

Abstract
Congenital disorders of glycosylation type I (CDG‐I) are inborn errors of metabolism, generally characterized by multisystem clinical manifestations, including developmental delay, hepatopathy, hypotonia, and skin, skeletal, and neurological abnormalities. Among others, dolichol‐phosphate‐mannose (DPM) is the mannose donor for N‐glycosylation as well as O‐mannosylation. DOLK‐CDG, DPM1‐CDG, DPM2‐CDG, and DPM3‐CDG are defects in the DPM synthesis showing both CDG‐I abnormalities and reduced O‐mannosylation of alpha‐dystroglycan (αDG), which leads to muscular dystrophy‐dystroglycanopathy. Mannose‐phosphate‐dolichol utilization defect 1 (MPDU1) plays a role in the utilization of DPM. Here, we report two MPDU1‐CDG patients without skin involvement, but with massive dilatation of the biliary duct system and dystroglycanopathy characteristics including hypotonia, elevated creatine kinase, dilated cardiomyopathy, buphthalmos, and congenital glaucoma. Biochemical analyses revealed elevated disialotransferrin in serum, and analyses in fibroblasts showed shortened lipid linked oligosaccharides and DPM, and reduced O‐mannosylation of αDG. Thus, MPDU1‐CDG can be added to the list of disorders with overlapping biochemical and clinical abnormalities of CDG‐I and dystroglycanopathy.
Synopsis
Mannose‐phosphate‐dolichol utilization defect 1 patients can have overlapping biochemical and clinical abnormalities of congenital disorders of glycosylation type I and dystroglycanopathy.
Keywords: congenital disorders of glycosylation, dolichol‐phosphate‐mannose, dystroglycanopathy, MPDU1‐CDG
1. INTRODUCTION
The congenital disorders of glycosylation (CDG) are inborn errors of metabolism with a great genetic heterogeneity. CDG‐I defects are located in the assembly of the lipid linked oligosaccharide (LLO) glucose3mannose9 N‐acetylglucosamine2‐PP‐dolichol (Glc3Man9GlcNAc2‐PP‐Dol) or the transfer of its oligosaccharide to proteins in the endoplasmic reticulum (ER). CDG‐I leads to multiorgan phenotypes, including disorders of the brain and neuromuscular system, hepatopathy, skin, and skeletal abnormalities.1
Genetic defects in the biosynthesis of dolichol‐phosphate‐mannose (DPM; NUS1‐CDG, DHDDS‐CDG, PMM2‐CDG, SRD5A3‐CDG, DOLK‐CDG, DPM1‐CDG, DPM2‐CDG, DPM3‐CDG) lead to CDG‐I profiles of transferrin. However, the clinical phenotypes associated with these defects are very different, which is partly explained by that DPM is required for N‐glycosylation, O‐mannosylation, C‐mannosylation, and GPI‐anchor synthesis. In DOLK‐CDG, DPM1‐CDG, DPM2‐CDG, and DPM3‐CDG, biochemical and phenotypic abnormalities overlap with CDG‐I and muscular dystrophy‐dystroglycanopathy, which is caused by reduced O‐mannosylation of alpha‐dystroglycan (αDG).2, 3, 4, 5
Mannose‐phosphate‐dolichol utilization defect 1 (MPDU1) is involved in the flipping of DPM and dolichol‐phosphate‐glucose (DPG) across the ER membrane and for efficient use of DPM and DPG within the ER lumen.6, 7 MPDU1‐CDG patients have CDG‐I with epilepsy, psychomotor retardation, and skin abnormalities.7, 8, 9 Here, we describe two siblings with a G73E substitution in MPDU1 without skin involvement, but with dilatation of the biliary ducts and dystroglycanopathy symptoms including elevated creatine kinase (CK), dilated cardiomyopathy (DCM), buphthalmos, and glaucoma.
2. MATERIALS AND METHODS
2.1. Subjects
Plasma and fibroblasts were obtained for CDG diagnostics in the Radboudumc Expertise Center for Disorders of Glycosylation in accordance with the Declaration of Helsinki. Informed consent was obtained from patients or their legal representatives. Written informed consent was obtained for inclusion of facial images.
2.2. CDG diagnostics
Serum transferrin isoelectric focusing (TIEF) and electrospray ionization mass spectrometry (ESI‐MS) were performed as described.10, 11, 12 Whole exome sequencing (WES), high‐performance liquid chromatography (HPLC), and thin‐layer chromatography (TLC) of LLOs and DPM were performed as described elsewhere.13, 14
2.3. Immunoblotting and laminin overlay assay
Fibroblasts were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal calf serum (Gibco) and 1% penicillin‐streptomycin (Gibco). Fibroblast lysates were enriched for glycoproteins using agarose‐conjugated wheat germ agglutinin (Sigma). Proteins were loaded on 10% polyacrylamide gels and transferred to nitrocellulose membranes by western blotting. Membranes were used for laminin overlay (LO)4, 15 or incubated with primary antibodies against glycosylated αDG (IIH6C4, 1:2500, Merck 05‐593), the dystroglycan core‐protein (DAG1, 1:333, Genetex), or βDG (1:250, Novacastra) and with HRP‐conjugated polyclonal goat anti‐rabbit or goat anti‐mouse antibodies (1:5000, DAKO).
3. RESULTS
3.1. Clinical description
The two patients are brother and sister from Iraqi origin. They were the third and fourth child of consanguineous parents (first cousins), and had two healthy sisters (Figure 1A). Table 1 summarizes the clinical features of these two patients and four previously reported MPDU1‐CDG patients.7, 8, 9
Figure 1.
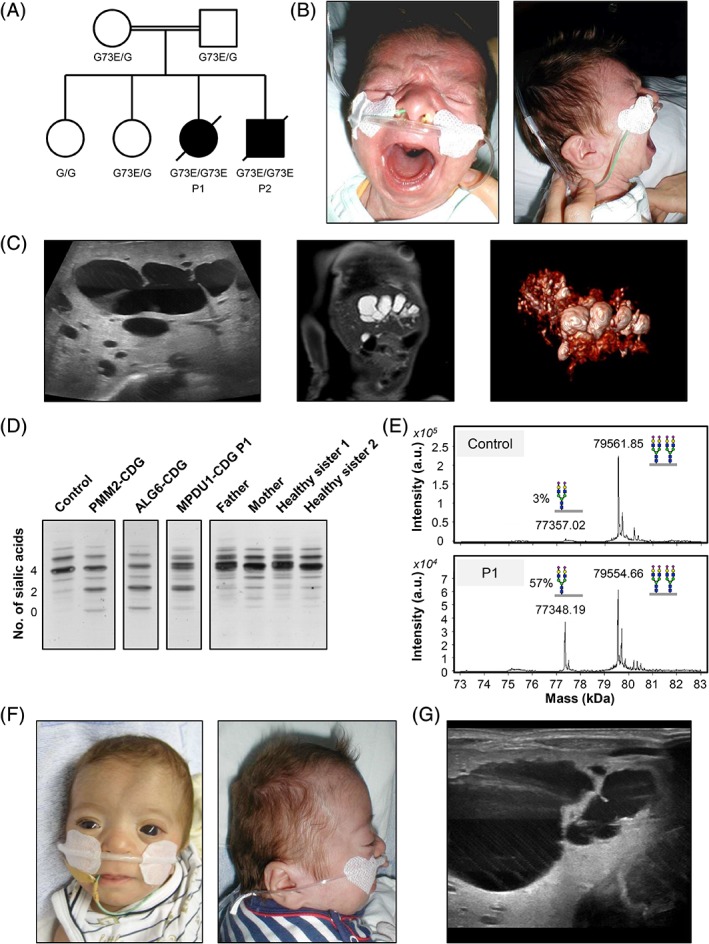
Family pedigree and clinical images of affected patients. A, Family pedigree of MPDU1 patients (P1 = patient 1, P2 = patient 2). B, Front and side facial view of patient 1. C, Abdominal sonography (left), magnetic resonance imaging (middle), and 3D image from magnetic resonance cholangiopancreatography (right) of patient 1 showing a vast dilatation of the complete intrahepatic biliary duct system. D, Serum transferrin isoelectric focusing (TIEF) analysis of P1, her parents and two healthy sisters. E, ESI‐MS of serum transferrin. Disialotransferrin levels are expressed as a percentage of tetrasialotransferrin. F, Front and side facial view of patient 2 at 4 months of age. G, Abdominal sonography of patient 2, showing dilatation of the intrahepatic biliary duct. CDG, congenital disorders of glycosylation; ESI‐MS, electrospray ionization mass spectrometry; MPDU1, mannose‐phosphate‐dolichol utilization defect 1
Table 1.
Clinical and laboratory data of the two presented patients and the patients from the literature
| Patient | Patient 1 | Patient 2 | Patient girl | Patient S | Patient L | Patient A | Patient boy |
|---|---|---|---|---|---|---|---|
| Described in | This paper | This paper | Thiel et al9 | Schenk et al7 | Schenk et al7 | Schenk et al7 | Kranz et al8 |
| Sex | Female | Male | Female | Male | Female | Male | Male |
| Zygosity | Homozygous | Homozygous | Homozygous | Homozygous | Compound heterozygous | Homozygous | Homozygous |
| Nucleotide change | |||||||
| Chr17[GRCh38] | g.7585994G>A | g.7585994G>A | g.7585994G>A | g.7585994G>A | g.7583864T>C | g.7586745T>C | g.7585997T>C |
| NM_004870.3 | c.218G>A | c.218G>A | c.218G>A | c.218G>A | c.2T>C g.7587164del c.511delC |
c.356T>C | c.221T>C |
| Protein change | p.G73E | p.G73E | p.G73E | p.G73E | p.M1T (loss of start codon) p.L171Sfs*42 |
p.L119P | p.L74S |
| Affected exon(s) | 3 | 3 | 3 | 3 | 1 and 6 | 4 | 3 |
| Parental consanguinity | + | + | + | + | No | + | No |
| Family | Two healthy older sisters | Brother of patient 1, two healthy older sisters | Healthy older sister and twin brother, older brother showed similar disease and died in the neonatal period | Normal | Brother died at 2 months with similar disease | Normal | n.a. |
| Pregnancy (weeks) | 36 + 2 weeks | 30 + 4 weeks placental hypertrophy |
30 + 5 weeks | 37 weeks | 40 weeks | 40 weeks | 39 weeks |
| Birth weight (g) | 2390 | 1420 | 2370 | 2485 | 3200 | 3200 | 2770 |
| Perinatal problems | Apneas and bradycardias, respiratory failure | Cyanotic, breathless, hypotonia, respiratory failure | Hypotonia, insufficient breathing | Hypotonia, seizures | Hypertonia | No | Hypotonia |
| Dysmorphology | Smooth philtrum, retrognathia, low‐set, posterior‐rotated ears, hypertelorism | Smooth philtrum, retrognathia, low‐set, posterior‐rotated ears, hypertelorism, micropenis | Hypertelorism, broad‐based nose, thin lips | Large anterior fontanel, bilateral parietal bossing, thin lips | No | No | Contractures |
| Psychomotor development | Absent | Absent | Absent | Absent | Severe retardation | Severe retardation | Ataxia, profound psychomotor retardation, unable to communicate |
| Feeding problems | Dysphagia | Dysphagia | Dysphagia | From 4 months | No | No | Deceased food intake, abdominal pain, and frequent vomiting |
| Hypotonia | + | + | + | +++ | No | + | + |
| Seizures | Seizures with apneas | Seizures with apneas, hypertonic attacks | No | Severe, with apnea | Hypertonic attacks in infancy | Generalized febrile seizures at 15 months | Seizures at 5 months |
| Electroencephalogram | Multifocal sharp waves | Parieto‐temporo‐occipital and multiregional spikes | Generalized background slowing in the theta and delta frequency ranges but no epileptiform activity | Hypsarrhythmia | Abnormal β‐activity | Generalized dysrhythmia | Hypersynchronic activity |
| Magnetic resonance imaging of the head | Enlarged subarachnoid space | n.a. | No abnormalities | Normal myelination, enlarged subarachnoid space, enlarged ventricles | Normal myelination | Normal myelination, enlarged frontal spaces | General cerebral atrophy |
| Tendon reflexes | Normal | Normal | n.a. | Normal | Normal | Normal | Normal |
| Nerve conduction velocity | n.a. | n.a. | n.a. | Normal | Normal | Normal | n.a. |
| Ophthalmoscopy | Buphthalmos with congenital glaucoma, pupillary iris pigment epithelial cysts | Buphthalmos with severe congenital glaucoma | n.a. | Optic atrophy | Pale papillae | Normal | Nystagmus, amaurosis, strabismus morphology of the retina normal |
| Visual and acoustic responses | BERA: pathological | BERA: pathological | n.a. | Absent | Normal | Normal | Amaurosis, BERA normal |
| Skin disorder | No | No | Small hands with scleroderma‐like consistency and loss of dermatoglyphic patterns, ichthyosis | Deneralized patchy desquamation | Ichthyosis | Transient eczema | Dry, transient hyperkeratotic and scaling with erythroderma |
| Somatic development | At 4 months | At 9 months | n.a. | At 4 months | At 16 years | At 10 years | Dystrophy with W, H, and HC below P3 |
| W: 4 kg | W: 5.6 kg | W: P3–1.1 kg | W: P3 | W: P97 | |||
| HC: P3–2 cm | H: P3–16 cm | H: P10–25 | |||||
| HC: P3 normal pubertal development |
HC: P75–97 | ||||||
| Other clinical features | Apneas with desaturations, respiratory insufficiency, death at 4 months | Apneas with desaturations, respiratory insufficiency, death at 11 months | >1.5 Months: microcephaly. Apneas, cyanosis, oxygen dependency, death at 6 months | No weight gain, oxygen dependency, ascites, death at 10 months | n.a. | n.a. | n.a. |
| Other organ features | Enhanced echogenicity of the marrow pyramids, small renal cysts Massive dilatation of the biliary duct system Postpartual pulmonary hypertension, later dilated cardiomyopathy |
Enhanced echogenicity of the marrow pyramids, small renal cysts Massive dilatation of the biliary duct system Dilated aorta ascendens, hypertrophic cardiomyopathy, arterial hypertension |
Initial electrocardiogram (ECG): Moderate focal hypertrophy of the basal interventricular septum but no cardiac malformations. ECG at 3 months: severe noncompaction cardiomyopathy |
Mild pericardial effusion bilateral small cortical renal cysts |
n.a. | n.a. | n.a. |
| Serum transaminases | Normal | Normal | n.a. | Normal | Normal | Normal | Normal |
| Other laboratory abnormalities | Thrombocytopenia CK elevated, fibrinogen low, ATIII not measurable | Thrombocytopenia CK elevated, ATIII low | CK elevated | Mild persistent thrombocytopenia, periodic elevation of CK | Transient deficiency of growth hormone and IGF‐I | No | Slightly reduced ATIII |
Abbreviations: ATIII, antithrombin III; BERA, brainstem evoked response audiometry; CK, creatine kinase; IGF‐I, insulin‐like growth factor 1; n.a., not available.
3.1.1. Patient 1
Patient P1, a girl, was born spontaneously in the 37th gestational week after an uneventful pregnancy. At birth, her length was 43 cm (standard deviation score, (SDS), −2.08), weight 2390 g (SDS, −0.81), and head circumference 32 cm. Because of postnatal apneas and bradycardias with deep desaturations, followed by respiratory failure, she was resuscitated and referred to the neonatal intensive care unit. Slight dysmorphic features were noted including a smooth philtrum, retrognathia, low‐set, posterior‐rotated ears, and hypertelorism with megalocorneae (Figure 1B).
At the age of 2 months, P1 showed tonic‐clonic seizures with multifocal sharp waves on electroencephalography (EEG). Brain magnetic resonance imaging (MRI) revealed enlarged outer fronto‐temporo‐parietal cerebrospinal fluid spaces. Abdominal sonography as well as magnetic resonance cholangiopancreatography showed a vast dilatation of the intrahepatic biliary ducts, especially within the left lobe of the liver (Figure 1C). There were no further signs of biliary duct obstruction or liver enlargement. Initially, kidney sonography showed enhanced echogenicity of the marrow pyramids without corresponding MRI abnormalities. However, at the age of 7 weeks, small renal cysts were suspected in an ultrasound scan.
Initially, electrocardiography and echocardiography were normal, except for a mild temporary pulmonary hypertension. At the age of 3 months, the patient evolved DCM with low output and a shortening fraction of at least 9%.
The megalocorneae possessed a diameter of 13 mm (normal in newborn = 9.5 mm). Further ophthalmologic examination revealed a buphthalmos with congenital glaucoma. The eye lenses were slightly cloudy and pupillary iris showed pigment epithelial cysts. A sensorineural hearing loss was identified with brainstem evoked response audiometry (BERA) evaluation. There were no abnormalities of the skin.
Blood CK levels were elevated up to 3090 U/L without substantial elevation of aspartate amino transferases (maximal 175 U/L) and alanine amino transferases (maximal 163 U/L), biochemical signs of cholestasis or icterus. There was a marked congenital thrombocytopenia (minimal 45 000 platelets/μL), a slightly decreased fibrinogen (minimal 96 mg/dL), and a nonmeasurable antithrombin III (ATIII, <20%).
Prometaphase karyotype analysis showed a normal 46,XX female karyotype and a normal array‐comparative genomic hybridization (array‐CGH), without any microdeletion or duplication. Congenital toxoplasmosis, other (syphilis, varicella‐zoster, parvovirus B19), rubella, cytomegalovirus, and herpes (TORCH) infections, peroxisomal diseases as well as mucopolysaccharidoses were ruled out by serology and metabolic screening. The serum amino acids, acylcarnitine profile, very long chain fatty acids, 7‐dehydrocholesterol, and urine organic acids were normal. In view of the clinical symptoms of multiorgan involvement with seizures, cardiomyopathy, eye, and liver abnormalities as well as thrombocytopenia and ATIII deficiency, a CDG was suspected and TIEF and ESI‐MS of serum transferrin were performed. These revealed reduced tetrasialotransferrin, with increased asialo‐ and disialotransferrin indicative of a CDG‐I (Figure 1D,E).
In her short life, the patient did not exhibit any psychomotor development, and had to be fed parenterally or by a gastric tube. The clinical course was complicated by an increasing frequency and severity of seizures with epileptic apneas followed by respiratory insufficiency. Congestive heart failure as a consequence of the DCM led to the termination of therapeutic interventions, and she died at the age of nearly 4 months.
3.1.2. Patient 2
This patient exhibited nearly the same clinical features and course as his affected sister (P1). He was born premature per cesarean section in the 31st gestational week because of a rupture of the placental membrane and uterine bleeding. His birth length was 38 cm (SDS, −0.38), his weight was 1420 g (SDS, −0.09), and he had a head circumference of 28 cm. He showed a smooth philtrum, retrognathia, and low‐set, posterior‐rotated ears. He had hypertelorism with prominent appearing eyes with enlarged and cloudy corneae (Figure 1F). Musculature was slightly hypotonic, but otherwise the child appeared to be normal and without skin abnormalities, except a micropenis. After birth, he was cyanotic, breathless, and without muscular tonus, requiring immediate cardiopulmonary resuscitation and controlled ventilation.
Echocardiography detected a dilated aorta ascendens, a transient pulmonary hypertension, and a hypertrophic cardiomyopathy (HCM) at the age of 3 weeks, which developed in combination with an arterial hypertension.
At the age of 6 months, he had generalized seizures accompanied by desaturations, whereas EEG showed parieto‐temporo‐occipital and multiregional spikes over both hemispheres. Sonography of the brain showed no gross abnormalities. Abdominal sonography revealed dilatation of the intrahepatic biliary duct system (Figure 1G). Kidney sonography exhibited enhanced echogenicity of the marrow pyramids. At the age of 4 months, he developed multiple small cysts subcapsular within the renal parenchyma.
The ophthalmological examination revealed a buphthalmos with slightly opaque corneae with a diameter of 11 mm and severe congenital glaucoma, requiring prompt trabeculectomy intervention. The BERA showed a sensorineural hearing loss.
Chromosomal analysis showed a normal 46,XY karyotype, and array‐CGH as well as TORCH serology were normal. He also exhibited thrombocytopenia (minimal 13 000 platelets/μL), elevated CK levels (up to 905 U/L), and low ATIII (maximal 40%), but transaminases, fibrinogen, thyroxin‐binding globulin, and bilirubin were normal.
Like his sister, P2 hardly presented any psychomotor development, and had to be fed parenterally or by a gastric tube. He showed severe apneas and bradycardias which were not treatable, and he died at the age of 11 months from respiratory failure.
3.2. Genetic analysis revealed a homozygous missense mutation in MPDU1
Whole exome sequencing revealed a homozygous missense mutation Chr17(GRCh38): g.7585994G>A; NM_004870.3(MPDU1): c.218G>A; p.(G73E) in both patients. This mutation has been reported in two other MPDU1‐CDG patients.7, 9 WES was also performed of the patients' healthy sisters and parents and confirmed parental segregation of the mutation (Figure 1A).
3.3. Biochemical analysis revealed elevated LLO intermediates, DPM, and reduced O‐mannosylation of αDG
Next, we analyzed the LLO composition in fibroblasts from patient 1. Cells were incubated with [2‐3H]mannose and [3H]oligosaccharides were extracted and analyzed using HPLC. Increased levels of dolichol‐linked Man5GlcNAc2 and Man9GlcNAc2 accompanied by reduced amounts of Glc3Man9GlcNAc2 were detected (Figure 2A) which indicated shortage of DPM and DPG in the ER lumen. Since TLC analysis further showed that DPM is synthesized in the patient's fibroblasts (Figure 2B), a lack of transport of DPM from the cytosol into the ER can be assumed.
Figure 2.
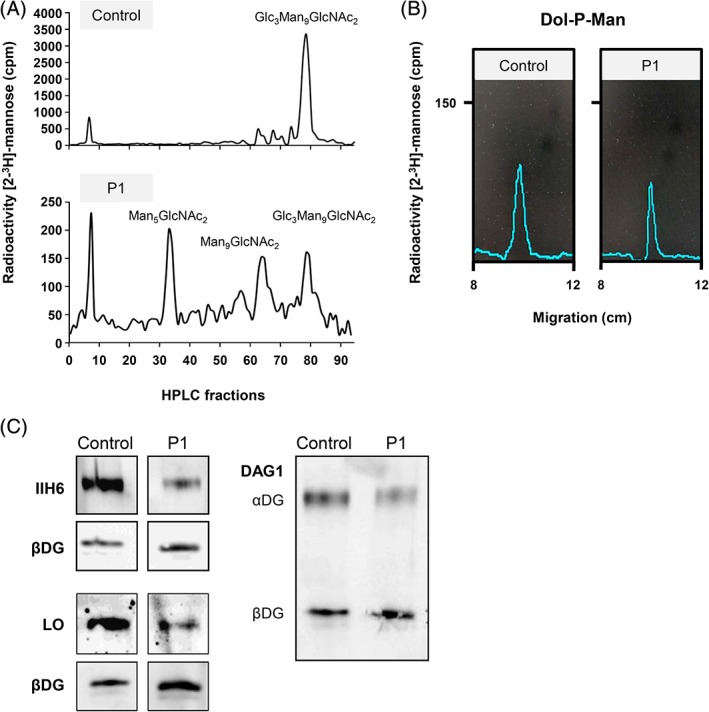
CDG diagnostics and biochemical analyses of MPDU1‐CDG P1. A, HPLC analysis of LLO from fibroblasts of patient 1 (P1) and a control revealed the accumulation of the shortened dolichol‐linked oligosaccharides Man5GlcNAc2 and Man9GlcNAc2. B, Thin‐layer chromatography (TLC) analysis of hydrophobic LLO extracts further revealed that dolichol‐phosphate‐mannose (Dol‐P‐Man) is synthesized in patient 1 fibroblasts. C, Analysis of O‐mannosylated αDG in P1 and control fibroblasts. IIH6 and laminin (laminin overlay, LO) only bind to fully functional O‐mannosyl glycans of αDG. DAG1 binds to the core of the dystroglycan protein, showing expression of αDG and βDG proteins. CDG, congenital disorders of glycosylation; HPLC, High‐performance liquid chromatography; LLO, lipid linked oligosaccharide; MPDU1, mannose‐phosphate‐dolichol utilization defect 1
Subsequently, we analyzed the O‐mannosylation of αDG in fibroblasts of patient P1 by IIH6 immunolabeling and a LO assay. We found that the signals for both IIH6 and LO were reduced in patient fibroblasts as compared to the signal of control fibroblasts (Figure 2C), suggesting reduced glycosylation of αDG.
4. DISCUSSION
Here, we describe two patients with mutations in MPDU1, causing MPDU1‐CDG (CDG‐1f) with overlapping symptoms of CDG‐I and dystroglycanopathy. MPDU1‐CDG is the fifth disorder related to DPM biosynthesis or utilization that bridges CDG‐I and the O‐mannosylation disorders.
So far, seven MPDU1‐CDG patients have been described. All patients showed psychomotor retardation and most patients had hypotonia, facial dysmorphism, eye defects, apnea, and skin abnormalities such as ichthyosis.7, 8, 9 Including the two patients described here, four patients have been described with the same G73E substitution.7, 9 Hypertelorism, dysphagia, small renal cysts, thrombocytopenia, cardiomyopathy, and respiratory problems characterized these patients, whereas these symptoms were not reported in the other three MPDU1‐CDG patients. In addition, all G73E patients died at an early age (<11 months), whereas the other MPDU1‐CDG patients at least reached their teenage years. Taken together, this suggests that the G73E substitution affects MPDU1 function more severely, and the additional clinical features can aid in the prediction of the disease progression when new MPDU1‐CDG patients are identified.
Interestingly, the two siblings described here showed a very similar clinical pattern, including very similar facial features, whereas siblings with other CDG disorders do not necessarily share as many clinical characteristics, for example, in ALG3‐CDG.16 The MPDU1‐CDG siblings shared the following abnormalities: massive dilatation of the intrahepatic biliary duct system, small renal cysts, buphthalmos with glaucoma, DCM, thrombocytopenia, elevated CK, and low ATIII. MPDU1 has been associated with the flipping of DPM over the ER membrane and thereby is part of the DPM biosynthesis defects.6 DPM is required for multiple glycosylation pathways. Thus, different symptoms can be caused by dysfunction of different glycosylation pathways. Buphthalmos, glaucoma, DCM, and elevated CK are clinical features that overlap with the disease spectrum of the dystroglycanopathies. Buphthalmos has so far not been described in any of the other DPM disorders, but is associated with Walker‐Warburg syndrome (WWS) and muscle‐eye‐brain (MEB) disease, which are severe variants of dystroglycanopathy.17 Glaucoma is a common feature of WWS and MEB, and has also been reported in SRD5A3‐CDG.17, 18
Unfortunately, the role of glycosylation in cardiac disease is not completely understood. Therefore, there is no clear explanation why patient 1 shows DCM, whereas patient 2 developed HCM. HCM has been reported in PMM2‐CDG, ATP6V1A‐CDG, and ATP6V1E1‐CDG.19 Recent studies have shown that abnormal glycosylation, for example, abnormal sialylation or reduced hybrid/complex N‐glycosylation, is related to heart disease.19, 20, 21 However, future investigations are required to understand the clinical relevance of these findings. So far, DCM has been described in FKRP‐CDG, FKTN‐CDG, POMT1‐CDG, POMT2‐CDG, DOLK‐CDG, DPM3‐CDG, and PGM1‐CDG.19, 22 With the exception of PGM1‐CDG, these CDGs are associated with abnormal O‐mannosylation of αDG. Hence, the cardiac pathomechanism in MPDU1‐CDG patient 1 could be related to abnormal O‐mannosylation, although studies in heart biopsies are warranted to study this. In line with the clinical symptoms overlapping with dystroglycanopathies, we showed reduced O‐mannosylation of αDG in patient fibroblasts.
In addition, we reported biliary duct abnormalities and renal cysts in our MPDU1‐CDG patients. Biliary duct abnormalities have also been observed in patients with mutations in mannose‐6‐phosphate isomerase (MPI), which interconverts fructose‐6‐phosphate to mannose‐6‐phosphate.23 Sabry et al24 reported a dehydrodolichyl diphosphate synthase (DHDDS‐CDG) patient with dilations of the biliary duct and renal failure, and Schenk et al7 reported renal cysts in a MPDU1‐CDG patient with the same G73E substitution. Biliary duct abnormalities and renal cysts are clinical symptoms associated with Caroli syndrome. In some cases, this syndrome has been associated with PKHD1 mutations, which are also known to cause autosomal recessive polycystic kidney disease. PKHD1 is extensively glycosylated,25 and it is tempting to speculate that abnormal glycosylation of PKHD1 causes the biliary duct abnormalities and renal cysts.
In summary, we reported on two newly identified MPDU1‐CDG patients and showed reduced N‐glycosylation and O‐mannosylation in patient material. Together with the muscular, eye, and heart abnormalities, this adds MPDU1‐CDG to the list of DPM disorders causing biochemical and clinical abnormalities overlapping CDG‐I and dystroglycanopathy. Identification of additional patients with defects in DPM availability, and extensive O‐mannosylation and N‐glycosylation analysis of patient material is required to further increase our understanding of the pathophysiology of the DPM disorders.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
W.V.T. has set up the study, performed laboratory experiments, acquired and interpreted data, and written the manuscript. A.A. has performed WES analysis and critically reviewed the results and revised the manuscript. E.K. has collected, analyzed, and reviewed clinical data and written the manuscript. N.A.B. has performed ESI‐MS analysis and critically reviewed the manuscript. M.A.W. has critically reviewed results and revised the manuscript. C.T. has provided LLO analysis and revised the manuscript. D.J.L. contributed to the setup of the study, has interpreted data, critically reviewed the results and the manuscript, and supervised the study.
PATIENT CONSENT
All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in this study or from their legal representatives. Written informed consent was obtained for inclusion of facial images.
ACKNOWLEDGMENTS
This work was supported by the Netherlands Organization for Scientific Research (VIDI Grant 91713359 to D.J.L.), the Prinses Beatrix Spierfonds (Grant W.OR17‐15 to D.J.L.), the Deutsche Forschungsgemeinschaft (FOR2509: TH1461/7‐1 to C.T.), and the European Union's Horizon 2020 research and innovation program under the ERA‐NET Cofund action N° 643578 (EUROCDG‐2).
van Tol W, Ashikov A, Korsch E, et al. A mutation in mannose‐phosphate‐dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Reports. 2019;50:31–39. 10.1002/jmd2.12060
Communicating Editor: Eva Morava
Funding information Deutsche Forschungsgemeinschaft, Grant/Award Number: FOR2509: TH1461/7‐1; Horizon 2020, Grant/Award Number: 643578; Nederlandse Organisatie voor Wetenschappelijk Onderzoek, Grant/Award Number: 91713359; Prinses Beatrix Spierfonds, Grant/Award Number: W.OR17‐15
REFERENCES
- 1. Jaeken J, Péanne R. What is new in CDG? J Inherit Metab Dis. 2017;40:621‐625. [DOI] [PubMed] [Google Scholar]
- 2. Barone R, Aiello C, Race V, et al. DPM2‐CDG: a muscular dystrophy‐dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. 2012;72:550‐558. [DOI] [PubMed] [Google Scholar]
- 3. Lefeber DJ, de Brouwer APM, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O‐mannosylation. PLoS Genet. 2011;7:e1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lefeber DJ, Schönberger J, Morava E, et al. Deficiency of Dol‐P‐Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85:76‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang AC, Ng BG, Moore SA, et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy‐type congenital muscular dystrophy. Mol Genet Metab. 2013;110:345‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anand M, Rush JS, Ray S, et al. Requirement of the Lec35 gene for all known classes of monosaccharide‐P‐dolichol‐dependent glycosyltransferase reactions in mammals. Mol Biol Cell. 2001;12:487‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schenk B, Imbach T, Frank CG, et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest. 2001;108:1687‐1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kranz C, Denecke J, Lehrman MA, et al. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG‐If). J Clin Investig. 2001;108:1613‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiel C, Wortmann S, Riedhammer K, et al. Severe ichthyosis in MPDU1‐CDG. J Inherit Metab Dis. 2018;41:1293‐1294. [DOI] [PubMed] [Google Scholar]
- 10. Abu Bakar N, Voermans NC, Marquardt T, et al. Intact transferrin and total plasma glycoprofiling for diagnosis and therapy monitoring in phosphoglucomutase‐I deficiency. Transl Res. 2018;199:62‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Babovic‐Vuksanovic D, O'Brien JF. Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol Diagn Ther. 2007;11:303‐311. [DOI] [PubMed] [Google Scholar]
- 12. de Jong G, van Noort WL, van Eijk HG. Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv Exp Med Biol. 1994;356:51‐59. [DOI] [PubMed] [Google Scholar]
- 13. Ashikov A, Abu Bakar N, Wen XY, et al. Integrating glycomics and genomics uncovers SLC10A7 as essential factor for bone mineralization by regulating post‐Golgi protein transport and glycosylation. Hum Mol Genet. 2018;27:3029‐3045. [DOI] [PubMed] [Google Scholar]
- 14. Thiel C, Rind N, Popovici D, et al. Improved diagnostics lead to identification of three new patients with congenital disorder of glycosylation‐Ip. Hum Mutat. 2012;33:485‐487. [DOI] [PubMed] [Google Scholar]
- 15. Michele DE, Barresi R, Kanagawa M, et al. Post‐translational disruption of dystroglycan‐ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417‐421. [DOI] [PubMed] [Google Scholar]
- 16. Kranz C, Sun L, Eklund EA, Krasnewich D, Casey JR, Freeze HH. CDG‐Id in two siblings with partially different phenotypes. Am J Med Genet A. 2007;143A:1414‐1420. [DOI] [PubMed] [Google Scholar]
- 17. Martin PT. The dystroglycanopathies: the new disorders of O‐linked glycosylation. Semin Pediatr Neurol. 2005;12:152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morava E, Wevers RA, Cantagrel V, et al. A novel cerebello‐ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010;133:3210‐3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marques‐da‐Silva D, Francisco R, Webster D, Dos Reis Ferreira V, Jaeken J, Pulinilkunnil T. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis. 2017;40:657‐672. [DOI] [PubMed] [Google Scholar]
- 20. Deng W, Ednie AR, Qi J, Bennett ES. Aberrant sialylation causes dilated cardiomyopathy and stress‐induced heart failure. Basic Res Cardiol. 2016;111:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ednie AR, Deng W, Yip KP, Bennett ES. Reduced myocyte complex N‐glycosylation causes dilated cardiomyopathy. FASEB J. 2019;33:1248‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Tol W, Michelakakis H, Georgiadou E, et al. Toward understanding tissue‐specific symptoms in dolichol‐phosphate‐mannose synthesis disorders; insight from DPM3‐CDG. J Inherit Metab Dis. 2019;1–9. [DOI] [PubMed] [Google Scholar]
- 23. Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Ann Transl Med. 2018;6:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sabry S, Vuillaumier‐Barrot S, Mintet E, et al. A case of fatal type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J Rare Dis. 2016;11:84‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Outeda P, Menezes L, Hartung EA, et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C‐terminus in disease mechanism. Kidney Int. 2017;92:1130‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]