Turcot syndrome (type 1)
Turcot syndrome, type 1, is a disease in which Lynch syndrome-associated colorectal cancer is accompanied by brain tumors, mainly glioblastoma. Germline mutations in the MLH1 or PMS2 gene or hypermethylation of the MLH1 promoter have been found186). Caution should be exercised, since brain tumors are reported to be a major cause of death in patients with Lynch syndrome187). (Side Memo 3: Turcot syndrome, [type 2]).
■Universal tumor screening
Recently, universal tumor screening, in which MSI testing or immunohistochemistry for mismatch repair proteins is performed in all patients (or patients aged 70 years or less) with colorectal or endometrial cancer, is recommended as a highly sensitive and cost-effective method for the diagnosis of Lynch syndrome in Western countries. The incidence of Lynch syndrome detected by universal tumor screening is reported to be 2.4 to 3.7% of all colorectal cancers188,189).
2) Differential diagnosis
Sporadic colorectal cancer with MSI-H:
Sporadic colorectal cancer with MSI-H is commonly characterized by occurrence in elderly females, occurrence of poorly differentiated adenocarcinoma, right-sided preponderance, etc. The primary cause of MSI-H is considered as an acquired aberrant methylation of the promoter region of the MLH1 gene190). In these tumors, immunohistochemistry shows loss of expression of MLH1 protein. In addition, the BRAF V600E mutation is found in the tumor tissue in 35-43% of patients191,192). On the other hand, the BRAF V600E mutation is not detected in most colorectal cancers associated with Lynch syndrome, even if they show MSI-H193). Therefore, checking for the presence or absence of the BRAF V600E mutation is sometimes used to differentiate between these diseases.
Polymerase proofreading-associated polyposis (PPAP):
The phenotype of PPAP19-21) is sometimes like that of FAP (AFAP) or Lynch syndrome, and PPAP needs to be differentiated from Lynch syndrome (Chapter I. Familial adenomatous polyposis; (2) Diagnosis; 4) Diseases and pathological conditions that need to be differentiated from FAP; Polymerase-proofreading-associated polyposis.)
Familial colorectal cancer type X:
Patients who meet the Amsterdam criteria I,174)Note 3 but in whom no pathogenic germline mutations are detected in the mismatch repair genes or the colorectal cancer does not show MSI-H, are unlikely to have Lynch syndrome, and the term “familial colorectal cancer type X”194) was proposed for the condition. Familial colorectal cancer type X is speculated to comprise multiple diseases. Reports from both Western countries and Japan195) have shown that the risk of developing Lynch syndrome-associated tumors other than colorectal cancer is significantly lower in cases of familial colorectal cancer type X.
Note 3) Amsterdam criteria I: While colorectal cancer, endometrial cancer, renal pelvic/ureteral cancer and small-bowel cancer are included as Lynch syndrome (HNPCC)-associated tumors in the Amsterdam criteria II, only colorectal cancer is included as Lynch syndrome (HNPCC)-associated tumor in the Amsterdam criteria I174).
3. Treatment
1) Treatment of colorectal cancer
●The following options exist for the extent of resection of the colorectum (types of surgical procedures) in Lynch syndrome:
(1) Extent of resection equivalent to that adopted for sporadic colorectal cancer
(2) Total colectomy
(3) Total proctocolectomy
●No consensus has been reached on the usefulness of prophylactic colectomy, and it is not generally recommended.
●Because colorectal cancer tends to develop at multiple sites of the colorectum in Lynch syndrome, including synchronous or metachronous development, the entire colorectum should be examined before surgery.
●Some reports from Western countries have recommended extended operations, such as total colectomy for colonic cancer and total proctocolectomy for rectal cancer, for colorectal cancer in Lynch syndrome. However, no prospective studies on their usefulness have been conducted, and no consensus has been reached yet. (CQ25)
●Prophylactic colectomy for Lynch syndrome mutation carriers is not generally recommended, because its efficacy has not been assessed. (CQ25)
●Colorectal cancers in most cases of Lynch syndrome show MSI-H. Although 5-fluorouracil (FU)-based anticancer drugs have been reported to be generally ineffective in colorectal cancers showing MSI-H, the usefulness of chemotherapy specifically in Lynch syndrome-associated colorectal cancer has not yet been clarified. (CQ26)
2) Management of extracolonic tumors
(1) Gastrointestinal tumors (gastric cancer, small-bowel cancer, bile duct cancer, pancreatic cancer, etc.)
(2) Gynecologic tumors (endometrial cancer, ovarian cancer, etc.) (CQ19)
(3) Urological tumors (renal pelvic/ureteral cancer, etc.)
(4) Other tumors (brain tumor, skin tumor, etc.)
●There is no clear evidence of any special considerations required for the above-mentioned tumors (1) to (4), except for the case of gynecologic cancers, in patients with Lynch syndrome. At present, treatment like that for the corresponding sporadic cancers (tumors) is used.
●In Lynch syndrome patients with colorectal cancer, it is desirable to conduct screening for other Lynch syndrome-associated tumors (in particular, gynecologic cancers, urological cancers, and gastrointestinal cancers) prior to elective colectomy.
4. Postoperative surveillance
1) Surveillance for multiple colorectal cancers and resection of adenomas
●Attention should be paid to the possible development of metachronous cancer in the remaining colorectum after surgery for colorectal cancer in patients with Lynch syndrome, and lifelong regular colonoscopic examination is required. (CQ29)
●Surveillance for recurrence of colorectal cancer after resection should be in accordance with the protocol used for cases of sporadic colorectal cancer.
●Colorectal adenomas, if detected, should be resected, because they may develop into colorectal cancer.
2) Surveillance for Lynch syndrome-associated extracolonic tumors
●A specialist group in Europe proposed the following method of surveillance for the main Lynch syndrome-associated tumors (Table 9)196).
Table 9.
Recommended Surveillance Protocols for Common Lynch Syndrome-Associated Tumors.
| Sites | Examinations | Lower age limit (years) for starting surveillance |
Surveillance interval (years) |
|---|---|---|---|
| Colorectum | Colonoscopy | 20-25 | 1-2 |
| Uterus, ovary | Transvaginal ultrasound | 30-35 | 0.5-1 |
| Endometrial biopsy | |||
| Endometrial cytology | |||
| Serum CA125 measurement | |||
| Stomach, duodenum | Gastroduodenoscopy | 30-35 | 1-2 |
| Urinary tract | Urinalysis and urinary cytology | 30-35 | 1-2 |
Modifications with ref. 196)
●It was proposed that surveillance be conducted by upper gastrointestinal endoscopy every 1 to 2 years in areas where gastric cancer is common, such as East Asia, and in Lynch syndrome patients with a family history of gastric cancer and their relatives197).
●No consensus has been reached on the method or interval of regular surveillance for endometrial and ovarian cancers. (CQ19)
●Lynch syndrome-associated urological tumors include renal pelvic/ureteral cancer. This type of cancer has been reported to be common in patients with germline mutations in the MSH2 gene, but none of the surveillance methods, including regular urinalysis and urinary cytology, have been demonstrated to be useful in improving prognosis.
5. Surveillance of colorectal cancer patients without a genetic diagnosis of Lynch syndrome
●In patients who are suspected to have Lynch syndrome but have not been diagnosed yet by genetic testing, the possibility of Lynch syndrome should be individually evaluated based on clinical information and the results of MSI testing, which is covered by national health insurance in Japan, and surveillance for Lynch syndrome-associated tumors should be conducted (Figure 22).
Figure 22.
Management of individuals without a definitive diagnosis of Lynch syndrome. MSI testing can be used as a complement to or a surrogate for IHC.
●In cases where the patient “meets the Amsterdam criteria II” or “has a past or family history highly suggestive of Lynch syndrome,” in addition to the results of MSI testing showing MSI-H, the patient should be regarded as having Lynch syndrome and surveillance should be conducted even if no genetic testing has been performed.
●In cases where the patient “meets the Amsterdam criteria II” or “has a past or family history highly suggestive of Lynch syndrome,” but the results of MSI testing show MSS or MSI-L (there are no findings strongly suggestive of mismatch repair gene deficiency), Lynch syndrome can certainly not be ruled out (Side Memo 8: MSI testing method and evaluation of the results). In these cases, follow-up should be subsequently performed while paying attention to the personal and family history, with colonoscopic examination for colorectal cancer conducted at least every 3 to 5 years.
●In case the patient “meets the revised Bethesda guidelines, but not the Amsterdam criteria II, or does not have a personal or family history strongly suggestive of Lynch syndrome,” if the results of MSI testing show MSI-H, the patient may have Lynch syndrome (many patients are likely to have sporadic colorectal cancer). Follow-up should be performed while validating the past and family history.
●In MSS or MSI-L colorectal cancer patients who are unlikely to have Lynch syndrome based on the family and medical history, surveillance for Lynch syndrome-associated tumors is not conducted. When patients have symptoms of colorectal cancer, or Lynch syndrome-associated tumors are observed in the patients or their relatives, detailed examination and reevaluation for Lynch syndrome is recommended.
6. Genetic counseling and management of families (relatives)
●It is desirable to provide genetic counseling not only to the patients, but also to their relatives.
●After providing an adequate explanation about the disease to first-degree relatives (parents, children, and siblings) and obtaining their consent, surveillance for Lynch syndrome-associated tumors should be conducted according to the assessed risk.
●For genetic testing, the “Guidelines for Genetic Tests and Diagnosis in Medical Practice” of the Japanese Association of Medical Sciences, guidelines of the Japanese Society for Familial Tumors, “Ethical Guidelines for Human Genome/Gene Analysis Research,” etc., should be observed. In addition, records should be carefully stored, in consideration of the privacy of the subjects.
●In genetic counseling, information about the disease should be provided and the significance of genetic testing as one of the options and its methods, limitations, costs, etc., should be explained to help patients and their families make an independent choice about whether to undergo genetic testing or not. For patients who wish to undergo genetic testing, testing should be performed after obtaining informed consent from the patient. Genetic counseling should be provided not only before and after genetic testing, but also repeatedly, where necessary.
●In principle, because Lynch syndrome-associated tumors generally develop in adulthood, genetic testing should be performed in adulthood.
●It should be ascertained as to whether the patients (clients) want to be apprised of the results of genetic testing with their family members or not. If a patient does not wish to be informed about the results in the presence of family members, an opportunity should be provided for giving the information individually.
1) Management of patient families (relatives) who have been diagnosed by genetic testing to have Lynch syndrome (Figure 23)
Figure 23.

Management of the families (relatives) of patients who have been definitively diagnosed with Lynch syndrome defined by genetic testing.
●Relatives who are definite mutation carriers or who have not undergone genetic testing should be regarded as having Lynch syndrome and undergo surveillance for Lynch syndrome-associated tumors (Figure 23).
●Relatives who have been confirmed to have no pathogenic mutation should undergo general cancer screening (Figure 23). Information on the necessity of surveillance and significance of genetic diagnosis should be provided to relatives who have reached the age of surveillance for Lynch syndrome-associated tumors. Everyone should decide, of his/her own free will, whether he/she wishes to undergo genetic testing or not through genetic counseling.
2) Management of patient families (relatives) who are suspected to have Lynch syndrome, but for whom no definitive diagnosis has been made
●In relatives of patients who have not undergone genetic testing or in whom genetic testing has failed to yield a definitive diagnosis of Lynch syndrome, individual risk assessment should be carried out by reference to the age of onset, incidence, etc., of Lynch syndrome-associated tumors in family members, and surveillance for associated tumors should be conducted.
●In relatives of patients suspected as having Lynch syndrome, surveillance should be conducted according to the protocol shown in Table 9, or colonoscopy should be started at an age 5-15 years younger than that of the earliest age at diagnosis of colorectal cancer in the family.
Clinical Questions
CQ17: Do Lynch syndrome-associated tumors require different treatments according to the causative gene of Lynch syndrome?
Recommendation category: C
The risk of developing Lynch syndrome-associated tumors may vary greatly depending on the causative gene. Therefore, it has been proposed that surveillance be conducted according to the organ-specific cancer risk, which is related to the causative gene.
Many studies comparing the major causative genes MLH1 and MSH2 have reported that the risk of colorectal cancer is equivalent between patients with MLH1 mutations and those with MSH2 mutations, and that the risk of development of Lynch syndrome-associated tumors (in particular, those of the urinary system) other than those of the large intestine is higher in patients with MSH2 mutations198). The risk of colorectal cancer development is lower in patients with MSH6 mutations than in those with MLH1 or MSH2 mutations, but the risk of endometrial cancer development in patients with MSH6 mutations is equivalent to or higher than that in those with MLH1 or MSH2 mutations (Table 10). Therefore, it is desirable to bear in mind, while conducting surveillance, that the incidence of Lynch syndrome-associated tumors varies greatly depending on the causative gene in Lynch syndrome. However, the risk of development of Lynch syndrome-associated tumors for each causative gene has not yet been fully assessed in Japanese.
Table 10.
Lifetime Risk of Lynch Syndrome-Associated Tumors According to the Kind of Causative Genes (up to the Age of 70 Years).
| MLH1 | MSH2 | MSH6 | PMS2 | |
|---|---|---|---|---|
| Colorectal cancer | 41% | 48% | 10%-22% | 15%-20% |
| Endometrial cancer | 18%-54% | 21%-30% | 16%-71% | 15% |
| Gastric cancer | 3%-6% | 0.2%-7% | ≤3% | - |
| Ovarian Cancer | 13%-20% | 9.5%-24% | 1%-11% | - |
| Renal pelvic/ureteral caner | 0.2%-2.9% | 2.2%-12% | <1% | - |
CQ18: What are the important histologic findings that need to be screened for in cases of colorectal cancer associated with Lynch syndrome?
Recommendation category: C
Tumor-infiltrating lymphocytes, a medullary growth pattern, mucinous/signet-ring differentiation and Crohn’s-like lymphocytic reaction are useful in screening not only for sporadic MSI-H colorectal cancer, but also for Lynch syndrome.
Several histological features are significantly more common in MSI-H colorectal cancers than in non-MSI-H (MSI-L or MSS) colorectal cancers, and these findings are useful to screen for patients with suspected Lynch syndrome. Four histologic findings, namely, (1) tumor-infiltrating lymphocytes (TIL), (2) a medullary growth pattern, (3) mucinous/signet-ring differentiation, and (4) Crohn’s-like lymphocytic reaction, are listed in the revised Bethesda guidelines179) (Figure 24A, 24B, 24C, 24D). However, these histological features are not necessarily specific to Lynch syndrome; they are commonly seen in both Lynch syndrome-associated and sporadic MSI-H colorectal cancers199).
Figure 24.
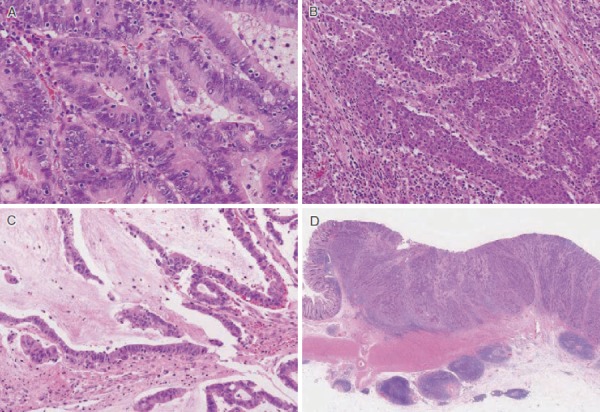
Pathological features of MSI-H colorectal cancer A: Tumor-infiltrating lymphocytes Numerous intra-epithelial lymphocytes showing clear halos. B: Medullary carcinoma Tumor showing a solid growth pattern without glandular structure. C: Mucinous adenocarcinoma showing prominent extracellular mucin. D: Crohn’s-like lymphoid reaction characterized by peritumoral lymphocytic aggregates.
CQ19: How should gynecologic cancers be treated in patients with Lynch syndrome (including mutation carriers who have not developed colorectal cancer)?
Recommendation category: C
No surveillance method for endometrial and ovarian cancers in patients with Lynch syndrome has been established. Some experts have proposed that transvaginal ultrasonography must be performed in addition to endometrial cytology or endometrial biopsy as part of surveillance for endometrial cancer.
The Lynch syndrome-associated gynecologic cancers are endometrial cancer and ovarian cancer.
Endometrial cancer is the second most common cancer after colorectal cancer in Lynch syndrome and it is regarded as a “sentinel cancer.” Dysfunctional uterine bleeding is the most common subjective symptom. Surveillance methods include endometrial cytology, endometrial biopsy and transvaginal ultrasonography, and definitive diagnosis is made by histopathological examination. Note that routine Pap smear is performed to screen for cervical cancer, but not for endometrial or ovarian cancer.
At present, risk-reducing surgery is the most effective method for the primary prevention of ovarian cancer. If risk-reducing surgery is not selected, surveillance should be conducted, but its usefulness has not been established. Transvaginal ultrasonography and CA125 testing have not been shown to be sufficiently sensitive or specific for hereditary breast and ovarian cancer syndrome, which is characterized by an increased risk of development of ovarian cancer, like in Lynch syndrome. Transvaginal ultrasonography and CA125 testing approximately every 6 months are the secondary prophylaxis in clinical practice. However, there is a risk of so-called interval cancer, in which cancer is detected by the appearance of subjective symptoms before the scheduled medical examination, although the previous medical examination was negative for cancer.
Risk-reducing surgery with hysterectomy and bilateral salpingo-oophorectomy can be considered as options for primary prevention of gynecologic cancer in Lynch syndrome patients who have completed childbearing or who are postmenopausal200). In addition, the option of simultaneous hysterectomy and bilateral salpingo-oophorectomy with surgery for colorectal cancer should also be considered for patients with colorectal cancer. Risk-reducing surgery should be performed after obtaining approval from the local ethics committee and carefully considering the medical care system in advance.
The surveillance methods shown in Table 9 ((4) Postoperative surveillance) should be followed in patients who do not wish to undergo risk-reducing surgery and choose to undergo surveillance instead.
CQ20: What are the points that should be kept in mind while conducting screening tests (MSI testing and immunohistochemistry) for Lynch syndrome?
Recommendation category: None
Sensitivity and specificity are equivalent between MSI testing and immunohistochemistry, and immunohistochemistry helps identify the putative causative genes. The costs and convenience of performance of the tests vary from institution to institution, and comprehensive consideration should be given to selection of one of the tests.
It has been reported that more than 90% of colorectal cancers in patients with Lynch syndrome show MSI-H201). On the other hand, according to reports from Western countries201-203) and Japan191,204), MSI-H colorectal cancer accounts for 12-16% and 6-7% of all colorectal cancers, respectively. Therefore, MSI testing is a useful screening test to shortlist patients with suspected Lynch syndrome. MSI testing has been covered by the national health insurance program since 2006 in Japan as a genetic test for malignancies in colorectal cancer patients with suspected Lynch syndrome. When MSI testing is performed, the possibility of hereditary cancer should be fully explained to the individuals and informed consent should be obtained. Related webpages are linked to the website of the Japanese Society for Familial Tumors (http://jsft.umin.jp/).
Clinicians must be aware that some Lynch syndrome patients with germline mutations in the MSH6 gene do not show MSI-H205,206). Therefore, if the patient shows MSI-L or MSS, but meets the Amsterdam criteria II175) or if the patient has clinical features (early-onset cancer or multiple cancers) strongly suggestive of Lynch syndrome, genetic testing of mismatch repair genes should be considered33). MSI testing mainly using mononucleotide repeat markers, which have recently been increasingly used, has also been reported to be highly sensitive in patients with MSH6 mutations (Side Memo 8: Method for MSI testing and evaluation of the results).
On the other hand, immunohistochemistry for mismatch repair proteins has rapidly become popular as a secondary screening test for Lynch syndrome. Immunohistochemistry can be performed at many institutions. In addition, when immunohistochemistry is performed, a thorough explanation should be provided to individuals and informed consent should be obtained, as for MSI testing. Related webpages are linked to the website of the Japanese Society for Familial Tumors (http://jsft.umin.jp/).
MSI testing and immunohistochemistry use different methods, but have equivalent sensitivity and specificity, and immunohistochemistry allows the possible causative gene among 4 MMR genes to be identified. The costs and convenience of performance of these tests vary from institution to institution, and comprehensive consideration, including the examination system at the institution, should be given to the selection of one of the tests. However, if the patient is clinically suspected as having Lynch syndrome even if one test is negative, implementation of the other test allows complementary screening to be performed.
Side Memo 8
■Method for MSI testing and evaluation of the results
Clinical testing companies provide MSI testing. Frozen samples or formalin-fixed paraffin-embedded specimens of tumor and non-tumor normal tissues are required for MSI testing (blood samples can be used instead of normal tissues). DNA is extracted from tumor and normal tissues to compare the microsatellite lengths between these tissues. In general, 5 markers (known as the Bethesda markers or NCI panel, consisting of 2 mononucleotide repeat markers and 3 dinucleotide repeat markers) have been used to assess MSI (Step 2. Tests used for secondary screening). Sometimes, when more markers are used, cases in which 30% or more markers show changes in the microsatellite lengths in tumor tissue are defined as having MSI-H. In addition, because mononucleotide repeat markers have high sensitivity, MSI-H is sometimes defined by the presence of instability detected using 3 or more markers. Some MSI tests with mononucleotide repeat markers allow MSI to be assessed using tumor tissue alone, and have rapidly become popular in Japan as well as in other countries.
CQ21: What are the points that must be borne in mind while conducting immunohistochemistry for mismatch repair gene products (proteins)?
Recommendation category: None
From the pattern of loss of the mismatch repair proteins in cell nuclei, genes causing the mismatch repair deficiency can be deduced. When evaluating the staining results, the adequacy of staining should be confirmed using internal positive controls.
1. Internal positive controls
Mismatch repair proteins are localized in the nuclei and are more strongly expressed in proliferating cells. The base of the colonic mucosal glands and germinal centers of lymph follicles are good positive controls in non-tumor tissues (Figure 25). Because tumor tissues generally have high proliferative activity, confirmation of staining of internal positive controls often makes evaluation easier.
Figure 25.
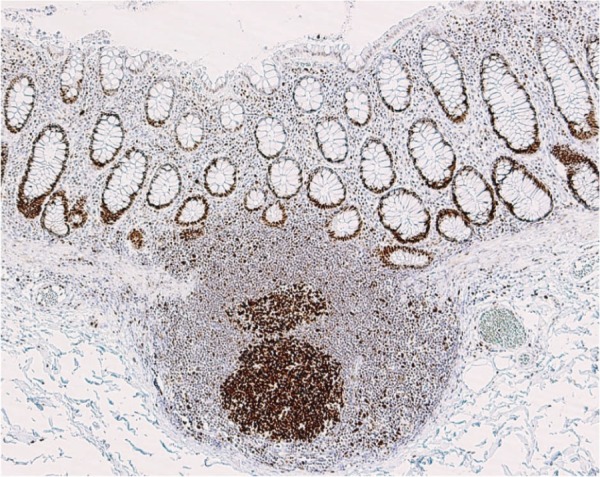
MSH2 expression in normal colon mucosa. Strong staining is seen in the germinal center of a lymphoid follicle and at the bottom of glands.
2. Staining patterns and evaluation
In tumors without mismatch repair deficiency, all 4 proteins are expressed. In tumors with mismatch repair deficiency, protein expression is lost reflecting the deficiency of mismatch repair genes, but individual mismatch repair gene deficiency does not correspond 1:1 to loss of protein expression (Table 11, Figure 26). Most cases exhibit one of the staining patterns shown in Table 11. If a staining pattern different from any of those shown in Table 11 is obtained, the validity of staining should be checked before considering the possibility of an exceptional case. In principle, invasive cancers show diffuse loss of expression.
Table 11.
Immunohistochemical Expression Patterns of the Mismatch Repair Proteins Associated with Each Suspected Causative Gene.
| Expressions of immunohistochemical staining | |||||
|---|---|---|---|---|---|
| MLH1 | MSH2 | PMS2 | MSH6 | ||
| Causative genes | MLH1 | - | + | - | + |
| MSH2 | + | - | + | - | |
| PMS2 | + | + | - | + | |
| MSH6 | + | + | + | - | |
Figure 26.
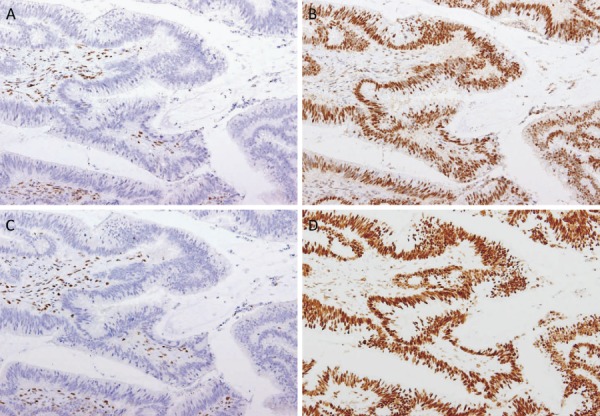
Immunohistochemistry for mismatch repair proteins in the colorectal cancer specimen resected from in a Lynch syndrome patient with a germline MLH1 mutation. Loss of MLH1 (A) and PMS2 (C) and retention of MSH2 (B) and MSH6 (D). Stromal cells served as internal positive controls.
The expression of PMS2 in addition to MLH1 is lost in tumors with MLH1 mutations, and the expression of MSH6 in addition to MSH2 is lost in tumors with MSH2 mutations (Table 11). Therefore, the use of only 2 antibodies, anti-PMS2, and anti-MSH6 antibodies, allows the screening of Lynch syndrome to be performed with a sensitivity equivalent to that using 4 antibodies207). If the expression of PMS2 or MSH6 is lost, staining for MLH1 or MSH2, respectively, should be added to deduce the mutated gene.
Side Memo 9
Exceptional staining results
■Expression of abnormal proteins due to missense mutations
In some cases of missense mutations (Appendix: II. Method for describing genomic variants; 3. Types of changes), non-functional proteins are expressed. This is known to be relatively common in Lynch syndrome patients with MLH1 mutations, and in most of these cases, only PMS2 expression is lost207). However, there are rare cases in which no abnormalities are detected by immunohistochemistry. Even if no abnormalities are detected by immunohistochemistry, if the patient is clinically strongly suspected as having Lynch syndrome, the addition of MSI testing may allow accurate diagnosis.
■Secondary mutations in mismatch repair genes due to microsatellite instability
Some mismatch repair genes have repeat sequences, and secondary mutations may occur in these genes. In some patients with MLH1 gene mutations (loss of MLH1/PMS2), MSH6 expression is lost diffusely or regionally208).
■Loss of MSH6 expression due to preoperative chemoradiotherapy
It has been reported that in colorectal cancer patients administered preoperative chemoradiotherapy, the tumor MSH6 expression may be lost even in the absence of abnormality in the MSH6 gene208).
CQ22: What is the significance of genetic diagnosis of Lynch syndrome and what are the important points that should be kept in mind?
Recommendation category: B
Genetic testing of mismatch repair genes is required for a definitive diagnosis of Lynch syndrome. Individuals for genetic testing should be selected carefully, and after genetic counseling is provided, it should be checked with them whether they wish to undergo the testing or not. The results of genetic testing should be evaluated for the genetic diagnosis of individuals’ relatives and medical management of the individuals and their relatives.
Pathogenic germline mutations in mismatch repair genes should be identified to make a definitive diagnosis of Lynch syndrome. Individuals (patients/relatives) for genetic testing should be appropriately selected, and genetic counseling should be provided before and after the genetic testing. Genetic testing sometimes fails to determine whether the individual has Lynch syndrome or not, and the results should be interpreted carefully. Testing for mismatch repair genes is not covered by the national health insurance program, but can be outsourced to testing companies.
1. Genetic testing of mismatch repair genes
Genetic counseling should be provided before and after genetic testing, because there are hereditary disease-specific precautions and considerations. In principle, an explanation about the genetic testing should be given by a physician, but the individuals can be referred to institutions with expertise in providing genetic counseling. If the individuals agree to undergo genetic testing, approximately 2 to 3 mL of blood should be collected and sent to the testing company to perform genetic testing. Direct sequencing is generally used for analysis, but if no mutations are identified, multiplex ligation-dependent probe amplification, Southern blotting, etc., should be used, because a part of a gene may be deleted, duplicated, or rearranged.
2. Evaluation of the results of genetic testing
If a gene mutation obviously causing the disease is found, a definitive diagnosis of Lynch syndrome can be made. After disclosing the results of genetic testing, genetic counseling, future surveillance planning and implementation, genetic testing of relatives, etc., should be considered. On the other hand, even if a genetic mutation is found, its causal relationship to the disease is unclear (a variant of uncertain significance [VUS]) in some cases (CQ23). In these cases, subsequent surveillance should be conducted as if no genetic testing had been performed. Even if no mutations are detected by genetic testing, Lynch syndrome cannot be completely ruled out, because there may be gene alterations that are undetectable by current testing methods or unknown causative genes. Risk assessment should be carried out according to the past medical and family history, and surveillance, as appropriate, should be conducted. If genetic testing of the proband reveals no mutation, genetic testing of other relatives is of little significance.
3. Genetic testing of relatives
Detection of a pathogenic gene mutation in the proband makes it possible to check whether his/her relatives have the same mutation. In this case, only the region containing the identified mutation should be tested. Genetic counseling should also be provided before and after genetic testing of the relatives. Detection of a mutation in one of the causative genes should lead to future surveillance planning and implementation. Unless there is a family history of early-onset (teens to 20s) cancer, genetic testing should generally be performed in adulthood. Everyone should decide, of his/her own free will, about whether he/she should undergo genetic testing or not.
The JSCCR multicenter study showed that in addition to colorectal cancer and endometrial cancer, which are common in Western countries, Lynch syndrome-associated tumors such as gastric cancer, ovarian cancer, and bile duct cancer, were the major causes of death in first-degree relatives of Japanese Lynch syndrome patients. In close relatives with definite Lynch syndrome, it is important to conduct surveillance bearing these malignancies in mind209).
CQ23: How should Lynch syndrome patients in whom genetic testing shows a variant of uncertain significance (VUS) be managed?
Recommendation category: C
Patients with VUS should be managed as if no genetic testing had been performed (Figure 22). If possible, the presence or absence of the variant should be assessed in relatives to investigate its association with tumor development.
The results of genetic testing show a “variant of uncertain significance (VUS)” in some cases. The interpretation of the clinical significance of VUS sometimes changes because of database updating, etc. There are programs to predict the influence of variants on the protein function (Sorting Intolerant From Tolerant [SIFT], http://sift.jcvi.org/index.html; Polymorphism Phenotyping version 2 [PolyPhen-2], http://genetics.bwh.harvard.edu/pph2, etc.). In cases where the presence or absence of the variant in relatives is strongly associated with tumorigenesis and in cases where the pattern of loss of mismatch repair proteins as assessed by immunohistochemistry is consistent with the VUS, the variant is most likely a pathogenic mutation, but further examination is required to confirm this.
If no definite information is available, the patient should be managed as follows.
●If a patient who has an MSI-H tumor or in whom immunohistochemistry shows loss of expression of one of the mismatch repair proteins has the clinical features of Lynch syndrome, he/she should be managed under the assumption that he/she has Lynch syndrome.
●If a patient who has an MSI-H tumor or in whom immunohistochemistry shows loss of expression of one of the mismatch repair proteins has no clinical findings suggestive of Lynch syndrome, periodic follow-up should be continued based on the family/past medical history.
CQ24: What are the points that should be considered while providing genetic counseling to patients with Lynch syndrome and their families?
Recommendation category: B
When genetic counseling is provided to patients with suspected Lynch syndrome and their families (relatives), the explanation provided about the disease should include an outline of Lynch syndrome, the mode of inheritance, the tests required for diagnosis, the risk of development of Lynch syndrome-associated tumors, including colorectal cancer, surveillance, etc., information resources on the disease, psychosocial support, etc.
It is important to keep in mind the following points (1) to (4) while providing genetic counseling to patients/their families for Lynch syndrome:
(1) The medical and family histories of the individuals should be taken.
(2) The following information should be provided.
An outline of Lynch syndrome (clinical symptoms, penetrance, natural course, incidence, causative genes, diagnosis, treatment, prophylaxis, etc.), its mode of inheritance (autosomal dominant inheritance), the risk of development of various cancers in the individuals (and their relatives) with or without a pathogenic mutation, the possibility of detecting gene mutations, the risk of various cancers when there is a pathogenic mutation, an outline of tests for Lynch syndrome (MSI testing, immunohistochemistry, genetic testing for mismatch repair genes, etc.), preventive measures (in particular, surveillance) based on the risk, information resources such as websites and books, information on patient groups, and current state of research in Japan and overseas.
(3) The surveillance protocols for Lynch syndrome-associated tumors according to the risk should be presented.
(4) Psychosocial support should be provided (patients should be asked to give voice to their concerns and anxieties about the disease, conflicts among family members, etc., and should receive empathy from the physician).
CQ25: Which are the surgical procedures that should be selected for colorectal cancer in patients with Lynch syndrome?
Recommendation category: C
No consensus has been reached on whether the same surgical procedures as those for sporadic colorectal cancer should be selected, or extended operations should be adopted, considering the risk of multiple colorectal cancers in patients with Lynch syndrome.
A retrospective cohort study of colorectal cancer in Lynch syndrome reported that the 10-, 20- and 30-year cumulative incidences of development of metachronous colorectal cancer after partial colectomy (segmental resection) were 16%, 41%, and 62%, respectively, and that the risk of development of metachronous colorectal cancer was lower when a longer segment of the intestine was resected210).
In addition, according to another retrospective cohort study, approximately 15% of primary colorectal cancers in patients with Lynch syndrome were rectal cancers, and most metachronous colorectal cancers were right colon cancers in patients who underwent proctectomy. Furthermore, endoscopic surveillance at mean intervals of 14 months revealed 10-, 20-, and 30-year cumulative incidences of development of metachronous colorectal cancers of 19%, 47%, and 69%, respectively211). There are insufficient data on whether total proctocolectomy should be selected or not for primary rectal cancer.
Also, no consensus has been reached on whether prophylactic colectomy should be performed in mutation carriers who have been genetically diagnosed as having a pathogenic mutation in one of the mismatch repair genes, but have not developed colorectal cancer. The lifetime risk of colorectal cancer development in male patients with Lynch syndrome is 54-74%, and that in female patients with Lynch syndrome is 30-52% (Table 6). Furthermore, a substantial number of mutation carriers do not develop colorectal cancer throughout their lifetimes. Therefore, prophylactic colectomy cannot be uniformly recommended, as in the case of patients with FAP. Accordingly, it is desirable to allow mutation carriers to decide for themselves the course of treatment they would wish to receive, after they are provided an explanation about the risk of development of metachronous colorectal cancer in Lynch syndrome, the necessity and limitations of surveillance, the significance of prophylactic surgery, the postoperative QOL, etc.
CQ26-1: Which are the adjuvant chemotherapy regimens effective for colorectal cancer in patients with Lynch syndrome?
Recommendation category: C
There is no clear evidence of the efficacy of postoperative adjuvant chemotherapy specifically for colorectal cancer in patients with Lynch syndrome. Stage III colonic cancer (colorectal cancer) in Lynch syndrome could be an indication for postoperative adjuvant chemotherapy.
Because there is little evidence of chemotherapy specific to colorectal cancer in patients with Lynch syndrome, chemotherapy is often considered in accordance with that for sporadic MSI-H colorectal cancers. However, it has been reported that postoperative 5-fluorouracil (FU)-based adjuvant chemotherapy is not useful in patients with sporadic MSI-H colorectal cancer, but is useful in MSI-H colorectal cancer patients aged less than 50 years with suspected Lynch syndrome212), suggesting that colorectal cancer in Lynch syndrome should be considered differently from sporadic MSI-H colorectal cancers. There are almost no useful data on postoperative adjuvant chemotherapy for sporadic MSI-H rectal cancer or Lynch syndrome-associated rectal cancer.
A meta-analysis of the MSI status and efficacy of postoperative adjuvant chemotherapy including 5-FU in cases with stage II/III sporadic colorectal cancer showed that MSI-H colorectal cancer had a better prognosis than MSS colorectal cancer, but that postoperative adjuvant chemotherapy did not improve the survival or recurrence-free survival in patients with MSI-H colorectal cancer213,214). However, the National Surgical Adjuvant Breast and Bowel Project (NSABP)-C07 trial and the Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) trial showed that oxaliplatin had an additive effect in postoperative adjuvant therapy for both MSI-H and MSS colonic cancers215). Therefore, at present, it is not recommended to determine whether stage III colonic cancer is an indication for postoperative adjuvant chemotherapy according to the MSI status. The usefulness of postoperative adjuvant chemotherapy has not been established for stage II colorectal cancer, and it is thought to be less useful, particularly in MSI-H cancers, because these cancers have favorable prognoses.
CQ26-2: What are the chemotherapy regimens effective against metastatic colorectal cancer in Lynch syndrome?
Recommendation category: C
There is no clear evidence of the efficacy of chemotherapy exclusively against metastatic colorectal cancer in Lynch syndrome. The same management strategies as those for sporadic colorectal cancer should be selected.
The incidence of MSI-H has been shown to be lower in stage IV than in stage II/III sporadic colorectal cancers216,217). Chemotherapy specific to metastatic colorectal cancers associated with Lynch syndrome or colorectal cancers showing MSI-H has not yet been clearly investigated and no conclusion has been reached. Therefore, regimens generally selected for sporadic colorectal cancers could be indicated for these cancers as well. The response rate to irinotecan as a second-line treatment in cases with acquired resistance to 5-FU was reported to be significantly higher in MSI-H cancers than in other sporadic colorectal cancers218). Recent reports on immune checkpoint inhibitors have attracted attention (Side Memo 10: MSI-H tumors and anti-PD-1 antibody drugs).
Side Memo 10
■MSI-H tumors and anti-PD-1 antibody drugs
Recently, anti-programmed death (PD)-1 antibodies, which are immune checkpoint inhibitors, have been shown to be effective in the treatment of metastatic colorectal cancers, particularly those showing MSI-H, and these antibodies are expected to also be effective against Lynch syndrome-associated colorectal cancer219). The response rate to anti-PD-1 antibodies was significantly higher in MSI-H colorectal cancer (40%) and other MSI-H cancers (71%) than in MSS colorectal cancer (0%). However, among the MSI-H cancer cases, a subgroup of 11 patients with Lynch syndrome showed a response rate of only 27%, lower than the response rate in patients with sporadic MSI-H cancers. Anyway, these results suggest that there is much scope for innovation in pharmacotherapy for MSI-H cancers, including those associated with Lynch syndrome.
CQ27: Are there any lifestyle remedies that are effective in preventing carcinogenesis in Lynch syndrome?
Recommendation category: B
Smoking cessation and proper body fat control are recommended to prevent colorectal carcinogenesis in Lynch syndrome.
The effects of smoking, body mass index, alcohol consumption, diets (red meat, processed meat, vegetables, fruits, fish, dairy products, and dietary fiber), etc., have been investigated. Studies, including a prospective cohort study, have shown an association of smoking and body mass index with the risk of carcinogenesis220,221).
The results of the JSCCR multicenter study suggested that smoking is a risk factor for the development of synchronous/metachronous multiple colorectal cancers in Lynch syndrome221).
Based on current evidence, smoking cessation and proper body fat control are recommended to prevent colorectal carcinogenesis in patients with Lynch syndrome.
CQ28: Is there any chemoprophylaxis that is effective in preventing carcinogenesis in Lynch syndrome?
Recommendation category: C
Aspirin may prevent the development of Lynch syndrome-associated tumors.
The Colorectal Adenoma/Carcinoma Prevention Programme 2 (CAPP2) was the first double-blind study to evaluate the efficacy of aspirin in preventing Lynch syndrome-associated tumors and colorectal adenomas. Long-term follow-up revealed that aspirin was significantly effective in preventing the development of colorectal cancer (primary endpoint) and other Lynch syndrome-associated tumors (secondary endpoint)222). No useful data with respect to the prevention of colorectal adenomas were obtained. The aspirin dose used in this study was 600 mg/day, a dose which is considered difficult to use widely in Japanese patients, who are more susceptible to gastrointestinal disorders caused by aspirin as compared to Europeans and Americans. On the other hand, a study conducted on a large patient cohort, although retrospective, showed the efficacy of aspirin and ibuprofen in preventing the development of colorectal cancer in Lynch syndrome223). It remains to be investigated in the future as to what dose and for what duration aspirin should be administered for obtaining this effect. Several studies have reported that low-dose aspirin may prevent the development of sporadic colorectal cancer224), and a clinical trial of low-dose aspirin in Lynch syndrome patients is ongoing.
CQ29: Is surveillance by colonoscopy effective in patients with Lynch syndrome?
Recommendation category: B
Endoscopic surveillance and resection of adenomas reduce the development of and death from colorectal cancer in cases of Lynch syndrome.
Patients with Lynch syndrome have been shown to be at a high risk of developing colorectal cancer, including those with remaining large intestine after surgery for colorectal cancer, and regular and lifelong endoscopic surveillance with the aim of any resecting precancerous adenomas and early detection of colorectal cancer is required225,226). Many studies have recommended that surveillance be started at the age of 20-25 years196). Regarding the intervals at which the examinations should be conducted, a prospective study by Järvinen et al.227) reported that endoscopic surveillance at 3-year intervals decreased the mortality from colorectal cancer by 65%. However, observational studies have confirmed the development of advanced cancer over 3-year periods in endoscopic surveillance, and several studies recommend annual surveillance228-230).
In Lynch syndrome patients, colorectal adenomas often develop at a young age (less than 40 years)228,231), show MSI-H, show high-grade atypia even if smaller than ordinary adenomas, and undergo malignant transformation within a short period of time232,233). In addition, the cumulative number of colorectal adenomas developing over a lifetime has been reported to be up to approximately 20234). Therefore, tumorous lesions, if detected, should be actively resected endoscopically, irrespective of their size.
Appendix
I. Principles in drawing and reading pedigrees
1. Points that must be kept in mind while taking the family history (Appendix Figure 1)
Appendix Figure 1.
Symbols for family pedigrees.
●Information on at least 3 generations should be obtained.
●It should be checked whether there are any consanguineous marriages (such as cousin marriages).
●Not only the number of affected individuals, but also the number of unaffected individuals among siblings should be checked.
●Date of taking family history, name of the person providing the information, and name of the person taking the family history should be described in the pedigree.
●Maternal and paternal pedigrees should be separately evaluated.
2. Outline of how to draw pedigrees (Appendix Figure 2)
Appendix Figure 2.
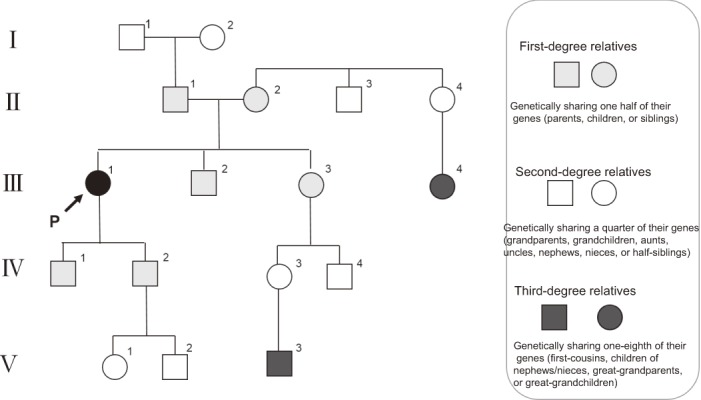
The first-, second-, or third-degree relatives of the proband.
●The proband (the affected individual leading to the detection of the affected family) should be indicated by P↗
●The client should be indicated by↗
●If possible, the husband (male partner) should be listed to the left of the wife (female partner).
●Siblings should be listed from left to right in order of birth.
●The generation number should be indicated in Roman numerals on the left side.
●Individual numbers should be given in Arabic numerals in order from left to right along generation lines.
●Necessary clinical information, such as the age at onset (age at diagnosis), affected site (left or right in the case of bilateral disease), course of treatment, surgical procedure, and pathological diagnosis, should be described.
Symbols generally used to draw pedigrees are shown below.
The first-, second-, or third-degree relatives of the proband are shown below.
II. Method for describing genomic variants
The description method proposed by the Human Genome Variation Society (http://varnomen.hgvs.org/) is generally used to describe genomic changes. Usually, information on reference sequences, their location, and any changes should be given in that order.
1. Symbols for reference sequences
Genomic reference sequence: g.
Coding DNA reference sequence#: c.
RNA reference sequence: r.
Protein reference sequence: p.
#Coding DNA sequence is a DNA sequence between the start and stop codons that serves as a template for the synthesis of mRNA, which is translated into protein.
2. Locations of variants
(1) Changes at the genomic DNA level should be indicated by “g.,” and the first nucleotide of the reference genome sequence should be numbered 1.
(2) Changes at the coding DNA level should be indicated by “c.,” and the A of the start codon ATG (translation start point) should be numbered 1 (in Appendix Figure 3, the last nucleotide of exon 1 is the 128th nucleotide from the A of the start codon ATG, and should be indicated as c. 128). Because coding DNA sequences are translated into proteins and contain no introns, when a nucleotide position in an intron is shown, the nucleotide number counted from an adjacent exon should be indicated using “+” or “-.” For example, in Appendix Figure 3, the 15th nucleotide from the start of intron 1 should be indicated as “c. 128 + 15,” and the second nucleotide upstream of the start of exon 2 (c. 129) should be indicated as “c. 129 - 2.”
Appendix Figure 3.

The gene structure and variant nomenclature.
(3) Changes at the RNA level should be indicated by “r,” in accordance with the method for describing the changes at DNA level.
(4) Changes at the protein level should be indicated by “p.,” and the translation initiation methionine should be numbered 1. Both three-letter and one-letter amino acid codes can be used.
3. Types of changes and their descriptions
(1) Changes at the DNA level should be described as follows. Substitution: >; deletion: del; insertion: ins; deletion-insertion: delins; duplication: dup; inversion: inv; conversion: con.
(2) For changes at the protein level, “>” is not used in the case of substitution, but the original and changed amino acids are shown before and after the amino acid position (number), respectively. Other changes, such as deletion (del), insertion (ins), deletion-insertion (delins), duplication (dup), inversion (inv) and conversion (con) are described in a similar manner to those at the DNA level.
In general, changes are commonly described at the coding DNA (c.) or protein (p.) level.
Specific examples are given below.
Example 1) Missense mutation
●c. 146T > A (p. Val49Glu)
The T nucleotide at the 146th position from the A of the start codon ATG is substituted with an A. This is associated with change of the 49th amino acid from valine (Val) to glutamic acid (Glu).
Example 2) Nonsense mutation
●c. 184C > T (p. Glu62Ter or p. Glu62*)
The C nucleotide at the 184th position is substituted with a T. This is associated with the 62nd codon becoming a stop codon, resulting in a stop of protein biosynthesis.
Example 3) Duplication and its associated frame-shift mutation
●c. 175dupA (p. Ile59Asnfs*20)
The A nucleotide at the 175th position is duplicated and the 176th nucleotide becomes A with the shift in the codon reading frame (this shift of the reading frame is called frame shift and designated as “fs”). This is associated with change of the 59th amino acid from isoleucine (Ile) to asparagine (Asn), and furthermore, with the 20th codon from this site becoming a stop codon (fs*20), resulting in the termination of protein biosynthesis.
Example 4) Deletion and its associated frame-shift mutation
●c. 3927_3931delAAAGA (p. Glu1309Aspfs*4)
The nucleotides the 3927th to 3931th position, AAAGA, are deleted. This is associated with change of the 1309th amino acid from glutamic acid (Glu) to aspartic acid (Asp) and the 4th codon from this site becoming a stop codon (fs*4).
Example 5) A mutation in an intron
●c. 792 + 1G > A
The G nucleotide at the first position following the last (792nd) of the exon is substituted with an A. This is speculated to be associated with abnormal splicing.
Example 6) Exon deletion
●c. 458-?_627 +?del
At least one exon (DNA sequence from C. 458 to C. 627) is deleted (unknown nucleotides in the deleted intron region are indicated by “?”).
In addition, to assess whether the variant obtained causes disease or not, registration of the variant in databases such as InSiGHT (http://insight-group.org/variants/database/) and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) should be checked, and a comprehensive assessment based on the results is sometimes required.
It cannot be necessarily said that variants listed in databases cause disease, and careful management is required. Variants are usually classified into 5 categories according to whether they can potentially be associated with disease or not (Attachment Table 1).
Attachment Table 1.
Number of Colorectal Cancers According to the Phenotype of Familial Adenomatous Polyposis.
| Number of colorectal cancers | Profuse type | Sparse type | AFAP | Total |
|---|---|---|---|---|
| 1 | 18 (62.1%) | 50 (73.5%) | 15 (83.3%) | 83 (72.2%) |
| 2 | 4 (13.8%) | 13 (19.1%) | 3 (16.7%) | 20 (17.4%) |
| 3 | 6 (20.7%) | 3 (4.4%) | 0 (0.0%) | 9 (7.8%) |
| 4 | 0 (0.0%) | 1 (1.5%) | 0 (0.0%) | 1 (0.9%) |
| 5 | 1 (3.4%) | 1 (1.5%) | 0 (0.0%) | 2 (1.7%) |
AFAP: attenuated FAP
Attachment
I. Familial adenomatous polyposis
Familial adenomatous polyposis in Japan: Data from the JSCCR “A retrospective multicenter study of familial adenomatous polyposis” (n = 303) (Attachment Table 1-5)
Attachment Table 2.
Stage of Colorectal Cancer According to the Phenotype of Familial Adenomatous Polyposis.
| Stage* | Profuse type | Sparse type | AFAP | Total |
|---|---|---|---|---|
| Stage I | 8 (27.6%) | 18 (26.9%) | 5 (27.8%) | 31 (27.2%) |
| Stage II | 4 (13.8%) | 14 (20.9%) | 6 (33.3%) | 24 (21.1%) |
| Stage III a | 5 (17.2%) | 14 (20.9%) | 2 (11.1%) | 21 (18.4%) |
| Stage III b | 8 (27.6%) | 7 (10.4%) | 3 (16.7%) | 18 (15.8%) |
| Stage IV | 4 (13.8%) | 14 (20.9%) | 2 (11.1%) | 20 (17.5%) |
*: according to the classification proposed by JSSCR
Attachment Table 3.
Cumulative Incidence of Development of Colorectal Cancer and Duodenal Adenoma.
| Colorectal cancer | Duodenal cancer | ||||||
|---|---|---|---|---|---|---|---|
| Age (Years) | Profuse type | Sparse type | AFAP | Profuse type | Sparse type | AFAP | |
| 20 | 0% | 1.1% | 0% | 1.3% | 1.1% | 0% | |
| 30 | 21.4% | 9.6% | 13.1% | 7.8% | 8.4% | 5.8% | |
| 40 | 47.7% | 41.1% | 20.4% | 32.4% | 28.4% | 18.4% | |
| 50 | 68.3% | 54.8% | 31.1% | 52.1% | 38.3% | 23.5% | |
| 60 | - | 80.2% | 55.3% | 71.3% | 48.9% | 23.5% | |
Attachment Table 4.
Cumulative Risk of Development of Desmoid Tumors after Colorectal Resection.
| 1y | 2y | 3y | 4y | 5y | |
|---|---|---|---|---|---|
| Cumulative risk (%) | 5.20% | 10.20% | 11.90% | 12.90% | 13.40% |
Attachment Table 5.
Postoperative Complications after Restorative Proctocolectomy+Ileal-Pouch Anal Anastomosis.
| Conventional open surgery | Stapled anastomosis | Hand-sewn anastomosis | |
|---|---|---|---|
| Intestinal obstruction | 6.5% | 8.1% | 5.7% |
| Anastomotic leakage | 0% | 0% | 0.9% |
| Anastomotic stricture | 3.4% | 0% | 5.7% |
| Intra-abdominal abscess | 3.5% | 0% | 4.0% |
| Wound infection | 2.2% | 0% | 4.0% |
| Dysuria | 1.3% | 0% | 1.1% |
| Erectile dysfunction | 0% | 0% | 1.5% |
| Ejaculatory dysfunction | 12.5% | 4.3% | 4.7% |
II. Lynch syndrome
Germline mutations in Lynch syndrome in Japan: Data from the JSCCR “Registration and genetic analysis of HNPCC (secondary study)” (Attachment Table 6)
Attachtment Table 6.
Germline Mutations in Lynch Syndrome in Japan: Data from the JSCCR “Registration and Genetic Analysis of HNPCC (Secondary Study) ”.
| Mismatch repair gene | Type of mutation | Regions (Sites) of mutation | Germline nucleotide change |
|---|---|---|---|
| MLH1 | Substitution | Exon1
Exon2 Exon2 Intron2, splice acceptor site Exon8 Intron9, splice donor site Intron9, splice donor site Exon13 Intron13, splice donor site Exon15 Intron15, splice donor site Exon19 |
c.37G>T (p.E13*)
c.122A>G (p.D41G) c.199G>A (p.G67R) c.208-2A>G (aberrant splicing) c.677G>A (p.R226Q, aberrant splicing) c.790+5G>T (aberrant splicing) c.790+1G>A (aberrant splicing) c.1459C>T (p.R487*) c.1558+1G>A (aberrant splicing) c.1731G>A (aberrant splicing) c.1731+5zG>A (aberrant splicing) c.2250C>G (p.Y750*) |
| Deletion | Intron2, splice donor site
Exon3 Exon4 Exon6 Exon6 Intron6, splice donor site Exon16 Exon1-5 Exon5 |
c.207+1_c.207+2delGT (aberrant splicing)
c.209_211delAAG (aberrant splicing) c.319_320delAT (p.I107Kfs*14) c.472delA (p.N158Tfs*2) c.523delA (p.I176Ffs*26) c.545+3delAC (aberrant splicing) c.1846_1848delAAG (p.K616del) c.1-94968_c.453+696del109180 (exon1-5 deletion) c.381-431_c.453+717del1221 |
|
| Insertion | Exon5
Exon15 Exon19 |
c.440_441insT (p.T148Dfs*24)
c.1672_1673insAACT (p.F560Tfs*8) c.2198_2199insTT (p.T735Sfs*49) |
|
| Deletion/insertion | Exon12 | c.1039-4215_c.1409+2347del6933ins101 (exon12 deletion) | |
| Duplication | Exon6
Intron9, splice donor site Exon12-13 |
c.464dupT (p.Y157Lfs*15)
c.790+2dupT (aberrant splicing) c.1039-?_1558+? (exon12-13 duplication) |
|
| MSH2 | Substitution | Intron1, splice donor site
Intron5, splice donor site Exon7 Exon7 Exon7 Exon7 Intron9, splice acceptor site Intron10, splice donor site Exon12 Exon12 Exon12 Exon14 Exon15 |
c.211+1G>C (aberrant splicing)
c.942+3A>T (aberrant splicing) c.1165C>T (p.R389*) c.1204C>T (p.Q402*) c.1225C>T (p.Q409*) c.1255C>T (p.Q419*) c1511-1G>A (aberrant splicing) c.1661+1G>A (aberrant splicing) c.1861C>T (p.R621*) c.1865C>T (p.P622L) c.1915C>T (p.H639Y, aberrant splicing) c.2455A>T (p.K819*) c.2563C>T (p.Q855*) |
| Deletion | Exon2
Exon7 Exon11 Exon11 Exon13 Exon14 Exon1 Exon1-6 Exon6-7 |
c.274_276delCTT (p.L92del)
c.1226_1227delAG (p.Q409Rfs*7) c.1705_1706delGA (p.E569Ifs*2) c.1744delG (p.V582Sfs*8) c.2031_2032delAT (p.I679Sfs*19) c.2309delT (p.I770Mfs*42) c.1-7550_c211+2019del9780 (exon1 deletion) c.1-19640_c1076+10104del42982 (exon1-6 deletion) c.943-596_c1276+12033del26275 (exon6-7 deletion) |
|
| Deletion/insertion | Exon14 | c.2300_2303delCAGAinsATATATAT (p.S767Yfs*20) | |
| Duplication | Exon5-6
Exon7 Exon2 Exon13 |
c.793-455_c1076+5894dup8510 (exon5-6 duplication)
c.1077-10584_c1276+207dup10991 (exon7 duplication) Exon2 duplication Exon13 duplication |
|
| MSH6 | Duplication | Exon5
Exon5 |
c.3261dupC (p.F1088Lfs*5)
c.3403dupC (p.N1136Lfs*31) |
[Translated from Japanese235) to English with a permission from the publisher of the original version]
Conflicts of Interest
Kei Muro received lecture fees from Chugai, Takeda, and Eli Lilly and research fundings from MSD, Daiichi Sankyo, Kyowa Hakko Kirin, Ono, Shionogi, and Gilead Sciences. Takako Nakajima received lecture fees from Taiho Pharm., Chugai Pharm and also received research funding from Taiho Pharm., Chugai Pharm., Takeda Pharm., MSD, Ono Pharm., AstraZeneca, and Dainippon Sumitomo Pharm. Takayuki Yoshino received a research funding from MSD K.K. and GlaxoSmithKline K.K. Shinji Tanaka received research funding from EA Pharma Co., Ltd., Otsuka Pharmaceutical Co., Ltd., JIMRO Pharmaceutical Co., Ltd., Takeda Pharmaceutical Company Ltd., Daiichi Sankyo, Ltd., Sumitomo Bakelite Co., Ltd., Asahi Kasei Medical CO., Ltd., Mochida Pharmaceutical Co., Ltd., Janssen Pharmaceutical K.K., Zeria Pharmaceutical Co., Ltd. and also received lecture fees: EA Pharma Co., Ltd., Olympus Co., Ltd. Narikazu Boku received research funds from Taiho Pharmaceutical and Ono Pharmaceutical. Yasuhiro Shimada received research funding from Taiho, Lilly, Merk-Serono, and MSD.
The other authors have no conflicts of interest and funding that should be declared.
Source of Funding
Development of the JSCCR Guidelines 2016 for HCRC was funded by the JSCCR.
References
- 1.Bussey HJR. Familial polyposis coli. Baltimore and London: Johns Hopkins University Press; 1975. Chapter 3, Genetic and epidemiological features of familial polyposis coli; p.9-17. [Google Scholar]
- 2.Iwama T, Tamura K, Morita T, et al. A clinical overview of familial adenomatous polyposis derived from the database of the Polyposis Registry of Japan. Int J Clin Oncol. 2004 Aug;9(4):308-16. [DOI] [PubMed] [Google Scholar]
- 3.Takayama T, Ohi M, Hayashi T, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001 Sep;121(3):599-611. [DOI] [PubMed] [Google Scholar]
- 4.Vögelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-development. N Engl J Med. 1988 Sep;319(9):525-32. [DOI] [PubMed] [Google Scholar]
- 5.Murata M, Utsunomiya J, Iwama T, et al. Frequency of adenomatosis coli in Japan. J Human Genet. 1981 Mar;26(1):19-30. [DOI] [PubMed] [Google Scholar]
- 6.Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis(FAP). Gut. 2008 May;57(5):704-13. [DOI] [PubMed] [Google Scholar]
- 7.Japanese Society for Cancer of the Colon and Rectum: General rules for clinical and pathological studies on cancer of the colon, rectum and anus. Tokyo Kanahara Publ Corp; 2013 [Google Scholar]
- 8.Hamilton SR, Liu B, Parson RE, et al. The molecular basis of Turcot's syndrome. N Engl J Med. 1995 Mar;332(13):839-47. [DOI] [PubMed] [Google Scholar]
- 9.Grover S, Kastrinos F, Steyerberg EW, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012 Aug;308(5):485-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagase H, Miyoshi Y, Horii A, et al. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res. 1992 Jul;52(14):4055-7. [PubMed] [Google Scholar]
- 11.Nugent KP, Phillips RK, Hodgson SV, et al. Phenotypic expression in familial adenomatous polyposis: partial prediction by mutation analysis. Gut. 1994 Nov;35(11):1622-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knudsen AN, Bisgaard ML, Bülow S. Attenuated familial adenomatous polyposis (AFAP): A review of the literature. Fam Cancer. 2003 Apr;2(1):43-55. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi LK, Burke CA, Bennett AE, et al. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008 Feb;6(2):180-5. [DOI] [PubMed] [Google Scholar]
- 14.Aretz S, Stienen D, Friedrichs N, et al. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP). Hum Mutat. 2007 Oct;28(10):985-92. [DOI] [PubMed] [Google Scholar]
- 15.Hes FJ, Nielsen M, Bik EC, et al. Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut. 2008 Jan;57(1):71-6. [DOI] [PubMed] [Google Scholar]
- 16.Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G: C→T: A mutations in colorectal tumors. Nat Genet. 2002 Feb;30(2):227-32. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen M, Franken PF, Reinards THC, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet. 2005 Sep;42(9):e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubbe SJ, Di Bernardo MC, Chandler IP, et al. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009 Aug;27(24):3975-80. [DOI] [PubMed] [Google Scholar]
- 19.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013 Feb;45(2):136-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015 Jul;137(2):320-31. [DOI] [PubMed] [Google Scholar]
- 21.Bellido F, Pineda M, Aiza G, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016 Apr;18(4):325-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasen HF, van Duijvendijk P, Buskens E, et al. Decision analysis in the surgical treatment of patients with familial adenomatous polyposis: A Dutch-Scandinavian collaborative study including 659 patients. Gut. 2001 Aug;49(2):231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kartheuser A, Stangherlin P, Brandt D, et al. Restorative proctocolectomy and ileal pouch-anal anastomosis for familial adenomatous polyposis revisited. Fam Cancer. 2006 Sep;5(3):241-60. [DOI] [PubMed] [Google Scholar]
- 24.Ueno H, Kobayashi H, Konishi T, et al. Prevalence of laparoscopic surgical treatment and its clinical outcomes in patients with familial adenomatous polyposis in Japan. Int J Clin Oncol. 2016 Aug;21(4):713-22. [DOI] [PubMed] [Google Scholar]
- 25.Konishi T, Ishida H, Ueno H, et al. Feasibility of laparoscopic total proctocolectomy with ileal pouch-anal anastomosis and total colectomy with ileorectal anastomosis for familial adenomatous polyposis: results of a nationwide multicenter study. Int J Clin Oncol. 2016 Oct;21(5):953-61. [DOI] [PubMed] [Google Scholar]
- 26.Parc Y, Piquard A, Dozois RR, et al. Long-term outcome of familial adenomatous polyposis patients after restorative coloproctectomy. Ann Surg. 2004 Mar;239(3):378-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson-Fawcett MW, Marcus VA, Redston M, et al. Adenomatous polyps develop commonly in the ileal pouch of patients with familial adenomatous polyposis. Dis Colon Rectum. 2001 Mar;44(3):347-53. [DOI] [PubMed] [Google Scholar]
- 28.Groves CJ, Beveridge IG, Swain DJ, et al. Prevalence and morphology of pouch and ileal adenomas in familial adenomatous polyposis. Dis Colon Rectum. 2005 Apr;48(4):816-23. [DOI] [PubMed] [Google Scholar]
- 29.Tajika M, Niwa Y, Bhatia V, et al. Risk of ileal pouch neoplasms in patients with familial adenomatous polyposis. World J Gastroenterol. 2013 Oct;19(40):6774-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoehner JC, Metcalf AM. Development of invasive adenocarcinoma following colectomy with ileoanal anastomosis for familial polyposis coli. Dis Colon Rectum. 1994 Aug;37(8):824-8. [DOI] [PubMed] [Google Scholar]
- 31.Ault GT, Nunoo-Mensah JW, Johnson L, et al. Adenocarcinoma arising in the middle of ileoanal pouches: report of five cases. Dis Colon Rectum. 2009 Mar;52(3):538-41. [DOI] [PubMed] [Google Scholar]
- 32.Angriman I, Scarpa M, Castagliuolo I. Relationship between pouch microbiota and pouchitis following restorative proctocolectomy for ulcerative colitis. World J Gastroenterol. 2014 Aug;20(29):9665-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal. Version 2.2015. [Internet]. 2015 Oct 7 [cited 2016 Jan 14] Available from: http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf [DOI] [PubMed] [Google Scholar]
- 34.Speake D, Evans DG, Lalloo F, et al. Desmoid tumours in patients with familial adenomatous polyposis and desmoids region adenomatous polyposis coli mutations. Br J Surg. 2007 Aug;94(8):1009-13. [DOI] [PubMed] [Google Scholar]
- 35.Bennett RL, Steinhaus KA, Uhrich SB, et al. Recommendations for standardized human pedigree nomenclature. Pedigree Standardization Task Force of the National Society of Genetic Counselors. Am J Hum Genet. 1995 Mar;56(3):745-52. [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett RL, French KS, Resta RG, et al. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Couns. 2008 Oct;17(5):424-33. [DOI] [PubMed] [Google Scholar]
- 37.Nieuwenhuis MH, Bülow S, Björk J, et al. Genotype predicting phenotype in familial adenomatous polyposis: A practical application to the choice of surgery. Dis Colon Rectum. 2009 Jul;52(7):1259-63. [DOI] [PubMed] [Google Scholar]
- 38.The Japanese Association of Medical Sciences. Guidelines for genetic tests and diagnoses in medical practice. J Jpn Med Assoc. 2011 Mar;140(2):355-61. Japanese. [Google Scholar]
- 39.Matsubara N, Isozaki H, Tanaka N. The farthest 3'distant end APC mutation identified in attenuated adenomatous polyposis coli with extracolonic manifestations. Dis Colon Rectum. 2000 May;43(5):720-1. [DOI] [PubMed] [Google Scholar]
- 40.Evans DG, Guy SP, Thakker N, et al. Non-penetrance and late appearance of polyps in families with familial adenomatous polyposis. Gut. 1993 Oct;34(10):1389-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004 Aug;127(2):444-51. [DOI] [PubMed] [Google Scholar]
- 42.Utsunomiya J, Iwama T, Imajo M, et al. Total colectomy, mucosal proctectomy, and ileoanal anastomosis. Dis Colon Rectum. 1980 Oct;23(7):459-66. [DOI] [PubMed] [Google Scholar]
- 43.Bertario L, Russo A, Radice P, et al. Genotype and phenotype factors as determinants for rectal stump cancer in patients with familial adenomatous polyposis. Ann Surg. 2000 Apr;231(4):538-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Church J. In which patients do I perform IRA, and why? Fam Cancer. 2006 Sep;5(3):237-40. [DOI] [PubMed] [Google Scholar]
- 45.Aziz O, Athanasiou T, Fazio VW, et al. Meta-analysis of observational studies of ileorectal versus ileal pouch-anal anastomosis for familial adenomatous polyposis. Br J Surg. 2006 Apr;93(4):407-17. [DOI] [PubMed] [Google Scholar]
- 46.McMullen K, Hicks TC, Ray JE, et al. Complications associated with ileal pouch-anal anastomosis. World J Surg. 1991 Nov-Dec;15(6):763-6. [DOI] [PubMed] [Google Scholar]
- 47.Weston-Petrides GK, Lovegrove RE, et al. Comparison of outcomes after restorative proctocolectomy with or without defunctioning ileostomy. Arch Surg. 2008 Apr;143(4):406-12. [DOI] [PubMed] [Google Scholar]
- 48.Remzi FH, Fazio VW, Gorgun E, et al. The outcome after restorative proctocolectomy with or without defunctioning ileostomy. Dis Colon Rectum. 2006 Apr;49(4):470-7. [DOI] [PubMed] [Google Scholar]
- 49.Ganschow P, Warth R, Hinz U, et al. Early postoperative complications after stapled vs handsewn restorative proctocolectomy with ileal pouch-anal anastomosis in 148 patients with familial adenomatous polyposis coli: a matched-pair analysis. Colorectal Dis. 2014 Feb;16(2):116-22. [DOI] [PubMed] [Google Scholar]
- 50.Mozafar M, Shateri K, Tabatabaey A, et al. Familial adenomatous polyposis: ileo-anal pouch versus ileo-rectal anastomosis. Gastroenterol Hepatol Bed Bench. 2014 Fall;7(4):206-10. [PMC free article] [PubMed] [Google Scholar]
- 51.Kennedy RD, Zarroug AE, Moir CR, et al. Ileal pouch anal anastomosis in pediatric familial adenomatous polyposis: A 24-year review of operative technique and patient outcomes. J Pediatr Surg. 2014 Sep;49(9):1409-12. [DOI] [PubMed] [Google Scholar]
- 52.Mennigen R, Sewald W, Senninger N, et al. Morbidity of loop ileostomy closure after restorative proctocolectomy for ulcerative colitis and familial adenomatous polyposis: A systematic review. J Gastrointestinal Surg. 2014 Dec;18(12):2192-200. [DOI] [PubMed] [Google Scholar]
- 53.Iwama T. [Pathological study of adenomatous coli.] Jpn J Surg. 1978 Jan;79(1):10-24. Japanese. [Google Scholar]
- 54.Sturt NJ, Gallagher MC, Bassett P, et al. Evidence for genetic predisposition to desmoid tumors in familial adenomatous polyposis independent of the germline APC mutation. Gut. 2004 Dec;53(12):1832-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rozen P, Macrae F. Familial adenomatous polyposis: the practical applications of clinical and molecular screening. Fam Cancer. 2006 Sep;5(3):227-35. [DOI] [PubMed] [Google Scholar]
- 56.Crabtree MD, Tomlinson IP, Talbot IC, et al. Variability in the severity of colonic disease infamilial adenomatous polyposis results from differences in tumour initiation rather than progression and depends relatively little on patient age. Gut. 2001 Oct;49(4):540-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olsen KØ, Juul S, Bülow S, et al. Female fecundity before and after operation for familial adenomatous polyposis. Br J Surg. 2003 Jun;90(6):227-31. [DOI] [PubMed] [Google Scholar]
- 58.Kartheuser AH, Parc R, Penna CP, et al. Ileal pouch-anal anastomosis as the first choice operation in patients with familial adenomatous polyposis: A ten-year experience. Surgery. 1996 Jun;119(6):615-23. [DOI] [PubMed] [Google Scholar]
- 59.Rozen P, Samuel Z, Rabau M, et al. Familial adenomatous polyposis at the Tel Aviv Medical Center: demographic and clinical features. Fam Cancer. 2001 Apr;1(2):75-82. [DOI] [PubMed] [Google Scholar]
- 60.Eccles DM, Lunt PW, Wallis Y, et al. An unusually severe phenotype for familial adenomatous polyposis. Arch Dis Child. 1997 Nov;77(5):431-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campos FG. Surgical treatment of familial adenomatous polyposis: dilemmas and current recommendations. World J Gastroenterol. 2014 Nov;20(44):16620-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kjaer MD, Laursen SB, Qvist N, et al. Sexual function and body image are similar after laparoscopy-assisted and open ileal pouch-anal anastomosis. World J Surg. 2014 Sep;38(9):2460-5. [DOI] [PubMed] [Google Scholar]
- 63.Fajardo AD, Dharmarajan S, George V, et al. Laparoscopic versus open 2-stage ileal pouch: Laparoscopic approach allows for faster restoration of intestinal continuity. J Am Coll Surg. 2010 Sep;211(3):377-83. [DOI] [PubMed] [Google Scholar]
- 64.Luz Moreira A, Church JM, Burke CA. The evolution of prophylactic colorectal surgery for familial adenomatous polyposis. Dis Colon Rectum. 2009 Aug;52(8):1481-6. [DOI] [PubMed] [Google Scholar]
- 65.Johansen C, Bitsch M, Bülow S. Fertility and pregnancy in women with familial adenomatous polyposis. Int J Colorectal Dis. 1990 Dec;5(4):203-6. [DOI] [PubMed] [Google Scholar]
- 66.Nieuwenhuis MH, Douma KF, Bleiker EM, et al. Female fertility after colorectal surgery for familial adenomatous polyposis: a nationwide cross-sectional study. Ann Surg. 2010 Aug;252(2):341-4. [DOI] [PubMed] [Google Scholar]
- 67.Oresland T, Palmblad S, Ellström M, et al. Gynaecological and sexual function related to anatomical changes in the female pelvis after restorative proctocolectomy. Int J Colorectal Dis. 1994 May;9(2):77-81. [DOI] [PubMed] [Google Scholar]
- 68.Bartels SA, D'Hoore A, Cuesta MA, et al. Significantly increased pregnancy rates after laparoscopic restorative proctocolectomy: a cross-sectional study. Ann Surg. 2012 Dec;256(6):1045-8. [DOI] [PubMed] [Google Scholar]
- 69.Remzi FH, Gorgun E, Bast J, et al. Vaginal delivery after ileal pouch-anal anastomosis: a word of caution. Dis Colon Rectum. 2005 Sep;48(9):1691-9. [DOI] [PubMed] [Google Scholar]
- 70.Juhasz ES, Fozard B, Dozois RR, et al. Ileal pouch-anal anastomosis function following childbirth. An extended evaluation. Dis Colon Rectum. 1995 Feb;38(2):159-65. [DOI] [PubMed] [Google Scholar]
- 71.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002 Apr;346(14):1054-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000 Jun;342(26):1946-52. [DOI] [PubMed] [Google Scholar]
- 73.Higuchi T, Iwama T, Yoshinaga K, et al. A randomized, double-blind, placebo-controlled trial of the effects of rofecoxib, a selective cyclooxygenase-2 inhibitor, on rectal polyps in familial adenomatous polyposis patients. Clin Cancer Res. 2003 Oct;9(13):4756-60. [PubMed] [Google Scholar]
- 74.Baron JA, Sandler RS, Bresalier RS, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006 Dec;131(6):1674-82. [DOI] [PubMed] [Google Scholar]
- 75.Koskenvuo L, Renkonen-Sinisalo L, Järvinen HJ, et al. Risk of cancer and secondary proctectomy after colectomy and ileorectal anastomosis in familial adenomatous polyposis. Int J Colorectal Dis. 2014 Feb;29(2):225-30. [DOI] [PubMed] [Google Scholar]
- 76.Campos FG, Perez RO, Imperiale AR, et al. Surgical treatment of familial adenomatous polyposis: ileorectal anastomosis or restorative proctolectomy? Arq Gastroenterol. 2009 Oct-Dec;46(4):294-9. [DOI] [PubMed] [Google Scholar]
- 77.Bülow S, Bülow C, Vasen H, et al. Colectomy and ileorectal anastomosis is still an option for selected patients with familial adenomatous polyposis. Dis Colon Rectum. 2008 Sep;51(9):1318-23. [DOI] [PubMed] [Google Scholar]
- 78.Church J, Simmang C. Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer). Dis Colon Rectum. 2003 Aug;46(8):1001-12. [DOI] [PubMed] [Google Scholar]
- 79.Goodman AJ, Dundas SAC, Scholefield JH, et al. Gastric carcinoma and familial adenomatous polyposis (FAP). Colorectal Dis. 1988 Nov;3(4):201-3. [DOI] [PubMed] [Google Scholar]
- 80.Utsunomiya J, Maki T, Iwama T, et al. Gastric lesion of familial polyposis coli. Cancer. 1974 Sep;34(3):745-54. [DOI] [PubMed] [Google Scholar]
- 81.Offerhaus GJ, Giardiello FM, Krush AJ, et al. The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology. 1992 Jun;102(6):1980-2. [DOI] [PubMed] [Google Scholar]
- 82.Park JG, Park KJ, Ahn YO, et al. Risk of gastric cancer among Korean familial adenomatous polyposis patients. Dis Colon Rectum. 1992 Oct;35(10):996-8. [DOI] [PubMed] [Google Scholar]
- 83.Iwama T, Mishima Y, Utsunomiya J. The impact of familial adenomatous polyposis on the tumorigenesis and mortality at the several organs; its rational treatment. Ann Surg. 1993 Feb;217(2):101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Galle TS, Juel K, Bülow S. Causes of death in familial adenomatous polyposis. Scand J Gastroenterol. 1999 Aug;34(8):808-12. [DOI] [PubMed] [Google Scholar]
- 85.Bülow S, Björk J, Christensen IJ, et al. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004 Mar;53(3):381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iida M, Yao T, Itoh H, et al. Natural history of duodenal lesion in Japanese patients with familial adenomatosis coli (Gardner's syndrome). Gastroenterology. 1989 May;96(5Pt 1):1301-6. [DOI] [PubMed] [Google Scholar]
- 87.Moozar KL, Madlensky L, Berk T, et al. Slow progression of periampullary neoplasia in familial adenomatous polyposis. J Gastrointest Surg. 2002 Nov-Dec;6(6):831-7. [DOI] [PubMed] [Google Scholar]
- 88.Brosens LA, Keller JJ, Offerhaus GJ, et al. Prevention and management of duodenal polyposis in familial adenomatous polyposis. Gut. 2005 Jul;54(7):1034-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iida M, Hizawa K, Matsumoto T, et al. [Long-term follow-up study of gastroduodenal lesions in patients with familial adenomatous polyposis.] Stomach and Intestine. 1997 Mar;32(4):563-76. Japanese. [Google Scholar]
- 90.Yamaguchi T, Ishida H, Ueno H, et al. Upper gastrointestinal tumours in Japanese familial adenomatous polyposis patients. Jpn J Clin Oncol. 2016 Apr;46(4):310-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spigelman AD, Williams CB, Talbot IC, et al. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet. 1989 Sep;334(8666):783-5. [DOI] [PubMed] [Google Scholar]
- 92.Saurin JC, Gutknecht C, Napoleon B, et al. Surveillance of duodenal adenomas in familial adenomatous polyposis reveals high cumulative risk of advanced disease. J Clin Oncol. 2004 Feb;22(3):493-8. [DOI] [PubMed] [Google Scholar]
- 93.Lopez-Ceron M, van den Broek FJ, Mathus-Vliegen EM, et al. The role of high-resolution endoscopy and narrow-band imaging in the evaluation of upper GI neoplasia in familial adenomatous polyposis. Gastrointest Endosc. 2013 Apr;77(4):542-50. [DOI] [PubMed] [Google Scholar]
- 94.Groves CJ, Saunders BP, Spigelman AD, et al. Duodenal cancer in patients with familial adenomatous polyposis(FAP): Results of a 10 year prospective study. Gut. 2002 May;50(5):636-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Penninga L, Svendsen LB. Pancreas-preserving total duodenectomy: A 10-year experience. J Hepatobiliary Pancreat Sci. 2011 Sep;18(5):717-23. [DOI] [PubMed] [Google Scholar]
- 96.van Heumen BW, Nieuwenhuis MH, van Goor H, et al. Surgical management for advanced duodenal adenomatosis and duodenal cancer in Dutch patients with familial adenomatous polyposis: a nationwide retrospective cohort study. Surgery. 2012 May;151(5):681-90. [DOI] [PubMed] [Google Scholar]
- 97.Schlemper RJ, Riddell RH, Kato Y, et al. The Vienna classification of gastrointestinal epithelial neoplasia. Gut. 2000 Aug;47(2):251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alexander JR, Andrews JM, Buchi KN, et al. High prevalence of adenomatous polyps of the duodenal papilla in familial adenomatous polyposis. Dig Dis Sci. 1989 Feb;34(2):167-70. [DOI] [PubMed] [Google Scholar]
- 99.Yao T, Iida M, Ohsato K, et al. Duodenal lesions in familial polyposis of the colon. Gastroenterology. 1977 Nov;73(5):1086-92. [PubMed] [Google Scholar]
- 100.Trimbath DJ, Griffin C, Romans K, et al. Attenuated familial adenomatous polyposis presenting as ampullary adenocarcinoma. Gut. 2003 Jun;52(6):903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iwama T, Tomita H, Kawachi Y, et al. Indications for local excision of ampullary lesions associated with familial adenomatous polyposis. J Am Coll Surg. 1994 Oct;179(4):462-4. [PubMed] [Google Scholar]
- 102.Ouaïssi M, Panis Y, Sielezneff I, et al. Long-term outcome after ampullectomy for ampullary lesions associated with familial adenomatous polyposis. Dis Colon Rectum. 2005 Dec;48(12):2192-6. [DOI] [PubMed] [Google Scholar]
- 103.Norton ID, Geller A, Petersen B, et al. Endoscopic surveillance and ablative therapy for periampullary adenomas. Am J Gastroenterol. 2001 Jan;96(1):101-6. [DOI] [PubMed] [Google Scholar]
- 104.Matsumoto T, Iida M, Nakamura S, et al. Natural history of ampullary adenoma in familial adenomatous polyposis: Reconfirmation of benign nature during extended surveillance. Am J Gastroenterol. 2000 Jun;95(6):1557-62. [DOI] [PubMed] [Google Scholar]
- 105.Ma T, Jang EJ, Zukerberg LR, et al. Recurrences are common after endoscopic ampullectomy for adenoma in the familial adenomatous polyposis(FAP)syndrome. Surg Endosc. 2014 Aug;28(8):2349-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gluck N, Strul H, Rozner G, et al. Endoscopy and EUS are key for effective surveillance and management of duodenal adenomas in familial adenomatous polyposis. Gastrointest Endosc. 2015 Apr;81(4):960-6. [DOI] [PubMed] [Google Scholar]
- 107.Chung RS, James M, van Stolk R. Pancreas-sparing duodenectomy: Indications, surgical technique, and results. Surgery. 1995 Mar;117(3):254-9. [DOI] [PubMed] [Google Scholar]
- 108.Burke CA, Santisi J, Church J, et al. The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am J Gastroenterol. 2005 Jul;100(7):1498-502. [DOI] [PubMed] [Google Scholar]
- 109.Matsumoto T, Esaki M, Yanaru-Fujisawa R, et al. Small-bowel involvement in familial adenomatous polyposis: Evaluation by double-baloon endoscopy and intraoperative enteroscopy. Gastrointest Endosc. 2008 Nov;68(5):911-9. [DOI] [PubMed] [Google Scholar]
- 110.Will OC, Mana RF, Phillips PSK, et al. Familial adenomatous polyposis and the small bowel: A loco-regional review and current management strategies. Pathol Res Pract. 2008 Jul;204(7):449-58. [DOI] [PubMed] [Google Scholar]
- 111.Schulz AC, Bojarski C, Buhr HJ, et al. Occurrence of adenomas in the pouch and small intestine of FAP patients after proctocolectomy with ileoanal pouch construction. Int J Colorectal Dis. 2008 Apr;23(4):437-41. [DOI] [PubMed] [Google Scholar]
- 112.Yamada A, Watabe H, Iwama T, et al. The prevalence of small intestinal polyps in patients with familial adenomatous polyposis: A prospective capsule endoscopy study. Fam Cancer. 2014 Mar;13(1):23-8. [DOI] [PubMed] [Google Scholar]
- 113.Iaquinto G, Fornasarig M, Quaia M, et al. Capsule endoscopy is useful and safe for small-bowel surveillance in familial adenomatous polyposis. Gastrointest Endosc. 2008 Jan;67(1):61-7. [DOI] [PubMed] [Google Scholar]
- 114.Wong RF, Tuteja AK, Haslem DS, et al. Video capsule endoscopy compared with standard endoscopy for the evaluation of small-bowel polyps in persons with familial adenomatous polyposis (with video). Gastrointest Endosc. 2006 Oct;64(4):530-7. [DOI] [PubMed] [Google Scholar]
- 115.Zuidema MF, Dekker W. A patient with a metastasizing jejunal carcinoma 17 years after colectomy for familial polyposis coli. Neth J Med. 1989 Jun;34(5-6):317-21. [PubMed] [Google Scholar]
- 116.Plum N, May A, Manner H, et al. Small-bowel diagnosis in patients with familial adenomatous polyposis: comparison of push enteroscopy, capsule endoscopy, ileoscopy, and enteroclysis. Z Gastroenterol. 2009 Apr;47(4):339-46. [DOI] [PubMed] [Google Scholar]
- 117.Knudsen AL, Bülow S. Desmoid tumor in familial adenomatous polyposis. Fam Cancer 2001 Apr;1(2):111-9. [DOI] [PubMed] [Google Scholar]
- 118.Nieuwenhuis MH, De Vos Tot Nederveen Cappel W, Botma A, et al. Desmoid tumors in a dutch cohort of patients with familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008 Feb;6(2):215-9. [DOI] [PubMed] [Google Scholar]
- 119.Bertario L, Presciuttini S, Sala P, et al. Causes of death and postsurgical survival in familial adenomatous polyposis: results from the Italian Registry. Italian Registry of Familial Polyposis Writing Committee. Semin Surg Oncol. 1994 May-Jun;10(3):225-34. [DOI] [PubMed] [Google Scholar]
- 120.Latchford AR, Sturt NJ, Neale K, et al. A 10-year review of surgery for desmoid disease associated with familial adenomatous polyposis. Br J Surg. 2006 Feb;93(2):1258-64. [DOI] [PubMed] [Google Scholar]
- 121.Church J, Lynch C, Neary P, et al. A desmoid tumor-staging system separates patients with intra-abdominal, familial adenomatous polyposis-associated desmoid disease by behavior and prognosis. Dis Colon Rectum. 2008 Jun;51(6):897-901. [DOI] [PubMed] [Google Scholar]
- 122.Stevenson JK, Reid BJ. Unfamiliar aspects of familial polyposis coli. Am J Surg. 1986 Jul;152(1):81-6. [DOI] [PubMed] [Google Scholar]
- 123.Benoit L, Faivre L, Cheynel N, et al. 3'Mutation of the APC gene and family history of FAP in a patient with apparently sporadic desmoid tumors. J Clin Gastroenterol. 2007 Mar;41(3):297-300. [DOI] [PubMed] [Google Scholar]
- 124.Tsukada K, Church JM, Jagelman DG, et al. Systemic cytotoxic chemotherapy and radiation therapy for desmoid in familial adenomatous polyposis. Dis Colon Rectum. 1991 Dec;34(12):1090-2. [DOI] [PubMed] [Google Scholar]
- 125.Hansmann A, Adolph C, Vogel T, et al. High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer. 2004 Feb;100 (3):612-20. [DOI] [PubMed] [Google Scholar]
- 126.Sturt NJ, Clark SK. Current ideas in desmoid tumours. Fam Cancer. 2006 Sep;5(3):275-85. [DOI] [PubMed] [Google Scholar]
- 127.Nieuwenhuis MH, Mathus-Vliegen EM, Baeten CG, et al. Evaluation of management of desmoid tumours associated with familial adenomatous polyposis in Dutch patients. Br J Cancer. 2011 Jan;104(1):37-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Desurmont T, Lefèvre JH, Shields C, et al. Desmoid tumour in familial adenomatous polyposis patients: responses to treatments. Fam Cancer. 2015 Mar;14(1):31-9. [DOI] [PubMed] [Google Scholar]
- 129.Clark SK, Neale KF, Landgrebe JC, Phillips RK, et al. Desmoid tumours complicating familial adenomatous polyposis. Br J Surg. 1999 Sep;86(9):1185-9. [DOI] [PubMed] [Google Scholar]
- 130.Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res. 2010 Oct;16(19):4884-91. [DOI] [PubMed] [Google Scholar]
- 131.Ishida H, Watanabe Y, Chika N, et al. [Management for extracolonic manifestations in familial adenomatous polyposis―gastro-duodenal lesions and desmoid tumors―.] J Jpn Soc Coloproctol. 2015 Oct;68(10):908-20. Japanese. [Google Scholar]
- 132.Gega M, Yanagi H, Yoshikawa R, et al. Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol. 2006 Jan;24(1):102-5. [DOI] [PubMed] [Google Scholar]
- 133.Kono T, Tomita I, Chisato N, et al. Successful low-dose chemotherapy using vinblastine and methotrexate for the treatment of an ileoanal pouch mesenteric desmoid tumor: report of a case. Dis Colon Rectum. 2004 Feb;47(2):246-9. [DOI] [PubMed] [Google Scholar]
- 134.Iwama T, Kuwabara K, Ushiama M, et al. Identification of somatic APC mutations in recurrent desmoid tumors in a patient with familial adenomatous polyposis to determine actual recurrence of the original tumor or de novo occurrence. Fam Cancer. 2009 Mar;8(1):51-4. [DOI] [PubMed] [Google Scholar]
- 135.Smith AJ, Lewis JJ, Merchant NB, et al. Surgical management of intra-abdominal desmoid tumours. Br J Surg. 2000 May;87(5):608-13. [DOI] [PubMed] [Google Scholar]
- 136.Giardiello FM, Offerhaus GJ, Lee DH, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993 Oct;34(10):1394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Steinhagen E, Guillem JG, Chang G, et al. The prevalence of thyroid cancer and benign thyroid disease in patients with familial adenomatous polyposis may be higher than previously recognized. Clin Colorectal Cancer. 2012 Dec;11(4):304-8. [DOI] [PubMed] [Google Scholar]
- 138.Bülow C, Bülow S. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group. Int J Colorectal Dis. 1997 Aug;12(4):240-2. [DOI] [PubMed] [Google Scholar]
- 139.Plail RO, Bussey HJ, Glazer G, et al. Adenomatous polyposis: An association with carcinoma of the thyroid. Br J Surg. 1987 May;74(5):377-80. [DOI] [PubMed] [Google Scholar]
- 140.Harach HR, Williams GT, Williams ED. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm. Histopathology. 1994 Dec;25(6):549-61. [DOI] [PubMed] [Google Scholar]
- 141.Cameselle-Teijeiro J, Chan JK. Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod Pathol. 1999 Apr;12(4):400-11. [PubMed] [Google Scholar]
- 142.Perrier ND, van Heerden JA, Goellner JR, et al. Thyroid cancer in patients with familial adenomatous polyposis. World J Surg. 1998 Jul;22(7):738-42. [DOI] [PubMed] [Google Scholar]
- 143.Truta B, Allen BA, Conrad PG, et al. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam Cancer. 2003 Jun;2(2):95-9. [DOI] [PubMed] [Google Scholar]
- 144.Herraiz M, Barbesino G, Faquin W, et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol. 2007 Mar;5(3):367-73. [DOI] [PubMed] [Google Scholar]
- 145.Will OC, Hansmann A, Phillips RK, et al. Adrenal incidentaloma in familial adenomatous polyposis: A long-term follow-up study and schema for management. Dis Colon Rectum. 2009 Sep;52(9):1637-44. [DOI] [PubMed] [Google Scholar]
- 146.Cetta F, Olschwang S, Petracci M, et al. Genetic alterations in thyroid carcinoma associated with familial adenomatous polyposis: Clinical implications and suggestions for early detection. World J Surg. 1998 Dec;22(12):1231-6. [DOI] [PubMed] [Google Scholar]
- 147.Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg. 2011 Mar;253(3):515-21. [DOI] [PubMed] [Google Scholar]
- 148.Attard TM, Giglio P, Koppula S, et al. Brain tumors in individuals with familial adenomatous polyposis: a cancer registry experience and pooled case report analysis. Cancer. 2007 Feb;109(4):761-6. [DOI] [PubMed] [Google Scholar]
- 149.Smith TG, Clark SK, Katz DE, et al. Adrenal masses are associated with familial adenomatous polyposis. Dis Colon Rectum. 2000 Dec;43(12):1739-42. [DOI] [PubMed] [Google Scholar]
- 150.Marchesa P, Fazio VW, Church JM, et al. Adrenal masses in patients with familial adenomatous polyposis. Dis Colon Rectum. 1997 Sep;40(9):1023-8. [DOI] [PubMed] [Google Scholar]
- 151.Hughes LJ, Michels VV. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet. 1992 Aug;43(6):1023-5. [DOI] [PubMed] [Google Scholar]
- 152.Iwama T, Mishima Y. Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer. 1994 Apr;73(8):2065-8. [DOI] [PubMed] [Google Scholar]
- 153.Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997 Jan;6(1):105-10. [DOI] [PubMed] [Google Scholar]
- 154.Phillips M, Dicks-Mireaux C, Kingston J, et al. Hepatoblastoma and polyposis coli (familial adenomatous polyposis). Med Pediatr Oncol. 1989 Nov;17(5):441-7. [DOI] [PubMed] [Google Scholar]
- 155.Thomas D, Pritchard J, Davidson R, et al. Familial hepatoblastoma and APC gene mutations: Renewed call for molecular research. Eur J Cancer. 2003 Oct;39(15):2200-4. [DOI] [PubMed] [Google Scholar]
- 156.Fuchs J, Rydzynski J, Von Schweinitz D, et al. Pretreatment prognostic factors and treatment results in children with hepatoblastoma: A report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer. 2002 Jul;95(1):172-82. [DOI] [PubMed] [Google Scholar]
- 157.Stringer MD, Hennayake S, Howard ER, et al. Improved outcome for children with hepatoblastoma. Br J Surg. 1995 Mar;82(3):386-91. [DOI] [PubMed] [Google Scholar]
- 158.Aretz S, Koch A, Uhlhaas S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer. 2006 Nov;47(6):811-8. [DOI] [PubMed] [Google Scholar]
- 159.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome(hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005 May;352(18):1851-60. [DOI] [PubMed] [Google Scholar]
- 160.Barrow E, Alduaij W, Robinson L, et al. Colorectal cancer in HNPCC: Cumulative lifetime incidence, survival and tumour distribution. A report of 121 families with proven mutations. Clin Genet. 2008 Sep;74(3):233-42. [DOI] [PubMed] [Google Scholar]
- 161.Grover S, Syngal S. Genetic testing in gastroenterology: Lynch syndrome. Best Pract Res Clin Gastroenterol. 2009 Apr;23(2):185-96. [DOI] [PubMed] [Google Scholar]
- 162.Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: A prospective cohort study. J Clin Oncol. 2012 Mar;30(9):958-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Raymond V, Mukherjee B, Wang F, et al. Elevated risk of prostate cancer among men Lynch syndrome. J Clin Oncol. 2013 May;31(14):1713-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: A report of 121 families with proven mutations. Clin Gene. 2009 Feb;75(2):141-9. [DOI] [PubMed] [Google Scholar]
- 165.Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: Later age of onset. Gastroenterology. 2005 Aug;129(2):415-21. [DOI] [PubMed] [Google Scholar]
- 166.Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009 Nov;137(5):1621-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Watson P, Vasen HF, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008. Jul;123(2):444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999 Apr;81(2):214-8. [DOI] [PubMed] [Google Scholar]
- 169.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009 Oct;302(16):1790-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kohlmann W, Gruber SB. Lynch syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews® [Internet]. Seattle, WA; University of Washington, Seattle; 1993-2016. Updated May 22, 2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1211/ [Google Scholar]
- 171.Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer. 2013 Jun;12(2):229-40. [DOI] [PubMed] [Google Scholar]
- 172.Lynch HT, Shaw MW, Magnuson CW, et al. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med. 1966 Feb;117(2):206-12. [PubMed] [Google Scholar]
- 173.Boland CR, Troncale FJ. Familial colonic cancer without antecedent polyposis. Ann Intern Med. 1984 May;100(5):700-1. [DOI] [PubMed] [Google Scholar]
- 174.Vasen HF, Mecklin JP, Khan PM, et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer(ICG-HNPCC). Dis Colon Rectum. 1991 May;34(5):424-5. [DOI] [PubMed] [Google Scholar]
- 175.Vasen HF. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J Clin Oncol. 2000 Nov;18(21 Suppl):81S-92. [PubMed] [Google Scholar]
- 176.Suter CM, Martin DI, Ward RL. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. 2004 May;36(5):497-501. [DOI] [PubMed] [Google Scholar]
- 177.Kempers MJ, Kuiper RP, Ockeloen CW, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: A cohort study. Lancet Oncol. 2011 Jan;12(1):49-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kuiper RP, Vissers LE, Venkatachalam R, et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat. 2011 Apr;32(4):407-14. [DOI] [PubMed] [Google Scholar]
- 179.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004 Feb;96(4):261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008 Aug;135(2):419-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012 Oct;308(15):1555-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Piñol V, Castells A, Andreu M, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005 Apr;293(16):1986-94. [DOI] [PubMed] [Google Scholar]
- 183.Furukawa Y. [Clinical and genetic characteristics of Japanese Lynch syndrome.] Frontiers in Colorectal Cancer. 2010 Jul;3(2):120-4. Japanese. [Google Scholar]
- 184.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008 Jul;10(4):293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yanaba K, Nakagawa H, Takeda Y, et al. Muir-Torre syndrome caused by partial duplication of MSH2 gene by Alu-mediated nonhomologous recombination. Br J Dermatol. 2008 Jan;158(1):150-6. [DOI] [PubMed] [Google Scholar]
- 186.Kanamori M, Kon H, Nobukuni T, et al. Microsatellite instability and the PTEN1 gene mutation in a subset of early onset gliomas carrying germline mutation or promoter methylation of the hMLH1 gene. Oncogene. 2000 Mar;19(12):1564-71. [DOI] [PubMed] [Google Scholar]
- 187.de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006 Mar;130(3):665-71. [DOI] [PubMed] [Google Scholar]
- 188.Canard G, Lefevre JH, Colas C, et al. Screening for Lynch syndrome in colorectal cancer: Are we doing enough? Ann Surg Oncol. 2012 Mar;19(3):809-16. [DOI] [PubMed] [Google Scholar]
- 189.Julié C, Trésallet C, Brouquet A, et al. Identification in daily practice of patients with Lynch syndrome (hereditary nonpolyposis colorectal cancer): Revised Bethesda guidelines-based approach versus molecular screening. Am J Gastroenterol. 2008 Nov;103(11):2825-35. [DOI] [PubMed] [Google Scholar]
- 190.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997 Mar;57(5):808-11. [PubMed] [Google Scholar]
- 191.Asaka S, Arai Y, Nishimura Y, et al. Microsatellite instability-low colorectal cancer acquires a KRAS mutation during the progression from Dukes' A to Dukes' B. Carcinogenesis, 2009 Mar;30(3):494-9. [DOI] [PubMed] [Google Scholar]
- 192.Koinuma K, Shitoh K, Miyakura Y, et al. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer. 2004 Jan;108(2):237-42. [DOI] [PubMed] [Google Scholar]
- 193.McGivern A, Wynter CV, Whitehall VL, et al. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer. 2004 Jun;3(2):101-7. [DOI] [PubMed] [Google Scholar]
- 194.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: Familial colorectal cancer type X. JAMA. 2005 Apr;293(16):1979-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yamaguchi T. Furukawa Y, Nakamura Y, et al. Comparison of clinical features between suspected familial colorectal cancer type X and Lynch syndrome in Japanese patients with colorectal cancer: a cross-sectional study conducted by the Japanese Society for Cancer of the Colon and Rectum. Jpn J Clin Oncol. 2015 Feb;45(2):153-9. [DOI] [PubMed] [Google Scholar]
- 196.Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut. 2013 Jun;62(6):812-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Arai M, Ogawa T, Chino A, et al. [Incidence and pathological features of tumors on surveillance by colonoscopy and upper gastroduodenal in patients with Lynch syndrome.] Fam Tumor. 2010 Jan;10(1):32-8. Japanese. [Google Scholar]
- 198.Dowty JG, Win AK, Buchanan DD, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. 2013 Mar;34(3):490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jenkins MA, Hayashi S, O'Shea AM, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology. 2007 Jul;133(1):48-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006 Jan;354(3):261-9. [DOI] [PubMed] [Google Scholar]
- 201.Aaltonen LA, Peltomäki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res. 1994 Apr;54(7):1645-8. [PubMed] [Google Scholar]
- 202.Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998 May;338(21):1481-7. [DOI] [PubMed] [Google Scholar]
- 203.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003 Mar;21(6):1174-9. [DOI] [PubMed] [Google Scholar]
- 204.Ishikubo T, Nishimura Y, Yamaguchi K, et al. The clinical features of rectal cancers with high-frequency microsatellite instability (MSI-H) in Japanese males. Cancer Lett. 2004 Dec;216(1):55-62. [DOI] [PubMed] [Google Scholar]
- 205.Jiricny J, Nyström-Lahti M. Mismatch repair defects in cancer. Curr Opin Genet Dev. 2000 Apr;10(2):157-61. [DOI] [PubMed] [Google Scholar]
- 206.Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006 Jun;354(26):2751-63. [DOI] [PubMed] [Google Scholar]
- 207.Shia J, Tang LH, Vakiani E, et al. Immunohistochemistry as first-line screening for detecting colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome: A 2-antibody panel may be as predictive as a 4-antibody panel. Am J Surg Pathol. 2009 Nov;33(11):1639-45. [DOI] [PubMed] [Google Scholar]
- 208.Shia J, Zhang L, Shike M, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1 / PMS2 deficiency. Mod Pathol. 2013 Jan;26(1):131-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tanakaya K, Yamaguchi T, Ishikawa H, et al. Causes of cancer death among first-degree relatives in Japanese families with Lynch Syndrome. Anticancer Res. 2016 Apr;36(3):1985-9. [PubMed] [Google Scholar]
- 210.Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: The advantage of more extensive colon surgery. Gut. 2011 Jul;60(7):950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Win AK, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol. 2013 Jun;20(6):1829-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sinicrope FA, Foster NR, Thibodeau SN, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011 Jun;103(11):863-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Des Guetz G, Schischmanoff O, Nicolas P, et al. Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur J Cancer. 2009 Jul;45(10):1890-6. [DOI] [PubMed] [Google Scholar]
- 214.Webber EM, Kauffman TL, O'Connor E, et al. Systematic review of the predictive effect of MSI status in colorectal cancer patients undergoing 5FU-based chemotherapy. BMC Cancer. 2015 Mar;15(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: Updated 10-year survival and outcomes according to braf mutation and mismatch repair status of the MOSAIC study. J Clin Oncol 2015 Dec;33(35):4176-87. [DOI] [PubMed] [Google Scholar]
- 216.Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011 Oct;117(20):4623-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014 Oct;20(20):5322-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Fallik D, Borrini F, Boige V, et al. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003 Sep;63(18):5738-44. [PubMed] [Google Scholar]
- 219.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015 Jun;372(26):2509-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.van Duijnhoven FJ, Botma A, Winkels R, et al. Do lifestyle factors influence colorectal cancer risk in Lynch syndrome? Fam Cancer. 2013 Jun;12(2):285-93. [DOI] [PubMed] [Google Scholar]
- 221.Tanakaya K, Furukawa Y, Nakamura Y, et al. Relationship between smoking and multiple colorectal cancers in patients with Japanese Lynch syndrome: A cross-sectional study conducted by the Japanese Society for Cancer of the Colon and Rectum. Jpn J Clin Oncol. 2015 Mar;45(3):307-10. [DOI] [PubMed] [Google Scholar]
- 222.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011 Dec;378(9809):2081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ait Ouakrim D, Dashti SG, Chau R, et al. Aspirin, ibuprofen, and the risk of colorectal cancer in Lynch syndrome. J Natl Cancer Inst. 2015 Jun;107(9). doi: 10.1093/jnci/djv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rothwell PM, Fowkes FG, Belch JF, et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011 Jan;377(9759):31-41. [DOI] [PubMed] [Google Scholar]
- 225.Rex DK, Kahi CJ, Levin B, et al. Guidelines for colonoscopy surveillance after cancer resection: A consensus update by the American Cancer Society and the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2006 May;130(6):1865-71. [DOI] [PubMed] [Google Scholar]
- 226.Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology. 2007 Oct;133(4):1093-8. [DOI] [PubMed] [Google Scholar]
- 227.Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000 May;118(5):829-34. [DOI] [PubMed] [Google Scholar]
- 228.Vasen HF, Nagengast FM, Khan PM. Interval cancers in hereditary non-polyposis colorectal cancer (Lynch syndrome). Lancet. 1995 May;345(8958):1183-4. [DOI] [PubMed] [Google Scholar]
- 229.Engel C, Rahner N, Schulmann K, et al. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol. 2010 Feb;8(2):174-82. [DOI] [PubMed] [Google Scholar]
- 230.Syngal S, Brand RE, Church JM, et al. American College of Gastroenterology: ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999 Jun;116(6):1453-6. [DOI] [PubMed] [Google Scholar]
- 232.Jass JR, Stewart SM. Evolution of hereditary non-polyposis colorectal cancer. Gut. 1992 Jun;33(6):783-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jass JR, Cottier DS, Pokos V, et al. Mixed epithelial polyps in association with hereditary non-polyposis colorectal cancer providing an alternative pathway of cancer histogenesis. Pathology. 1997 Feb;29(1):28-33. [DOI] [PubMed] [Google Scholar]
- 234.Kalady MF, Kravochuck SE, Heald B, et al. Defining the adenoma burden in Lynch syndrome. Dis Colon Rectum. 2015 Apr;58(4):388-92. [DOI] [PubMed] [Google Scholar]
- 235.Japanese Society for Cancer of the Colon and Rectum. [Japanese Society for Cancer of the Colon and Rectum (JSCCR) Guidelines 2016 for the clinical practice of hereditary colorectal cancer]. 1st ed. Tokyo: KANEHARA & CO., LTD.; 2016. 95 p. Japanese. [DOI] [PMC free article] [PubMed] [Google Scholar]