

Summary
Early gut microbial colonization is important for postnatal metabolic and immune development. However, little is known about the effects of different feeding modes (suckling versus bottle‐feeding) or microbial sources on this process in farm animals. We found that suckled and bottle‐fed newborn lambs had their own distinct gut microbiota. Results from 16S rRNA gene sequencing and qPCR showed that, compared with suckling, bottle feeding significantly increased the abundances of Escherichia/Shigella, Butyricicoccus, and Clostridium XlVa, while significantly decreased the abundance of Clostridium XI. The higher levels of Escherichia/Shigella in bottle‐fed lambs suggest that artificial feeding may increase the number of potential pathogens and delay the establishment of the anaerobic environment and anaerobic microbes. Feeding modes also affected the direct transmission of bacteria from the mother and the environment to newborns. The SourceTracker analysis estimated that the early gut microbes of suckled lambs were mainly derived from the mother's teats (43%) and ambient air (28%); whereas those of bottle‐fed lambs were dominated by bacteria from the mother's vagina (46%), ambient air (31%), and the sheep pen floor (12%). These findings advance our understanding of gut microbiota in early life and may help design techniques to improve gut microbiota and health.
Introduction
Mounting evidence suggests that the early development of the gut microbiota of newborns is essential for health by maintaining intestinal homeostasis and barrier function, stimulating development of the immune system, contributing to nutrient digestion, and protecting against pathogens (Sekirov et al., 2010; Maynard et al., 2012; Wopereis et al., 2014). The symbiotic microbe‐host interactions during the earliest days of life critically determine life‐long health (Rautava et al., 2012; Stinson et al., 2017; Korpela and de Vos, 2018). Disruption of the early gut microbiota has been linked to diseases in later life, including obesity, metabolic diseases, inflammatory bowel disease, irritable bowel syndrome, necrotizing enterocolitis, and autoimmune diseases and allergy (Rautava et al., 2012; Christian et al., 2017).
The maternal microbiota may affect the fetal immune development in utero, regardless of the actual colonization status of the fetus (de Agueero et al., 2016; Jenmalm, 2017). Extensive microbial colonization of the healthy newborn gut starts during and after birth by both vertical and horizontal transmission (Christian et al., 2017). The first phase of microbial colonization occurs via vertical transfer when the fetus passes through the birth canal and ingests maternal vaginal microorganisms (Funkhouser and Bordenstein, 2013). After birth, the newborn gut becomes further colonized with a wide array of microbes from maternal and environmental sources (Dominguez‐Bello et al., 2010; Shin et al., 2015; Ferretti et al., 2018). After 48 h, the number of intestinal bacteria reaches as high as 104–106 colony‐forming units per millilitre of intestinal content (Goulet, 2015). The natural pattern of postnatal microbiota acquisition and colonization is influenced by factors such as infant feeding mode, care environment, and use of antibiotics, leading to normal or aberrant establishment of the early gut microbiota (Biasucci et al., 2008; Tamburini et al., 2016).
Feeding mode is an important driver of early microbial colonization, influencing the structure and function of the neonatal gut microbiome (Azad et al., 2013; Martin et al., 2016). In humans, there are differences in gut microbial composition between breastfed and formula‐fed infants (Yatsunenko et al., 2012; O'Sullivan et al., 2015). Breastfed infants have more Bifidobacteria and Lactobacilli in their gut microbiota than formula‐fed infants (Bezirtzoglou et al., 2011; Wopereis et al., 2014; Backhed et al., 2015). The close physical contact during breastfeeding leads to vertical transfer of the bacterial community from the mother's teat and surrounding skin, stool, oral cavity, and breast milk to offspring (Mackie et al., 1999; Funkhouser and Bordenstein, 2013). More importantly, breast milk contains beneficial oligosaccharides, glycans, antimicrobial proteins, and secretory immunoglobulin A that contribute to shaping the infant gut microbiota (Wopereis et al., 2014). Breast milk oligosaccharides and glycans act as prebiotics and antimicrobials that selectively affect the growth of specific bacteria, for example, by stimulating the growth of Bifidobacterium and Lactobacillus spp., and reducing colonization and invasion of potential pathogenic bacteria by blocking their attachment to epithelial cell surfaces (Ward et al., 2006; Christian et al., 2017).
While the importance of feeding mode on the structure and function of the developing human microbiome is undoubted (Thompson et al., 2015), very little is known about the effects of suckling versus artificial feeding on the early gut microbiota of farm animals, especially for ruminants, which rely exclusively on their acquired gut microbes to digest their plant‐based feed. In ruminant farming, there are two main systems for managing young animals. In commercial dairy cattle or goat farms, calves or kids are typically separated from their mothers after birth and fed either milk replacer or whole milk; in contrast, in fattening systems, the newborns remain with their mothers until weaning (Yáñez‐Ruiz et al., 2015). Newborns fed artificially lack physical contact with their mothers and may not acquire maternal microbiota. Although a number of farm animal studies have investigated the composition, development, and functionality of gut microbiota in the early days of life (Jami et al., 2013; Rey et al., 2014; Jiao et al., 2015), the microbial sources of the early gut microbiota under different feeding modes is still poorly understood. Identifying the effects of feeding mode on composition and acquisition of the early gut microbiota of newborn farm animals is a key to understanding its contribution to modulating the newborn gut microbiota and to the immune system, metabolism, and development of the host. In this study, we investigated the impact of feeding mode (suckling versus artificial feeding) on early intestinal microbial colonization of three‐day old lambs using 16S rRNA gene sequencing and qPCR methods. We explored the microbial sources and the corresponding proportions of the early gut microbiota by source under the two feeding modes.
Results
Sequencing depth and alpha diversity
Sequence analysis of the 83 samples from the infant gut (n = 12) and maternal (n = 45) and environmental (n = 26) niches yielded 2,962,836 clean reads with an average of 35,697 ± 1,779 reads per sample. After removing the contaminating reads in the negative controls from the data set, the number of operational taxonomic units (OTUs) detected overall was 6,358 based on a threshold of ≥ 97% nucleotide sequence identity between reads. With a subsample of 18,146 reads, all the sample‐based rarefaction curves reached a plateau (Supporting Information Fig. S1), suggesting that our sequencing depth provided sufficient OTU coverage to describe the bacterial community composition of all samples accurately. All results are presented using median values, unless otherwise stated.
The number of OTUs and Chao1 estimator differed significantly (P < 0.05) between the suckled and bottle‐fed groups (Fig. 1), while the Shannon and Simpson indices did not (P > 0.05). In the maternal bacterial community, the number of OTUs and the Chao1 estimator for maternal ventral, teat, udder skin, and stool were significantly higher than those for oral cavity, vagina, and breast milk. Breast milk had the lowest alpha diversity indices among the maternal niches (Supporting Information Fig. S2). In the environmental bacterial community, there were no significant differences in any of the diversity indices between niches (Supporting Information Fig. S3).
Figure 1.
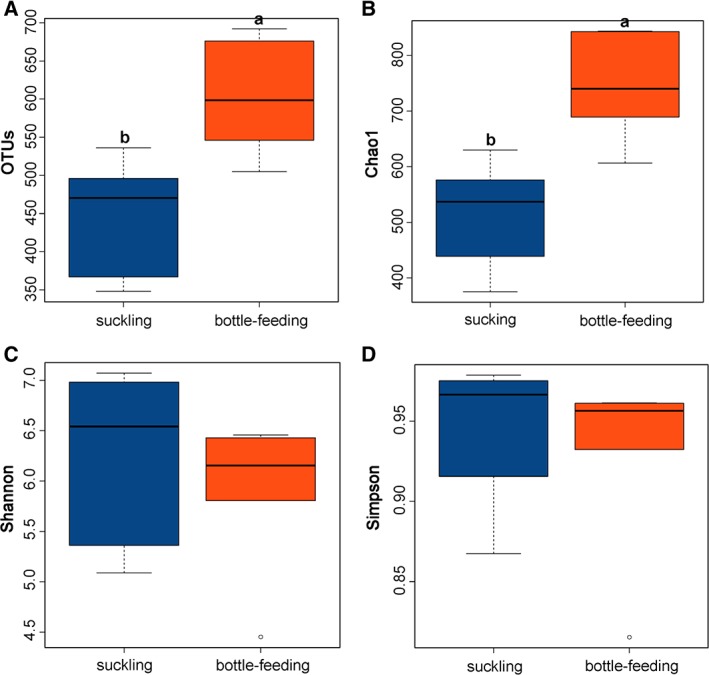
Box‐and‐whisker plots of alpha diversity indices (OTUs, Chao1, Shannon, and Simpson) for gut microbial communities in suckled (n = 6) and bottle‐fed (n = 6) newborn lambs. Values are median ± interquartile range. Boxes with different letters above their whiskers are significantly different at P < 0.05 using two‐tailed Wilcoxon signed‐rank test. [Color figure can be viewed at http://wileyonlinelibrary.com]
Bacterial community composition of maternal and environmental sources
Maternal bacterial communities were structured primarily by niche, with distinct oral, skin, stool, breast milk, and vaginal assemblages dominated by taxa typical for these sites (Fig. 2A). For example, at the phylum level these included: the Bacteroidetes and Proteobacteria in the oral cavity and vagina; the Firmicutes and Bacteroidetes in stool and on ventral, udder, and teat skin; and the Proteobacteria in breast milk (Fig. 3A). At the genus level, Prevotella, Bacteroides, and Paracoccus were prevalent in the vagina; Bacteroides, Prevotella, and Alysiella in the oral cavity; Bacteroides and Prevotella on the ventral, udder, and teat skin; Bacteroides and Clostridium XlVa, and Treponema in the stool; and Pseudomonas and Paracoccus in breast milk (Fig. 3B).
Figure 2.
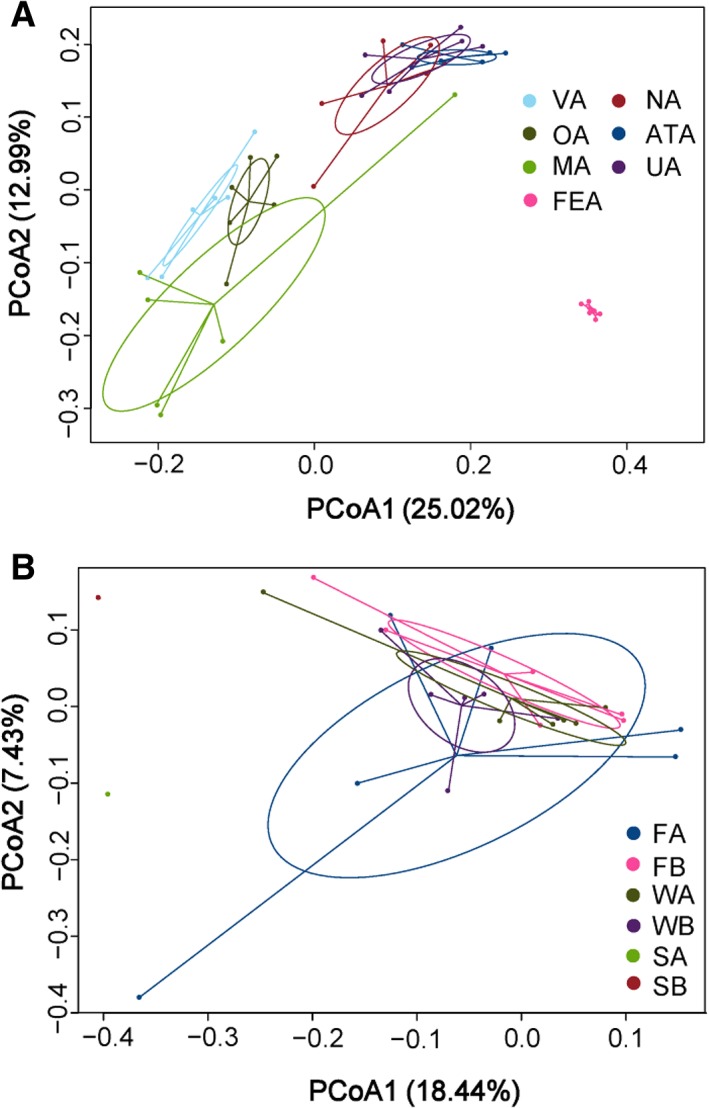
Principal coordinates analysis (PCoA) of the bacterial communities from maternal (A) and environmental (B) sources. PCoA was used to assess the differences in the composition of bacterial communities between samples and to visualize the potential clustering of samples. Each point represents a different sample calculated using unweighted UniFrac distance according to OTUs composition and abundance. The greater the distance between two points, the lower the similarity between them, whereas samples with more similar bacterial communities cluster closer together. VA: maternal vagina; OA: maternal oral cavity; MA: milk from birth mother; FEA: maternal stool; NA: maternal teat skin; ATA: maternal abdominal region skin; UA: maternal udder skin; FA: sheep pen floor of suckled group; WA: pen wall of suckled group; SA: ambient air of suckled group; FB: sheep pen floor of bottle‐fed group; WB: pen wall of bottle‐fed group; SB: ambient air of bottle‐fed group. Except for the SA and SB, the sample size of each maternal and environmental niche was 6. The ambient air microbes of each group were sampled using the same gelatin membrane filter for three consecutive days, so both SA and SB had a sample size of 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
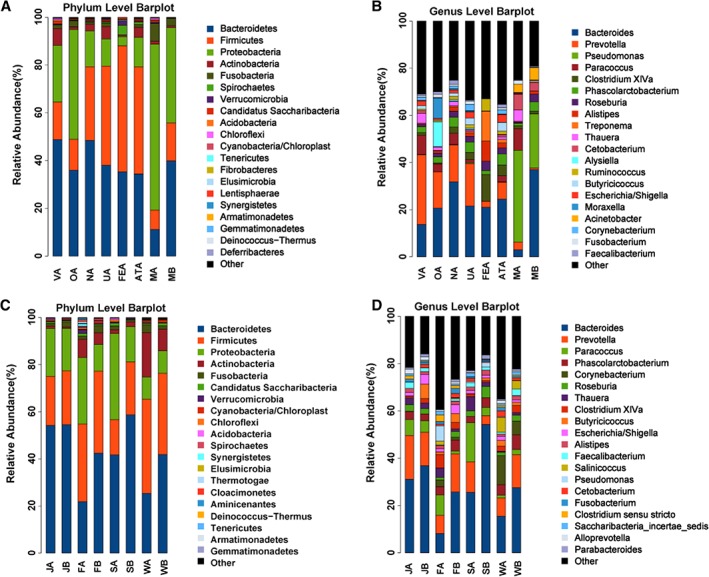
Phylum‐ and genus‐level bacterial composition of gut microbial communities in suckled and bottle‐fed lambs, and maternal and environmental sources of bacteria. Colour‐coded bar charts show the average relative abundances of different phyla and genera. (A, B) Phylum‐ and genus‐level bacterial composition of maternal sources of bacteria. VA: maternal vagina; OA: maternal oral cavity; NA: maternal teat skin; UA: maternal udder skin; ATA: maternal abdominal region skin; FEA: maternal stool; MA: milk from birth mother; MB: milk for bottle feeding; (C, D) Phylum‐ and genus‐level bacterial composition of gut microbial communities in suckled and bottle‐fed lambs, and environmental sources of bacteria. JA: jejunum content from suckled lambs, n = 6; JB: jejunum content from bottle‐fed lambs, n = 6; FA: sheep pen floor of suckled group; FB: sheep pen floor of bottle‐fed group; SA: ambient air of suckled group; SB: ambient air of bottle‐fed group; WA: pen wall of suckled group; WB: pen wall of bottle‐fed group. Except for the MB, SA, and SB, the sample size of each maternal and environmental niche was 6. The sample size of MB was 3. The ambient air microbes of each group were sampled using the same gelatin membrane filter for three consecutive days, so both SA and SB had a sample size of 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
The environmental sources harboured essentially undifferentiated bacterial communities between the two treatment groups (Fig. 2B, P > 0.05). At the phylum level, the pen floor and ambient air of both the suckled and bottle‐fed groups were dominated by the Bacteroidetes, Firmicutes, and Proteobacteria; the pen wall of both the suckled and bottle‐fed groups by Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria (Fig. 3C). At the genus level, the pen floor was dominated by Bacteroides, Prevotella, Paracoccus, and Pseudomonas in the suckled group and Bacteroides, Prevotella, and Phascolarctobacterium in the bottle‐fed group; the ambient air by Bacteroides, Prevotella, and Paracoccus in the suckled group and Bacteroides in the bottle‐fed group; the pen wall by Bacteroides, Corynebacterium, and Prevotella in both the suckled and bottle‐fed groups (Fig. 3D).
Gut bacterial community composition in suckled and bottle‐fed lambs
Twenty‐nine phyla were identified in the jejunum samples taken from newborn lambs. Among all samples, 10 phyla had a relative abundance of ≥ 0.1% and were dominated by the Bacteroidetes, Firmicutes, and Proteobacteria in the two groups (Supporting Information Table S1). Relative abundances of the dominant phyla were not significantly different between the two groups (P > 0.05, Supporting Information Table S1), but their genera composition varied considerably (Table 1, Fig. 3D).
Table 1.
Relative abundances of the 15 most abundant genera (relative abundance ≥ 1%) between the suckled and bottle‐fed groups. Values are median ± SD, n = 6 for each group.
| Phylum | Genus | Median relative abundance (%) | P‐value | |
|---|---|---|---|---|
| suckling | bottle‐feeding | |||
| Bacteroidetes | Bacteroides | 24.39 ± 15.35 | 35.30 ± 12.26 | 0.563 |
| Prevotella | 4.04 ± 2.36 | 3.18 ± 1.76 | 0.688 | |
| Alloprevotella | 1.65 ± 0.94 | 1.24 ± 0.71 | 0.688 | |
| Alistipes | 1.42 ± 0.65 | 0.96 ± 0.43 | 0.813 | |
| Parabacteroides | 0.58 ± 0.18 | 1.04 ± 0.33 | 0.031 | |
| Firmicutes | Phascolarctobacterium | 2.99 ± 0.88 | 2.28 ± 0.91 | 0.688 |
| Roseburia | 2.13 ± 1.03 | 2.10 ± 0.79 | 0.844 | |
| Faecalibacterium | 2.49 ± 0.55 | 1.22 ± 0.51 | 0.031 | |
| Clostridium XI | 1.22 ± 0.88 | 0.38 ± 0.40 | 0.031 | |
| Clostridium XlVa | 0.62 ± 0.22 | 1.50 ± 0.70 | 0.031 | |
| Butyricicoccus | 0.07 ± 0.05 | 5.48 ± 3.35 | 0.031 | |
| Proteobacteria | Paracoccus | 2.00 ± 3.34 | 2.58 ± 1.93 | 0.688 |
| Thauera | 1.15 ± 2.57 | 1.16 ± 2.01 | 0.844 | |
| Escherichia/Shigella | 0.75 ± 0.84 | 3.47 ± 1.02 | 0.031 | |
| Fusobacteria | Fusobacterium | 1.12 ± 0.43 | 0.87 ± 0.35 | 0.219 |
The two groups shared 15 genera with relative abundances of ≥ 1% in at least one group. Of these, the relative abundances of 6 genera differed significantly between the groups (Table 1). The Bacteroidetes, the most abundant phylum in all samples, was composed mainly of the genus Bacteroides (Table 1). Among the dominant Bacteroidetes genera, the abundance of only Parabacteroides was significantly different (P < 0.05) between the two groups. Genera composition of the second most abundant phylum, Firmicutes, differed considerably between the two groups: Faecalibacterium and Clostridium XI were significantly more abundant in the suckled group (P < 0.05), while Butyricicoccus and Clostridium XlVa were more abundant in the bottle‐fed group (P < 0.05; Table 1). The abundance of the Proteobacteria genus Escherichia/Shigella was significantly lower (P < 0.05) in the suckled group than in the bottle‐fed group.
Feeding modes and initial gut microbiota composition
The Venn diagram showed a large number of shared and unique OTUs between the two groups (Supporting Information Fig. S4), indicating that each group hosts its own distinct microbial community. PCoA revealed that the samples clustered together according to feeding mode, suggesting that different feeding modes significantly influenced the bacterial community in the gut (Fig. 4A). This was also confirmed by ANOSIM, which showed significantly high R values (R = 0.465, P < 0.05) between the two groups (Fig. 4B). PCoA and ANOSIM also showed high within‐group variability in gut microbiota (Fig. 4).
Figure 4.
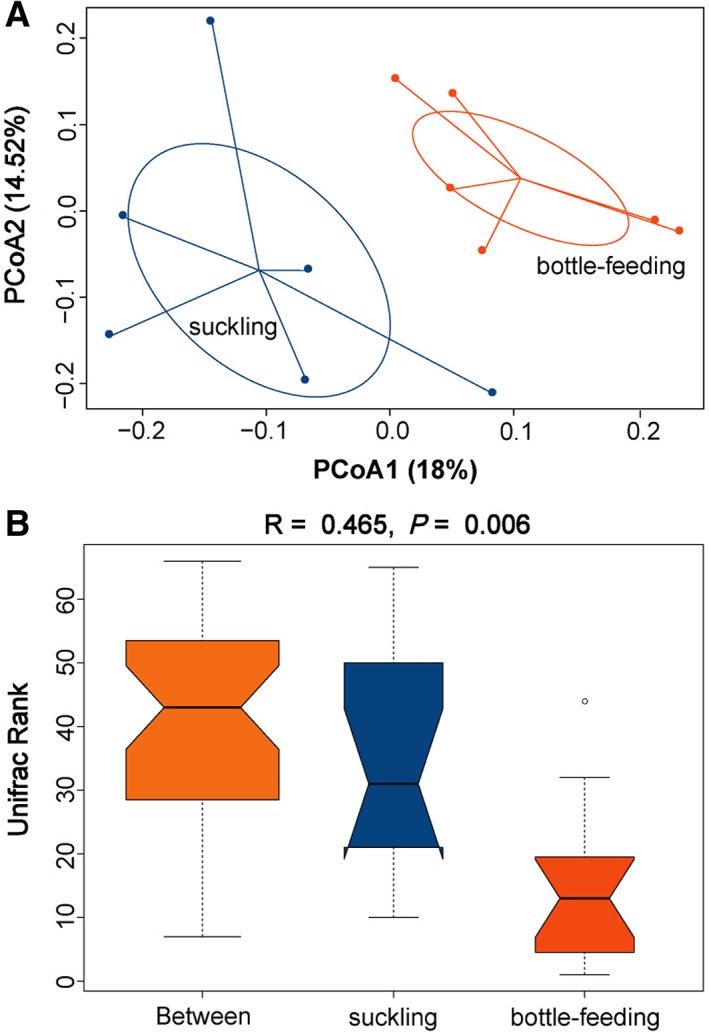
Beta diversity of the bacterial communities in guts of suckled (n = 6) and bottle‐fed (n = 6) lambs. (A) Principal coordinates analysis (PCoA) of bacterial communities in the two feeding groups. PCoA was used to assess the variation in composition of bacterial communities between samples and to visualize the potential clustering of samples. Each point represents a different sample calculated using Unweighted UniFrac distance according to the presence and absence of OTUs. A greater distance between two points indicates lower similarity between them, whereas samples with more similar bacterial communities cluster closer together. (B) Analysis of similarity (ANOSIM) of bacterial communities in the two feeding groups. The X‐axis denotes all samples (between) and each group. The Y‐axis shows the unweighted UniFrac rank. ANOSIM is a permutation‐based test of the null hypothesis that within‐group distances are not significantly smaller than between‐group distances. The test statistic R, which measures the strength of the results, ranging from −1 to 1, R > 0 indicates that the between‐group differences are greater than within‐group differences, whereas R < 0 indicates that the differences between groups are smaller than those within groups. When R ≈ 0, the null hypothesis is true, and there are no difference between all samples and groups. P < 0.05 is considered statistically significant. [Color figure can be viewed at http://wileyonlinelibrary.com]
Quantification of total bacteria and five selected bacterial genera in gut microbiota
Absolute quantitative real‐time PCR was used to quantify the 16S rRNA gene copy numbers of total bacteria and five selected bacterial genera that showed statistically significant differences in the sequencing results. The 16S rRNA gene copy numbers of Escherichia, Butyricicoccus, and Clostridium XlVa were significantly greater in the bottle‐fed group than in the suckled group, whereas the opposite was the case for Clostridium XI (Fig. 5). The 16S rRNA gene copy numbers of total bacteria and Faecalibacterium were not significantly different between the two groups. The relative abundances of Escherichia, Butyricicoccus, and Clostridium Xl showed similar statistical differences to results from Illumina MiSeq sequencing. The relative abundance of Faecalibacterium calculated by qPCR showed no significant group differences (Supporting Information Table S2).
Figure 5.
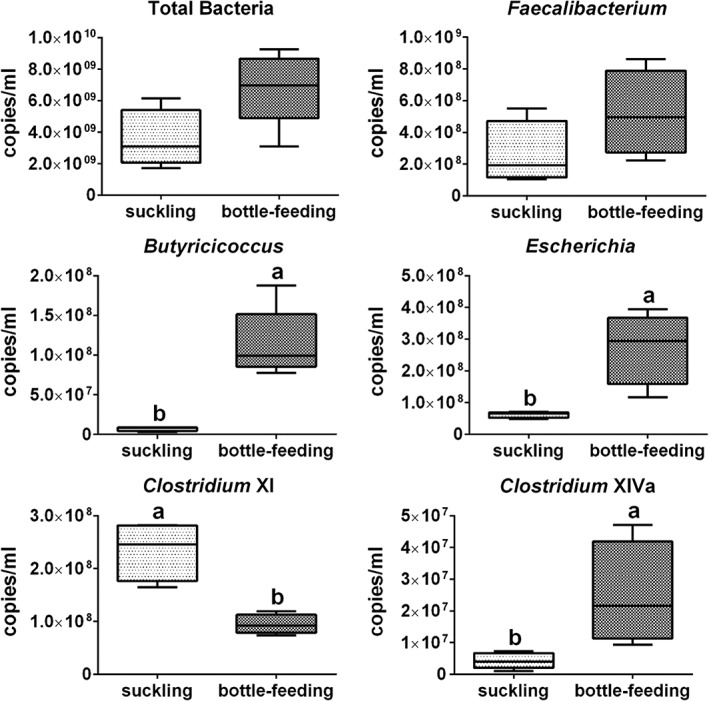
The 16S rRNA gene copy numbers of the total bacteria and selected bacterial genera per millilitre of jejunum content samples of suckled (n = 6) and bottle‐fed (n = 6) lambs. Values are median ± interquartile range. Boxes with different letters above their whiskers are significantly different at P < 0.05 using two‐tailed Wilcoxon signed‐rank test.
Microbial sources and proportion estimates of the early gut bacterial communities
Bray‐Curtis distance analysis showed good OTU‐level similarity between the newborn lamb gut and mother's teats, vagina, oral cavity, and ambient air in the suckled group (Fig. 6A), indicating that gut bacterial communities of suckled lambs originated mainly from these niches. Guts of newborn lambs in the bottle‐fed group showed good OTU‐level similarity to the mother's vagina and ambient air (Fig. 6B), demonstrating that gut bacterial communities of bottle‐fed newborn lambs were mainly derived from these two niches.
Figure 6.
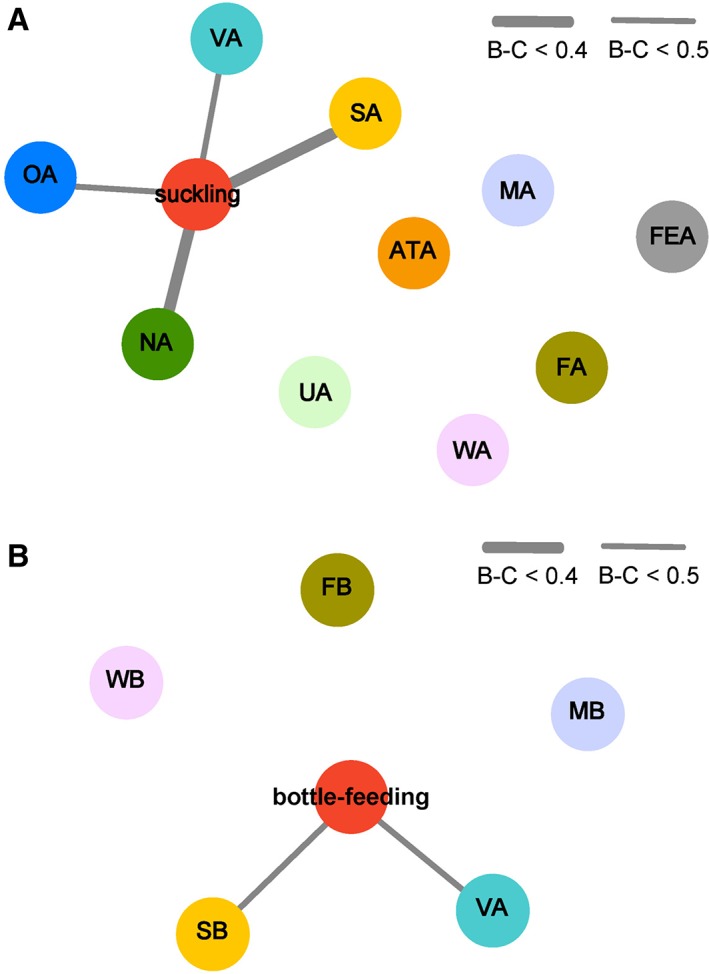
Similarity analysis of the bacterial communities in the gut of suckled (n = 6) and bottle‐fed (n = 6) lambs and the source environments. Bray‐Curtis (B‐C) dissimilarity was calculated using OTU‐level abundance of bacteria from the lamb gut, and maternal and environmental niches in this study. The different coloured circles represent bacterial communities of the gut and source environments. The thicker the connecting line and the closer the circles, the greater the similarity of the bacterial communities. (A) Similarity analysis of the bacterial communities in the gut of suckled lambs and the source environments. VA: maternal vagina; OA: maternal oral cavity; MA: milk from birth mother; FEA: maternal stool; NA: maternal teat skin; ATA: maternal abdominal region skin; UA: maternal udder skin; FA: sheep pen floor of suckled group; WA: pen wall of suckled group; SA: ambient air of suckled group. (B) Similarity analysis of the bacterial communities in the gut of bottle‐fed lambs and the source environments. MB: milk for bottle feeding; FB: sheep pen floor of bottle‐fed group; WB: pen wall of bottle‐fed group; SB: ambient air of bottle‐fed group. Except for the MB, SA, and SB, the sample size of each maternal and environmental niche was 6. The sample size of MB was 3. The ambient air microbes of each group were sampled using the same gelatin membrane filter for three consecutive days, so both SA and SB had a sample size of 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
SourceTracker analysis at OTU‐level predicted that the gut bacterial communities of suckled lambs tended to be composed mainly of bacteria from the mother's teat (43%), vagina (7%), ventral skin (6%), oral cavity (5%), and ambient air (28%), whereas those of bottle‐fed lambs were dominated by bacteria from the mother's vagina (46%), ambient air (31%), and the pen floor (12%) (Fig. 7A and B). SourceTracker also predicted several gut bacterial mixtures; for suckled lambs, the possible mixtures included “teat, air, vagina, ventral skin, and oral cavity”, and “teat, air, vagina, and ventral skin” as sources, with mother's teat and ambient air as the stable source. The bottle‐fed lambs had “vagina, air, and pen floor” and “vagina and air” components with vagina and ambient air as the stable sources (Fig. 7C). The most common source of the 15 most abundant bacterial genera (except Clostridium XI) in the gut of suckled lambs was the mother's teat and that of Clostridium XI was the mother's vagina. The most common source of the 15 most abundant genera of bottle‐fed lambs was the mother's vagina (Supporting Information Fig. S5).
Figure 7.
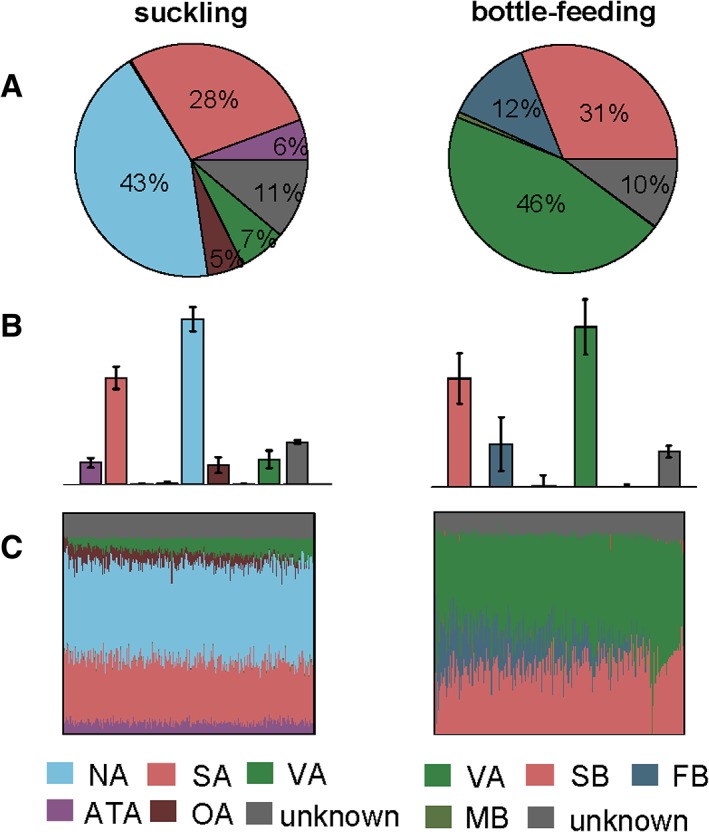
SourceTracker source environment proportion estimates for the gut bacterial communities in suckled (n = 6) and bottle‐fed (n = 6) lambs. (A) Pie charts of the mean proportions for 250 draws from Gibbs sampling; (B) bar charts of proportion estimates for the same samples; (C) visualization of the 250 Gibbs draws, with each column showing the mixture from one draw, and the columns ordered to keep similar mixtures together. VA: maternal vagina; OA: maternal oral cavity; FEA: maternal stool; NA: maternal teat skin; ATA: maternal abdominal region skin; UA: maternal udder skin; MA: milk from birth mother; FA: sheep pen floor of suckled group; WA: pen wall of suckled group; SA: ambient air of suckled group; MB: milk for bottle feeding; FB: sheep pen floor of bottle‐fed group; WB: pen wall of bottle‐fed group; SB: ambient air of bottle‐fed group. Except for the MB, SA, and SB, the sample size of each maternal and environmental niche was 6. The sample size of MB was 3. The ambient air microbes of each group were sampled using the same gelatin membrane filter for three consecutive days, so both SA and SB had a sample size of 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
Our results suggest that different feeding modes significantly affected the gut bacterial community composition in newborn lambs. The significantly higher OTU number and Chao1 estimator in the bottle‐fed group compared to the suckled group suggest that artificial feeding significantly increased bacterial richness in the initial gut microbiota of newborn lambs. This result is consistent with findings that bacterial richness are lower in breastfed infants than in artificially fed infants (Bezirtzoglou et al., 2011; Azad et al., 2013; Backhed et al., 2015). In our study, the bottle‐fed group had significantly higher bacterial community richness than the suckled group, but there were no statistically significant differences in Shannon and Simpson indices between the two groups, indicating that the bottle‐fed group had a larger number of rare species and the suckled group had greater community evenness than the bottle‐fed group.
The dominant phyla in the gut of the two groups were the Bacteroidetes, Firmicutes, and Proteobacteria. These dominant phyla and their shared genera represent the core microbiota in the gut of newborn lambs. These dominant phyla and genera are also the core microbiota in ruminant guts at different ages (Jami et al., 2013; Jiao et al., 2015; Wang et al., 2017). This suggests that these core bacterial communities in the gut of mature animals are established at a very early stage in life, in line with previous studies (Fonty et al., 1987; Minato et al., 1992; Jami et al., 2013) that detected bacterial species important for the proper functioning of the adult gut in the first week after birth.
The most abundant phylum Bacteroidetes accounted for more than 49% of the total reads in the two groups. The vast majority of reads belonged to the genus Bacteroides, with an abundance of 24.39% and 35.30% in the suckled and bottle‐fed group, respectively. It was also the most abundant genus in the gut of newborn calves, appearing within several weeks after birth (Li et al., 2012; Klein‐Joebstl et al., 2014; Rey et al., 2014). Bacteroides species, most of which are acetate and propionate producers, share important metabolic functions in the gut (Berry et al., 2013; Valles et al., 2014). Bacteroides has a remarkable ability to ferment a large assortment of plant oligosaccharides and polysaccharides, host‐derived glycans, and breast milk oligosaccharides (Marcobal et al., 2010; Adamberg et al., 2018), with some species being efficient users of mucin molecules found in the mucus gel layer of the intestinal epithelium (Salyers et al., 1977; Berry et al., 2013). Dietary composition is thought to be the primary factor affecting gut microbiota, with greater Bacteroides abundance associated with diets rich in protein, sugar, and starch while greater Prevotella abundance is associated with diets composed mainly of plant fibre (Jami et al., 2013). Bacteroides, one of the first anaerobes to appear in the gut (Le Huerou‐Luron et al., 2010), can establish itself quickly in the neonatal gut as it is one of the most efficient utilizers of milk oligosaccharides and mucin molecules (Berry et al., 2013; Valles et al., 2014).
The Firmicutes was the second most abundant phylum, accounting for more than 23% of the total sequences, with the vast majority of the reads belonging to the order Clostridiales. In this study, all genera with a relative abundance of more than 1% in the phylum Firmicutes belong to this order. Clostridiales abundance was significantly higher in non‐exclusively breastfed infants than in exclusively breastfed infants (Thompson et al., 2015). However, our study did not find significant differences in Clostridiales abundance between feeding groups (16.18% versus 18.00%, P > 0.05). Clostridiales is one of the six major microbiota functional groups that establish early in the infant gut, and many of its species display a wide range of phenotypes and functional diversity (Valles et al., 2014). We found that the differences in the Firmicutes between the two feeding groups were caused largely by the differences in Clostridiales genera. The suckled lambs had greater proportions of Faecalibacterium and Clostridium XI in their gut microbiota, whereas the bottle‐fed lambs had greater proportions of Butyricicoccus and Clostridium XlVa. The 16S rRNA gene copy numbers and relative abundances of these genera exhibited the same statistical differences, except for Faecalibacterium. Faecalibacterium, Butyricicoccus, and Clostridium XlVa are known as butyrate producers (Louis and Flint, 2009; Geirnaert et al., 2014), which are obligate anaerobic Gram‐positive bacteria. They represent a functional group, rather than a coherent phylogenetic group, originating from the Clostridium leptum (or clostridial cluster IV) cluster, for example, Faecalibacterium and Butyricicoccus, and the Clostridium coccoides (or clostridial cluster XIVa) cluster, for example, Clostridium XlVa and Roseburia (Louis and Flint, 2009).
In this study, both 16S rRNA gene sequencing and quantitative real‐time PCR detected a significant increase in the abundance of Escherichia in bottle‐fed lambs compared to suckled lambs, confirming results of previous studies of infants (Mackie et al., 1999; Penders et al., 2006). Escherichia is a well‐known member of the normal intestinal microflora of humans and many other animals (Kaper et al., 2004; Lukjancenko et al., 2010). The ability of Escherichia strains to survive in many ecological niches facilitates the colonization of different habitats, including abiotic environments. Escherichia colonizes the neonatal gut within hours of birth and establishes itself as the most abundant facultative anaerobe of the intestinal microflora (Leimbach et al., 2013). Facultative anaerobes consume oxygen and create the anaerobic environment that is required for proliferation of anaerobic microbes (Mackie et al., 1999; Jost et al., 2012). In contrast to suckled newborn lambs, bottle‐fed lambs had a higher abundance of Escherichia, suggesting that artificial feeding may delay the establishment of the anaerobic environment and anaerobic microbes (Le Huerou‐Luron et al., 2010).
Escherichia is a highly versatile genus comprising harmless commensal and pathogenic strains that have acquired the ability to cause intestinal (enteritis, diarrhoea, or dysentery) or extraintestinal (urinary tract infections, sepsis, or meningitis) diseases in their hosts by attaching to the epithelial cells and/or invading target host cells (Croxen et al., 2013; Leimbach et al., 2013; Kalita et al., 2014). Compared to artificial feeding, breastfeeding is associated with lower incidence of necrotizing enterocolitis and diarrhoea during early life (Aggett et al., 2006; Le Huerou‐Luron et al., 2010). For example, diarrhoea was reported to be reduced by half in breastfed infants compared with artificially fed infants during the period of breastfeeding (Dewey et al., 1995; Hoddinott et al., 2008). The higher abundance of Escherichia in bottle‐fed infants may, in part, explain why susceptibility to necrotizing enterocolitis and diarrhoea is often higher among them than in breastfed infants. Direct transmission of the maternal microbiota to the newborn may serve a defensive role, leading to occupation of niches and a reduction in colonization by Escherichia and other pathogens as site‐specific communities develop.
Our results also suggest that different feeding modes affected the direct initial transmission of bacteria from maternal and environmental niches to newborns. Both bacterial community similarity and SourceTracker analyses demonstrated that the gut microbes of suckled lambs were mainly derived from the mother's teats, vagina, and oral cavity, and ambient air, and that of bottle‐fed lambs were dominated by bacteria from the mother's vagina and ambient air. In addition, the mother's ventral skin was also an important source of gut bacteria for suckled lambs, as well as pen floor for bottle‐fed lambs.
The mother's vaginal microbiota is an important source of the lamb gut microbiota regardless of feeding mode, contributing 46% to bottle‐fed and 7% to suckled lamb gut microbiota, suggesting vertical transmission from the vaginal community to the newborn. The mother's vaginal microbiota provides the first natural microbial source for newborns. During vaginal delivery, direct contact of the newborn with the vaginal microbiota plays an important role in initiating colonization of its gut microbiome (Orrhage and Nord, 1999; Matsumiya et al., 2002). Vaginally delivered infants harboured primary gut bacterial communities similar to their own mothers' vaginal microbiota, with caesarean‐section‐delivered infants acquiring gut bacterial communities resembling those found on the skin surface (Dominguez‐Bello et al., 2010).
Our results show that almost half of the gut microbiota in suckled lambs might originate from their mothers' teat microbiota. Similar vertical transmission is also seen in humans. Bacteria originating from the nipple and surrounding skin are important sources of the infant gut microbiota (Mackie et al., 1999; Ferretti et al., 2018), demonstrating that direct contact of the newborn's mouth with the nipple during breastfeeding contributes substantially to the infant's early gut bacterial community. The mother's oral cavity is another important source of the suckled lamb gut microbiota, which is consistent with previous studies on humans (Mackie et al., 1999;Dominguez‐Bello et al., 2010; Ferretti et al., 2018). A recent study showed that the faecal microbiota of neonatal and 1‐day calves were more similar to their mother's oral microbiota, as compared to the vaginal or faecal microbiota, indicating that the mother's oral cavity is an important source of early gut microbiota (Alipour et al., 2018). After birth, the mother's oral cavity microbes were mechanically transferred to the newborns by licking the suckled lambs.
Bacterial communities from the environment are also important sources of early gut microbiota. The first exposure of infants born by caesarean delivery is most likely to environmental microbiota from equipment, ambient air, and nursing staff (Mackie et al., 1999), such as operating room microbiota containing deposits of human skin bacteria, which seeds the infant's microbiome (Shin et al., 2015). Vaginal microbiota is the only source of maternal microbes to which bottle‐fed newborn lambs are directly exposed. Therefore, horizontal transmission of environmental microorganisms from the ambient air, sheepfold, and facilities plays a key role in initial intestinal colonization. In our study, bacteria in ambient air was an important contributor to the early gut microbiota of both bottle‐fed and suckled lambs and is an easily overlooked long‐term source of newborn gut microbes (Dominguez‐Bello et al., 2010; Ferretti et al., 2018). We also show that microbiota of the pen floor is an important source of gut bacteria for bottle‐fed lambs. The proportion of early gut bacteria from environmental sources was more than 1.5 times higher for bottle‐fed lambs compared to suckled lambs (43% vs 28%), indicating that the former are more likely to become colonized by environmental microorganisms than the latter.
Breast milk contains abundant maternally derived commensal bacteria (Urbaniak et al., 2016; Milani et al., 2017) and its microbiota plays an important role in human infant gut microbial colonization and the newborn's immune system development (Heikkila and Saris, 2003; Daft et al., 2015). In contrast, we found that breast milk was not an important source for the gut microbiota of newborn lambs. This is likely due to the low bacterial community richness and diversity in breast milk, relative to both maternal and environmental bacterial communities (Supporting Information Fig. S2 and S3).
To our knowledge, our study is the first to compare the effects of two feeding modes (suckling versus artificial feeding) on the early gut microbiota composition of newborn farm animals. Unlike previous studies in humans that examined only the maternal sources of microbes, we believe our study is the first to assess both maternal and environmental sources and their contributions to the newborn gut microbiome. However, our study has several limitations. We used 16S rRNA gene amplicon‐based sequencing instead of metagenomics sequencing, which limited our understanding of differences in strain level and function of gut microbes between feeding modes. We did not investigate the long‐term effects of feeding mode and nor did we look at longitudinal data on sources of gut microbiota. In addition, the small sample size we used may have amplified differences between feeding groups. One further limitation of our study to note is that the SourceTracker analysis we applied to estimate the proportion of microbial sources in the early gut microbial community does not directly infer the microbial transfer from maternal and environmental niches to the gut of newborn lambs, but instead it suggests the relatedness between microbial sources and the gut microbial community.
In conclusion, we detected adult‐type gut bacteria in the gut of 3‐day old lambs, indicating that the core intestinal bacteria are established in the early stages of life. Intestinal bacterial composition of the newborn lambs was dominated by the phyla Bacteroidetes, Firmicutes, and Proteobacteria, and by the genus Bacteroides. Different feeding modes significantly affected the colonization of gut microbes at an early stage of life. Compared with suckling, artificial feeding significantly increased the abundances of Escherichia/Shigella, Butyricicoccus, Clostridium XlVa, and Parabacteroides in the early gut microbiota of newborn lambs, whereas it significantly decreased the abundance of Clostridium XI. Different feeding modes also affected direct initial transmission of bacteria from maternal and environmental niches to newborns. The SourceTracker analysis suggests that the gut microbes of suckled newborn lambs were mainly derived from the mother's teats, vagina, ventral skin, and oral cavity, and from ambient air, and that of bottle‐fed lambs mainly originate from the mother's vagina, ambient air, and the pen floor. Our results provide preliminary information for understanding the relationship between the early intestinal microbiota and type of feeding, the sources of the early gut microbiota of newborns, and the role of maternal and horizontal transmission in the initial establishment of the newborn gut microbiota. Moreover, our results provide important insights for other systems in which the interplay between the environment and diet plays an important role in shaping the acquisition of gut microbes during early life. However, further longitudinal studies with larger sample sizes and more detailed sequencing are required for investigating the long‐term effects of different feeding modes on gut microbiota, health, and development of animals, and microbial acquisition patterns.
Experimental procedures
Animal experiment and sample collection
Animal care and use for the experiments were approved by and performed according to the guidelines of the Animal Ethics Committee of the Chinese Academy of Agricultural Sciences, Beijing, China.
A flock of healthy pregnant ewes, each carrying two foetuses and with the same expected date of delivery, were selected from the same herd and raised individually in 1 × 3 m sheep pens. The ewes were fed the same diet without administration of antibiotics or probiotics during pregnancy. The ewes that gave birth to twins by vaginal delivery on the same day were selected in this trial. After birth, one of the twin lambs was taken immediately from the ewe, caged individually in a sheep pen to avoid direct contact with other adult animals and lambs, and fed fresh colostrum collected from nursing mothers within 2 h of birth. It was bottle‐fed twice daily with milk collected from other lactating ewes. The other lamb remained with the birth mother and was suckled ad libitum. Finally, 6 ewes and their twins were included in this trial with 6 lambs in each feeding group.
Microbial samples from maternal and environmental niches were taken using sterile swabs. The mother's vagina and oral cavity samples were swabbed when the ewes showed obvious signs of parturition, such as a swollen vulva and udders. The mother's faeces samples were collected by rectal stimulation near the time of delivery. Skin swabs were also taken ventrally (right and left ventral posterior) and from the teat and udder for three consecutive days after delivery. Milk, including colostrum, from the birth mothers was collected twice daily for three consecutive days. Milk, including colostrum, for bottle feeding was collected from nursing mothers every morning, and once the required volume was collected, milk was pooled and collected once daily for three consecutive days. Before milk collection, the teat skin of birth mothers and nursing mothers was disinfected with alcohol and the first three milkings were discarded. Swabs were taken from the pens wall and floor of both feeding groups for three consecutive days. The ambient air of sheep pens of both feeding groups was sampled for three consecutive days using an air sampler (AirPort MD8 No.16757, Sartorius Stedim Biotech GmbH, 37,070, Goettingen, Germany) and gelatin membrane filters (No.17528‐80‐ACD, Sartorius). Each group of the 6 adjacent sheep pens was placed in an isolation fence. The four corners and the central position of the isolation fence were sampled with 750 l air at each position at a rate of 40 l per min during a three‐minute sampling period.
All lambs were euthanized by penetrative captive bolt followed by exsanguination at 3 days of age. When a lamb suckles, milk bypasses the rumen and reticulum and enters directly into the abomasum. At this age, the jejunum is the main place for food digestion and absorption and the nutritionally important microbial community is intestinal. Therefore, samples were taken from the jejunum and not from the rumen, as at this age a lamb is monogastric, and ruminal samples may not contain the microbes from the milk. Samples of jejunum content were collected aseptically at the same site from all the lambs within 1 h of euthanasia. Samples were stored immediately in sterile tubes, frozen in liquid nitrogen, and sent for DNA extraction.
DNA extraction and PCR amplification
Genomic DNA was extracted from the swabs, milk, ambient air, and intestinal digesta in a biosafety cabinet using the MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories, Solana Beach, CA) according to the manufacturer's protocols. The samples collected for three consecutive days were pooled as one sample. The swabs were processed as previously described (Dominguez‐Bello et al., 2010). In brief, the cotton tips of frozen swabs were placed in bead tubes and 60 μl of Solution C1 was added. Tubes were then incubated at 65 °C for 10 min and shaken horizontally for 2 min using vortex adapter. The remaining steps were performed following the manufacturer. DNA quantity was measured on a Thermo NanoDrop 2000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE). The V3–V4 regions of the bacterial 16S rRNA gene were amplified using the universal primers 338F 5′‐barcode‐ACTCCTACGGGAGGCAGCAG‐3′ and 806R 5′‐barcode‐GGACTACHVGGGTWTCTAAT‐3′ (Dennis et al., 2013), which contain an eight‐base sequence barcode unique to each sample. PCR amplification and product processing were performed as previously described (Bi et al., 2017). PCR reactions were carried out in triplicate in 20 μl reaction mixtures containing 4 μl 5 × FastPfu Buffer, 2 μl 2.5 mM dNTPs, 0.8 μl forward and reverse primer (5 μM), 0.4 μl FastPfu Polymerase, and 10 ng template DNA. The PCR cycling parameters consisted of initial denaturation at 95 °C for 3 min, followed by 27 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 45 s, and a final extension at 72 °C for 10 min. PCR products were extracted from 2% agarose gels, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA), and quantified using the QuantiFluor™‐ST system (Promega, Madison, WI). Non‐sample blank extraction controls, empty sterile swab extraction controls, and PCR negative controls were included in this study.
16S rRNA gene sequencing and sequence analysis
Equimolar amounts of the barcoded V3–V4 amplicons were pooled and paired‐end sequenced (2 × 250 bp) on an Illumina HiSeq PE250 platform (Illumina, Inc., San Diego, CA) according to standard protocols. The sequencing data we generated have been deposited in the NCBI Sequence Read Archive (SRA) under accession numbers SRR7986812 to SRR7986894. DNA sequence data for all samples were processed and analysed according to Bi et al. (2017). Briefly, sequences were removed from the analysis, if they were < 50 bp, had a quality score < 20, contained ambiguous characters, contained mismatch in the barcode region, or contained two or more nucleotide mismatches in the primer sequence. Only sequences with an overlap of > 10 bp and < 10% mismatches were assembled, and the assembled sequences were then trimmed of primers and barcodes. Chimeric sequences were identified and removed. After quality control, the potential contaminating sequences in the negative controls were removed from the data set. The decontamination procedure was based on comparing the relative abundances of the sequences in the negative controls and in the samples. The shared sequences were classified as contaminants when their relative abundances in the samples were no more than 10 times of that in the controls (Lukasik et al., 2017). This type of decontamination procedure minimizes false‐positive observations but removes some sequences that are genuinely present in the samples. After data decontamination, the sequences were assigned to OTUs at a 97% identity threshold using UPARSE (Edgar, 2013).
Alpha diversity indices, including the number of OTUs, Chao1 estimator, and Shannon and Simpson indices, were calculated by normalizing the number of clean reads in all samples to 18,146 sequences using mothur software (Schloss et al., 2009). Rarefaction curves were analysed with mothur and plotted using R. A representative sequence was chosen from each OTU by selecting the sequence that had the largest number of hits in the OTU. Representative sequences were assigned against the SILVA bacteria alignment database (Quast et al., 2013) using the RDP classifier (Cole et al., 2009) with a confidence threshold of 80%. Sequences were aligned against PyNAST (DeSantis et al., 2006), and a phylogenetic tree was built using FastTree (Price et al., 2009).
Beta diversity was calculated for the normalized OTU table using UniFrac distance matrices (Lozupone and Knight, 2005; Lozupone et al., 2007) to determine the amount of bacterial diversity shared between the two groups. Principal coordinates analysis (PCoA) of bacterial communities was performed using unweighted UniFrac distances based on the presence and absence of OTUs, and the plot was generated using R. Analysis of similarity (ANOSIM) was also performed using unweighted UniFrac distance to test for differences in bacterial community composition in the samples from the two groups.
To identify possible sources and their proportions in the early gut bacterial community of newborn lambs, Bray–Curtis dissimilarity analysis, and SourceTracker were used. Bray–Curtis dissimilarity (Aagaard et al., 2014) was used to determine the similarities at OTU level between the gut and maternal and environmental microbial communities. Microbial SourceTracker developed by Knights et al. (2011) is a Bayesian approach that is more accurate than other methods for estimating the proportion of contamination from source environments within a sink environment (Knights et al., 2011). We applied SourceTracker to survey the proportion of the lamb gut microbial community originating from the maternal and environmental microbial communities.
qPCR
Quantitative real‐time PCR analysis was performed to detect the 16S rRNA gene copy numbers of the total bacteria and five selected bacterial genera that showed statistical significance in sequencing results between the two groups using the primers shown in Supporting Information Table S3. A standard curve was constructed from a reference plasmid containing the 16S rRNA gene of total bacteria and of each individual strain. Real‐time PCR was performed in triplicate in 20 μl reaction mixtures containing 10 μl 2 × PCR Master Mix (SinoGene, Beijing, China), 0.5 μl of each primer (10 μM), 8.0 μl ddH2O, and 1 μl of 10 ng DNA templates. The PCR cycling parameters consisted of initial denaturation at 94 °C for 3 min followed by 35 cycles of denaturation at 94 °C for 20 s, annealing at 58 °C for 20 s and extension at 72 °C for 30 s, and a final extension at 72 °C for 10 min. The 16S rRNA gene copy numbers of total bacteria and the 5 bacterial genera per millilitre of jejunum digesta were then calculated. Relative abundances of the 5 bacterial genera were calculated by dividing the 16S rRNA gene copy number of each bacterial genus by the 16S rRNA gene copy number of total bacteria.
Statistical analyses
Unless otherwise stated, statistical analyses were performed in R 3.4. Alpha diversity indices for the lamb gut bacterial community, relative abundances of communities at phylum and genus level, and qPCR results for total bacteria and five selected bacterial genera between the two groups were statistically analysed using a two‐tailed Wilcoxon signed‐rank test. Alpha diversity indices of the maternal and environmental bacterial communities were analysed by the Kruskal‐Wallis test. Differences in bacterial community composition between the environmental niches of the two groups and between maternal niches were examined by PERMANOVA. The P values were adjusted using the Benjamini‐Hochberg correction for multiple comparisons when required. Statistical significance was set at P < 0.05 level.
Supporting information
Fig. S1. Sample‐based rarefaction curves showing the increase in OTU numbers as a function of the number of reads sampled. Each curve represents one individual sample (n = 83) and its corresponding rarefaction carves.
Fig. S2. Alpha diversity indices (OTUs, Chao1, Shannon, and Simpson) of the bacterial communities from maternal sources. Values are median ± interquartile range. Boxes with different letters above their whiskers are significantly different at P < 0.05. VA: maternal vagina; OA: maternal oral cavity; MA: milk from birth mother; FEA: maternal stool; NA: maternal teat skin; ATA: maternal abdominal region ventral skin; UA: maternal udder skin. The sample size of each maternal niche was 6.
Fig. S3. Alpha diversity indices (OTUs, Chao1, Shannon, and Simpson) of the bacterial communities from environmental sources. Values are median ± interquartile range. WA: sheep pen wall of suckled group; FA: pen floor of suckled group; WB: pen wall of bottle‐fed group; FB: pen floor of bottle‐fed group. The sample size of each environmental niche was 6.
Fig. S4. Venn diagram showing the shared and unique OTUs between the suckled (n = 6) and bottle‐fed (n = 6) lambs.
Fig. S5. Average relative abundances of the 15 most abundant bacterial genera and their most common source environment in the gut of suckled (n = 6) and bottle‐fed (n = 6) lambs. SourceTracker analysis assigns a different source environment to each sequence of an OTU in the jejunum samples. The legend shows genus name, OTU identifier, and the most common source environment. NA: maternal teat skin; VA: maternal vagina.
Table S1. Relative abundances of the 10 most abundant phyla (relative abundance ≥ 0.1%) between the suckled and bottle‐fed groups. Values are median ± SD, n = 6 for each group.
Table S2. Relative abundances of 5 selected bacterial genera calculated by quantitative real‐time PCR for suckled and bottle‐fed lambs. Values are median ± SD, n = 6 for each group.
Table S3. 16S rRNA gene‐targeted specific primers used in this study.
ACKNOWLEDGEMENTS
This study was supported by the National Key Research and Development Program of China (2017YFD0500500) and Fundamental Research Funds for Central Non‐profit Scientific Institution. This study was also supported in part by a United States Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) HATCH grant WIS02007 to GS and a USDA Agriculture and Food Research Initiative Education and Literacy Initiative predoctoral fellowship no. 2018‐67011‐27997 to MSC. We thank Shanghai Realbio Technology Co., Ltd for providing the sequencing and analysis services in the research.
Contributor Information
Yan Tu, Email: diaoqiyu@caas.cn.
Qiyu Diao, Email: diaoqiyu@caas.cn.
References
- Aagaard, K. , Ma, J. , Antony, K.M. , Ganu, R. , Petrosino, J. , and Versalovic, J. (2014) The placenta harbors a unique microbiome. Sci Transl Med 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamberg, K. , Kolk, K. , Jaagura, M. , Vilu, R. , and Adamberg, S. (2018) The composition and metabolism of faecal microbiota is specifically modulated by different dietary polysaccharides and mucin: an isothermal microcalorimetry study. Benef Microbes 9: 21–34. [DOI] [PubMed] [Google Scholar]
- Aggett, P.J. , Agostoni, C. , Axelsson, I. , De Curtis, M. , Goulet, O. , Hernell, O. , et al (2006) Feeding preterm infants after hospital discharge—a commentary by the ESPGHAN committee on nutrition. J Pediatr Gastr Nutr 42: 596–603. [DOI] [PubMed] [Google Scholar]
- Alipour, M.J. , Jalanka, J. , Pessa‐Morikawa, T. , Kokkonen, T. , Satokari, R. , Hynonen, U. , et al (2018) The composition of the perinatal intestinal microbiota in cattle. Sci Rep‐Uk 8: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad, M.B. , Konya, T. , Maughan, H. , Guttman, D.S. , Field, C.J. , Chari, R.S. , et al (2013) Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. Can Med Assoc J 185: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed, F. , Roswall, J. , Peng, Y. , Feng, Q. , Jia, H. , Kovatcheva‐Datchary, P. , et al (2015) Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17: 690–703. [DOI] [PubMed] [Google Scholar]
- Berry, D. , Stecher, B. , Schintlmeister, A. , Reichert, J. , Brugiroux, S. , Wild, B. , et al (2013) Host‐compound foraging by intestinal microbiota revealed by single‐cell stable isotope probing. P Natl Acad Sci U S A 110: 4720–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezirtzoglou, E. , Tsiotsias, A. , and Welling, G.W. (2011) Microbiota profile in feces of breast‐ and formula‐fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe 17: 478–482. [DOI] [PubMed] [Google Scholar]
- Bi, Y. , Yang, C. , Diao, Q. , and Tu, Y. (2017) Effects of dietary supplementation with two alternatives to antibiotics on intestinal microbiota of preweaned calves challenged with Escherichia coli K99. Sci Rep‐Uk 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasucci, G. , Benenati, B. , Morelli, L. , Bessi, E. , and Boehm, G. (2008) Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr 138: 1796S–1800S. [DOI] [PubMed] [Google Scholar]
- Christian, M. , Sabrina, D. , Francesca, B. , Eoghan, C. , Francesca, T. , Jennifer, M. , et al (2017) The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol R 81: e00036–e00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J.R. , Wang, Q. , Cardenas, E. , Fish, J. , Chai, B. , Farris, R.J. , et al (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxen, M.A. , Law, R.J. , Scholz, R. , Keeney, K.M. , Wlodarska, M. , and Finlay, B.B. (2013) Recent advances in understanding enteric pathogenic Escherichia coli . Clin Microbiol Rev 26: 822–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daft, J.G. , Ptacek, T. , Kumar, R. , Morrow, C. , and Lorenz, R.G. (2015) Cross‐fostering immediately after birth induces a permanent microbiota shift that is shaped by the nursing mother. Microbiome 3: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Agueero, M.G. , Ganal‐Vonarburg, S.C. , Fuhrer, T. , Rupp, S. , Uchimura, Y. , Li, H. , et al (2016) The maternal microbiota drives early postnatal innate immune development. Science 351: 1296–1301. [DOI] [PubMed] [Google Scholar]
- Dennis, K.L. , Wang, Y. , Blatner, N.R. , Wang, S. , Saadalla, A. , Trudeau, E. , et al (2013) Adenomatous polyps are driven by microbe‐instigated focal inflammation and are controlled by IL‐10‐producing T cells. Cancer Res 73: 5905–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis, T.Z. , Hugenholtz, P. , Keller, K. , Brodie, E.L. , Larsen, N. , Piceno, Y.M. , et al (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey, K.G. , Heinig, J. , and Nommsenrivers, L.A. (1995) Differences in morbidity between breast‐fed and formula‐fed infants. J Pediatr‐Us 126: 696–702. [DOI] [PubMed] [Google Scholar]
- Dominguez‐Bello, M.G. , Costello, E.K. , Contreras, M. , Magris, M. , Hidalgo, G. , Fierer, N. , and Knight, R. (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. P Natl Acad Sci USA 107: 11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. [DOI] [PubMed] [Google Scholar]
- Ferretti, P. , Pasolli, E. , Tett, A. , Asnicar, F. , Gorfer, V. , Fedi, S. , et al (2018) Mother‐to‐infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24: 133–145.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonty, G. , Gouet, P. , Jouany, J.P. , and Senaud, J. (1987) Establishment of the microflora and anaerobic fungi in the rumen of lambs. Microbiology 133: 1835–1843. [Google Scholar]
- Funkhouser, L.J. , and Bordenstein, S.R. (2013) Mom knows best: the universality of maternal microbial transmission. PLoS Biol 11: e1001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geirnaert, A. , Steyaert, A. , Eeckhaut, V. , Debruyne, B. , Arends, J.B.A. , Van Immerseel, F. , et al (2014) Butyricicoccus pullicaecorum, a butyrate producer with probiotic potential, is intrinsically tolerant to stomach and small intestine conditions. Anaerobe 30: 70–74. [DOI] [PubMed] [Google Scholar]
- Goulet, O. (2015) Potential role of the intestinal microbiota in programming health and disease. Nutr Rev 731: 32–40. [DOI] [PubMed] [Google Scholar]
- Heikkila, M.P. , and Saris, P. (2003) Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J Appl Microbiol 95: 471–478. [DOI] [PubMed] [Google Scholar]
- Hoddinott, P. , Tappin, D. , and Wright, C. (2008) Breast feeding. BMJ‐Brit Med J 336: 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jami, E. , Israel, A. , Kotser, A. , and Mizrahi, I. (2013) Exploring the bovine rumen bacterial community from birth to adulthood. ISME J 7: 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenmalm, M.C. (2017) The mother‐offspring dyad: microbial transmission, immune interactions and allergy development. J Intern Med 282: 484–495. [DOI] [PubMed] [Google Scholar]
- Jiao, J. , Huang, J. , Zhou, C. , and Tan, Z. (2015) Taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Appl Environ Microb 81: 3502–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost, T. , Lacroix, C. , Braegger, C.P. , and Chassard, C. (2012) New insights in gut microbiota establishment in healthy breast fed neonates. PLoS One 7: e44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita, A. , Hu, J. , and Torres, A.G. (2014) Recent advances in adherence and invasion of pathogenic Escherichia coli . Curr Opin Infect Dis 27: 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper, J.B. , Nataro, J.P. , and Mobley, H. (2004) Pathogenic Escherichia coli . Nat Rev Microbiol 2: 123–140. [DOI] [PubMed] [Google Scholar]
- Klein‐Joebstl, D. , Schornsteiner, E. , Mann, E. , Wagner, M. , Drillich, M. , and Schmitz‐Esser, S. (2014) Pyrosequencing reveals diverse fecal microbiota in Simmental calves during early development. Front Microbiol 5: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights, D. , Kuczynski, J. , Charlson, E.S. , Zaneveld, J. , Mozer, M.C. , Collman, R.G. , et al (2011) Bayesian community‐wide culture‐independent microbial source tracking. Nat Methods 8: 761–U107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpela, K. , and de Vos, W.M. (2018) Early life colonization of the human gut: microbes matter everywhere. Curr Opin Microbiol 44: 70–78. [DOI] [PubMed] [Google Scholar]
- Le Huerou‐Luron, I. , Blat, S. , and Boudry, G. (2010) Breast‐ v. Formula‐feeding: impacts on the digestive tract and immediate and long‐term health effects. Nutr Res Rev 23: 23–36. [DOI] [PubMed] [Google Scholar]
- Leimbach, A. , Hacker, J. , and Dobrindt, U. (2013) E. coli as an all‐rounder: the thin line between commensalism and pathogenicity In Current Topics in Microbiology and Immunology, Dobrindt U., Hacker J.H., and Svanborg C. (eds). Berlin, Heidelberg: Springer‐Verlag, pp. 3–32. [DOI] [PubMed] [Google Scholar]
- Li, R.W. , Connor, E.E. , Li, C. , Baldwin, R.L. , and Sparks, M.E. (2012) Characterization of the rumen microbiota of pre‐ruminant calves using metagenomic tools. Environ Microbiol 14: 129–139. [DOI] [PubMed] [Google Scholar]
- Louis, P. , and Flint, H.J. (2009) Diversity, metabolism and microbial ecology of butyrate‐producing bacteria from the human large intestine. FEMS Microbiol Lett 294: 1–8. [DOI] [PubMed] [Google Scholar]
- Lozupone, C. , and Knight, R. (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microb 71: 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C.A. , Hamady, M. , Kelley, S.T. , and Knight, R. (2007) Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microb 73: 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasik, P. , Newton, J.A. , Sanders, J.G. , Hu, Y. , Moreau, C.S. , Kronauer, D.J.C. , et al (2017) The structured diversity of specialized gut symbionts of the New World army ants. Mol Ecol 26: 3808–3825. [DOI] [PubMed] [Google Scholar]
- Lukjancenko, O. , Wassenaar, T.M. , and Ussery, D.W. (2010) Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol 60: 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie, R.I. , Sghir, A. , and Gaskins, H.R. (1999) Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr 69: 1035S–1045S. [DOI] [PubMed] [Google Scholar]
- Marcobal, A. , Barboza, M. , Froehlich, J.W. , Block, D.E. , German, J.B. , Lebrilla, C.B. , and Mills, D.A. (2010) Consumption of human milk oligosaccharides by gut‐related microbes. J Agr Food Chem 58: 5334–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, R. , Makino, H. , Yavuz, A.C. , Ben‐Amor, K. , Roelofs, M. , Ishikawa, E. , et al (2016) Early‐life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS One 11: e0158498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumiya, Y. , Kato, N. , Watanabe, K. , and Kato, H. (2002) Molecular epidemiological study of vertical transmission of vaginal Lactobacillus species from mothers to newborn infants in Japanese, by arbitrarily primed polymerase chain reaction. J Infect Chemother 8: 43–49. [DOI] [PubMed] [Google Scholar]
- Maynard, C.L. , Elson, C.O. , Hatton, R.D. , and Weaver, C.T. (2012) Reciprocal interactions of the intestinal microbiota and immune system. Nature 489: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C. , Mangifesta, M. , Mancabelli, L. , Lugli, G.A. , James, K. , Duranti, S. , et al (2017) Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J 11: 2834–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato, H. , Otsuka, M. , Shirasaka, S. , Itabashi, H. , and Mitsumori, M. (1992) Colonization of microorganisms in the rumen of young calves. J Gen Appl Microbiol 38: 447–456. [Google Scholar]
- Orrhage, K. , and Nord, C.E. (1999) Factors controlling the bacterial colonization of the intestine in breastfed infants. Acta Paediatr 88430: 47–57. [DOI] [PubMed] [Google Scholar]
- O'Sullivan, A. , Farver, M. , and Smilowitz, J.T. (2015) The influence of early infant‐feeding practices on the intestinal microbiome and body composition in infants. Nutr Metab Insights 81: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penders, J. , Thijs, C. , Vink, C. , Stelma, F.F. , Snijders, B. , Kummeling, I. , et al (2006) Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118: 511–521. [DOI] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. , and Arkin, A.P. (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautava, S. , Luoto, R. , Salminen, S. , and Isolauri, E. (2012) Microbial contact during pregnancy, intestinal colonization and human disease. Nat Rev Gastro Hepat 9: 565–576. [DOI] [PubMed] [Google Scholar]
- Rey, M. , Enjalbert, F. , Combes, S. , Cauquil, L. , Bouchez, O. , and Monteils, V. (2014) Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J Appl Microbiol 116: 245–257. [DOI] [PubMed] [Google Scholar]
- Salyers, A.A. , Vercellotti, J.R. , West, S. , and Wilkins, T.D. (1977) Fermentation of mucin and plant polysaccharides by strains of bacteroides from human colon. Appl Environ Microb 33: 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , et al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microb 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov, I. , Russell, S.L. , Antunes, L.C.M. , and Finlay, B.B. (2010) Gut microbiota in health and disease. Physiol Rev 90: 859–904. [DOI] [PubMed] [Google Scholar]
- Shin, H. , Pei, Z. , Martinez, K.A.I. , Rivera‐Vinas, J.I. , Mendez, K. , Cavallin, H. , et al (2015) The first microbial environment of infants born by C‐section: the operating room microbes. Microbiome 3: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson, L.F. , Payne, M.S. , and Keelan, J.A. (2017) Planting the seed: origins, composition, and postnatal health significance of the fetal gastrointestinal microbiota. Crit Rev Microbiol 43: 352–369. [DOI] [PubMed] [Google Scholar]
- Tamburini, S. , Shen, N. , Wu, H.C. , and Clemente, J.C. (2016) The microbiome in early life: implications for health outcomes. Nat Med 22: 713–722. [DOI] [PubMed] [Google Scholar]
- Thompson, A.L. , Monteagudo‐Mere, A. , Cadenae, M.B. , Lampi, M.L. , and Azcarate‐Peril, M.A. (2015) Milk‐ and solid‐feeding practices and daycare attendance are associated with differences in bacterial diversity, predominant communities, and metabolic and immune function of the infant gut microbiome. Front Cell Infect Mi 5: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak, C. , Angelini, M. , Gloor, G.B. , and Reid, G. (2016) Human milk microbiota profiles in relation to birthing method, gestation and infant gender. Microbiome 4: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valles, Y. , Artacho, A. , Pascual‐Garcia, A. , Loreto Ferrus, M. , Jose Gosalbes, M. , Jose Abellan, J. , et al (2014) Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of spanish infants. PLoS Genet 10: e1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Elekwachi, C. , Jiao, J. , Wang, M. , Tang, S. , Zhou, C. , et al (2017) Changes in metabolically active bacterial community during rumen development, and their alteration by rhubarb root powder revealed by 16S rRNA amplicon sequencing. Front Microbiol 8: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, R.E. , Ninonuevo, M. , Mills, D.A. , Lebrilla, C.B. , and German, J.B. (2006) In vitro fermentation of breast milk oligosaccharides by Bifidobacterium infantis and Lactobacillus gasseri . Appl Environ Microb 72: 4497–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wopereis, H. , Oozeer, R. , Knipping, K. , Belzer, C. , and Knol, J. (2014) The first thousand days ‐ intestinal microbiology of early life: establishing a symbiosis. Pediat Allerg Imm‐Uk 25: 428–438. [DOI] [PubMed] [Google Scholar]
- Yáñez‐Ruiz, D.R. , Abecia, L. , and Newbold, C.J. (2015) Manipulating rumen microbiome and fermentation through interventions during early life: a review. Front Microbiol 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko, T. , Rey, F.E. , Manary, M.J. , Trehan, I. , Dominguez‐Bello, M.G. , Contreras, M. , et al (2012) Human gut microbiome viewed across age and geography. Nature 486: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Sample‐based rarefaction curves showing the increase in OTU numbers as a function of the number of reads sampled. Each curve represents one individual sample (n = 83) and its corresponding rarefaction carves.
Fig. S2. Alpha diversity indices (OTUs, Chao1, Shannon, and Simpson) of the bacterial communities from maternal sources. Values are median ± interquartile range. Boxes with different letters above their whiskers are significantly different at P < 0.05. VA: maternal vagina; OA: maternal oral cavity; MA: milk from birth mother; FEA: maternal stool; NA: maternal teat skin; ATA: maternal abdominal region ventral skin; UA: maternal udder skin. The sample size of each maternal niche was 6.
Fig. S3. Alpha diversity indices (OTUs, Chao1, Shannon, and Simpson) of the bacterial communities from environmental sources. Values are median ± interquartile range. WA: sheep pen wall of suckled group; FA: pen floor of suckled group; WB: pen wall of bottle‐fed group; FB: pen floor of bottle‐fed group. The sample size of each environmental niche was 6.
Fig. S4. Venn diagram showing the shared and unique OTUs between the suckled (n = 6) and bottle‐fed (n = 6) lambs.
Fig. S5. Average relative abundances of the 15 most abundant bacterial genera and their most common source environment in the gut of suckled (n = 6) and bottle‐fed (n = 6) lambs. SourceTracker analysis assigns a different source environment to each sequence of an OTU in the jejunum samples. The legend shows genus name, OTU identifier, and the most common source environment. NA: maternal teat skin; VA: maternal vagina.
Table S1. Relative abundances of the 10 most abundant phyla (relative abundance ≥ 0.1%) between the suckled and bottle‐fed groups. Values are median ± SD, n = 6 for each group.
Table S2. Relative abundances of 5 selected bacterial genera calculated by quantitative real‐time PCR for suckled and bottle‐fed lambs. Values are median ± SD, n = 6 for each group.
Table S3. 16S rRNA gene‐targeted specific primers used in this study.