

Abstract
DNMT3A (DNA methyltransferase 3A) is a de novo DNA methyltransferase responsible for establishing CpG methylation patterns within the genome. DNMT3A activity is essential for normal development, and its dysfunction has been linked to developmental disorders and cancer. DNMT3A is frequently mutated in myeloid malignancies with the majority of mutations occurring at Arg-882, where R882H mutations are most frequent. The R882H mutation causes a reduction in DNA methyltransferase activity and hypomethylation at differentially-methylated regions within the genome, ultimately preventing hematopoietic stem cell differentiation and leading to leukemogenesis. Although the means by which the R882H DNMT3A mutation reduces enzymatic activity has been the subject of several studies, the precise mechanism by which this occurs has been elusive. Herein, we demonstrate that in the context of the full-length DNMT3A protein, the R882H mutation stabilizes the formation of large oligomeric DNMT3A species to reduce the overall DNA methyltransferase activity of the mutant protein as well as the WT–R882H complex in a dominant-negative manner. This shift in the DNMT3A oligomeric equilibrium and the resulting reduced enzymatic activity can be partially rescued in the presence of oligomer-disrupting DNMT3L, as well as DNMT3A point mutations along the oligomer-forming interface of the catalytic domain. In addition to modulating the oligomeric state of DNMT3A, the R882H mutation also leads to a DNA-binding defect, which may further reduce enzymatic activity. These findings provide a mechanistic explanation for the observed loss of DNMT3A activity associated with the R882H hot spot mutation in cancer.
Keywords: cancer, cancer biology, DNA methyltransferase, DNA methylation, oligomerization, oligomer, DNMT3A, epigenetics, R882H
Introduction
In humans, DNA methylation patterns are established and maintained through the coordinated efforts of four genes in the DNA methyltransferase (DNMT)2 family: DNMT1, DNMT3A, DNMT3B, and DNMT3L (1). DNMT1 is a maintenance DNA methyltransferase with a strong substrate preference for hemimethylated DNA. DNMT1 copies CpG methylation marks from the parental strand to the daughter strand during DNA replication to ensure that methylation patterns are maintained after cell division (1). DNMT3A and DNMT3B are de novo DNMTs, required for establishing methylation patterns during embryonic development, but they can also contribute to the maintenance of DNA methylation (2, 3). Although these two de novo DNMT isoforms are highly similar and have some overlapping functions, they have also been shown to play unique roles in biology (2, 4). This is highlighted by the observation that DNMT3B KO mice are embryonic lethal, while DNMT3A KO mice die several weeks after birth (2). DNMT3L is catalytically inactive, lacking key amino acids for DNA-binding activity and methyltransferase activity (5–7). DNMT3L serves as a stabilizing and stimulating co-factor for the DNMT3A and DNMT3B methyltransferases during embryonic development but is not normally expressed in differentiated cell types (8–12). DNMT3L KO mice are viable, but fail to reproduce due to deficient germline DNA methylation, likely mediated by DNMT3A (13).
DNMT3A is one of the most frequently mutated genes in hematopoietic cancers, with mutations observed in as many as 20% of AML patients (14, 15). The most frequent DNMT3A mutation is the R882H hot spot mutation (Fig. 1A), which is associated with a lower overall probability of survival of AML patients (16). Malignant cells from cancer patients carrying this mutation display hypomethylation patterns at differentially-methylated regions (DMRs), although total genomic CpG methylation is nearly unchanged relative to WT cells (17, 18). In engineered mouse models, loss of DNMT3A function similarly causes hypomethylation at DMRs, which is associated with a failure of hematopoietic stem cell (HSC) differentiation, expansion of the HSC compartment, and leukemogenesis (4, 19–26). The R882H mutation has been proposed to have dominant-negative activity (18, 27), explaining why R882H is always found to be heterozygous in leukemia patients (28). Although the R882H DNMT3A mutation has been the subject of multiple biochemical studies, the mechanism by which this mutation reduces DNA methyltransferase activity is unclear. DNMT3A has been shown to form different-sized multimeric complexes, where tetramers are likely the most active species (18, 29, 30). The R882H mutation is located at the C-terminal catalytic domain of DNMT3A (Fig. 1A), near the interface of two DNMT3A monomers in the tetrameric complex (Fig. 1B). In prior studies, the R882H mutation has been proposed to prevent the formation of the active DNMT3A tetramer, with the isolated catalytic domain only forming dimers with low enzymatic activity in solution (18, 31, 32), but this has never been confirmed with the full-length protein. Although the dominant-negative behavior of the R882H mutation has been demonstrated in both mouse models (27) and in human cell lines upon overexpression of the mutant protein (18, 26), a recent biochemical study refuted these findings, claiming the purified mutant protein does not have dominant-negative activity (33).
Figure 1.
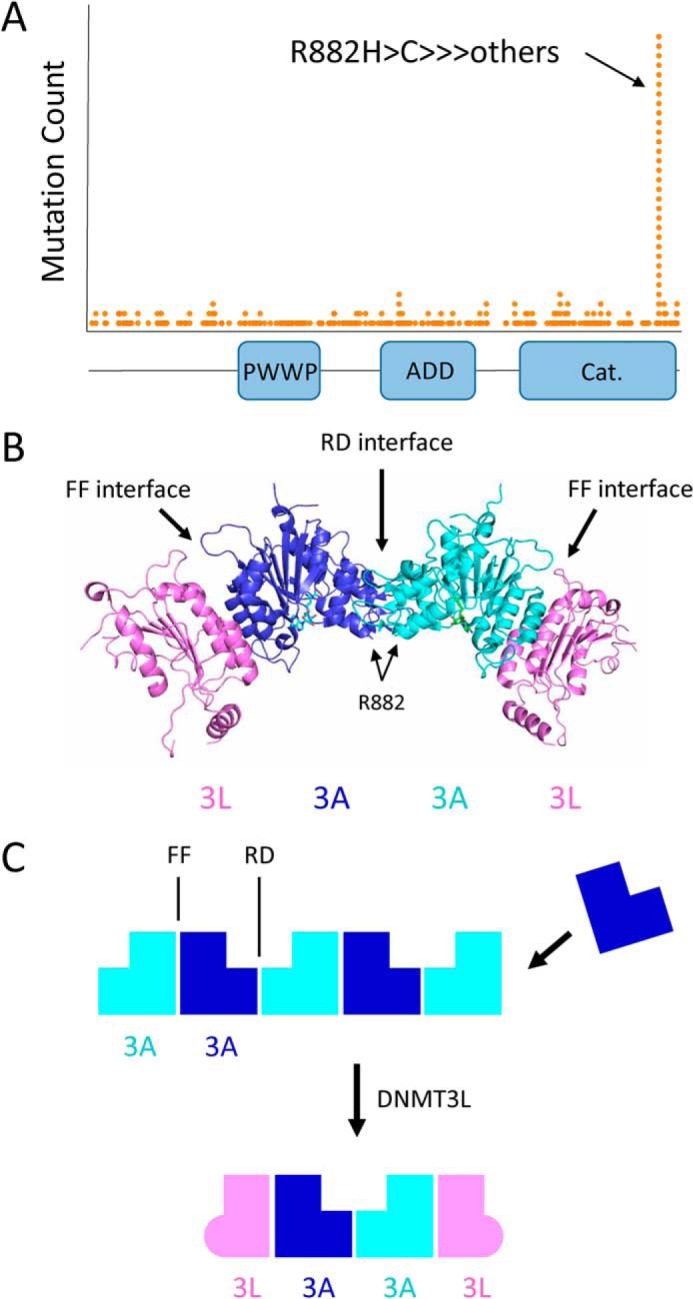
DNMT3A cancer mutations, domain architecture, and oligomer formation. A, histogram of DNMT3A mutations observed in cancer from the TCGA cohort. The R882H hot spot mutation is located at the C-terminal catalytic domain. B, crystal structure of the DNMT3L–DNMT3A–DNMT3A–DNMT3L complex (PDB code 2QRV). Arg-882 is located at the RD interface between two DNMT3A catalytic domains. C, DNMT3A catalytic domain mediates the formation of large oligomers through self-association along two binding surfaces, called the RD and FF interfaces. DNMT3L prevents the formation of large DNMT3A oligomers, instead forming a tetramer composed of two DNMT3A and two DNMT3L molecules.
In addition to tetramers, DNMT3A can form multimeric complexes of significantly larger sizes (30). This is facilitated by the methyltransferase domain, which contains two distinct surfaces each capable of binding to another catalytic domain monomer (34). The catalytic domain can therefore drive oligomerization of the DNMT3A protein (Fig. 1C). The two oligomer-forming interfaces of the catalytic domain are structurally different (34). One interface (referred to as the RD interface) is composed mostly of a series of electrostatic interactions and hydrogen bonds, with Arg-885 and Asp-876 making key interactions in the symmetrical binding event (34). Binding at the second interface (the FF interface) is driven by a central core of hydrophobic interactions, with Phe-732 making a key hydrophobic stacking interaction (34). Single-point mutations at Arg-885, Asp-876, and Phe-732 have been shown to disrupt DNMT3A oligomer formation to some extent (30, 31). DNMT3A has never been crystallized on its own, likely due to its propensity to oligomerize. Crystal structures of DNMT3A in a complex of DNMT3L have been solved (34–36), where DNMT3L serves as an oligomer-disrupting co-factor. The FF interface is conserved between DNMT3A and DNMT3L, whereas the RD interface is not. DNMT3L can therefore bind to DNMT3A at the FF interface only. This forces the formation of a DNMT3L–DNMT3A–DNMT3A–DNMT3L tetramer (Fig. 1, B and C). DNMT3L has been shown to activate DNMT3A methyltransferase activity in both biochemical and cellular assays, although the mechanism is unclear (8–10, 32, 37, 38). In addition to the catalytic domain, DNMT3A contains an N-terminal unstructured region and two histone recognition domains, the PWWP and ADD domains (Fig. 1A). The PWWP domain specifically recognizes trimethylated histone H3K36 and is important for recruiting DNMT3A to heterochromatin (39, 40). The ADD domain binds to unmodified histone H3K4, which regulates DNA binding and the methyltransferase activity of DNMT3A (35, 41).
In this work, we analyzed the solution behavior and biochemical properties of the full-length DNMT3A WT and mutant proteins. We find that the R882H hot spot mutation stabilizes the formation of large oligomeric DNMT3A species, which are intrinsically less active than smaller tetramers. When mixed with WT DNMT3A protein, the mutant protein shifts the equilibrium of WT–R882H complexes to larger and less active oligomers, contributing to the dominant-negative behavior of the mutant. DNMT3L disrupts the formation of large oligomers to activate DNMT3A and partially rescues the activity of the R882H mutant. In addition to influencing the multimeric state of DNMT3A, R882H also causes a defect in DNA binding, which is not rescued in the presence of DNMT3L. Together, these observations provide a mechanistic explanation for the observed dominant-negative loss of DNMT3A function in leukemia patients carrying the R882H mutation.
Results
The goal of our work was to interrogate the mechanism by which the R882H mutation impairs DNMT3A function. Prior to working with the mutant protein, we investigated whether the N-terminally truncated WT catalytic domain would be a suitable replacement for the WT full-length protein, which has lower expression yields in Escherichia coli. Previous studies have demonstrated that DNMT3A can form very large oligomeric species, as well as smaller complexes around the size of a tetramer (18, 29, 30). We confirmed this observation by size-exclusion chromatography with purified full-length and catalytic domain DNMT3A (Fig. S1). For both proteins, a peak is observed near the void volume consistent with some fraction of DNMT3A existing as a very large oligomer, whereas a second peak elutes around the expected elution volume of a tetramer (Fig. 2A). It has been proposed that the formation of very large oligomers enhances the DNA methyltransferase activity of the isolated catalytic domain (42). To verify that full-length and catalytic domains behave similarly in this regard, we measured the DNA methyltransferase activity of the full-length and catalytic domain proteins at varying concentrations in a radiometric assay, measuring the transfer of a tritium-labeled methyl group from S-adenosylmethionine (SAM) to a DNA substrate. At low concentrations, the oligomer equilibrium would be expected to shift to smaller species, whereas at higher protein concentrations, the equilibrium should favor larger multimers. Here, we find that the full-length protein and catalytic domain behave very differently. Although the catalytic domain demonstrates a linear relationship between enzymatic activity and protein concentration, the full-length protein does not (Fig. 2B). Instead, the specific activity of the full-length protein decreases at increasing concentrations (Fig. 2C). This demonstrates that although the activity of the catalytic domain is unaffected by the oligomeric state in this experiment, the enzymatic activity of the full-length protein is very sensitive, with larger oligomers being less active than smaller species. Given that the DNMT3A catalytic domain does not behave the same as the full-length protein in our biochemical assays, we decided to use only the biologically-relevant full-length protein for the remainder of our work.
Figure 2.
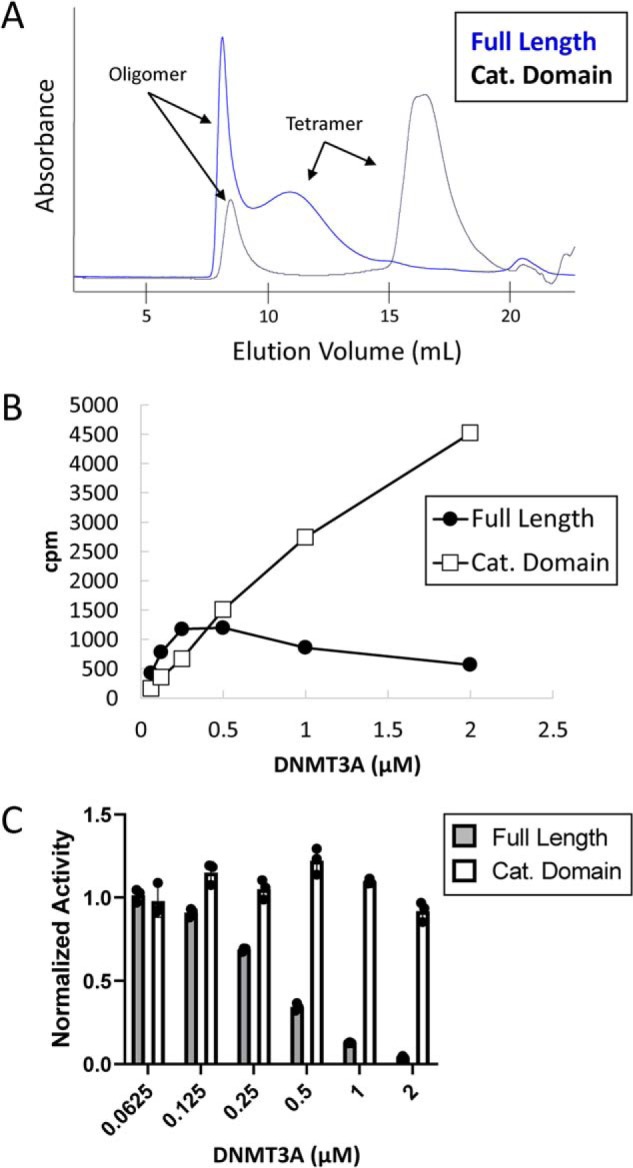
Comparison of full-length and catalytic domain oligomeric state and activity. A, chromatogram of purified full-length and catalytic domain DNMT3A injected onto a Superose 6 size-exclusion column. 500 μg of each protein was injected. B, total activity of DNMT3A full-length and catalytic domain proteins at the indicated concentrations measured using a radioactive end-point assay. C, specific activity of full-length and catalytic domain proteins at the indicated concentrations. Specific activity at the lowest DNMT3A concentration was normalized to one for full-length and catalytic domains, respectively. Error bars represent S.D., n = 3.
We next turned our attention to the mutant protein. Full-length WT and R882H DNMT3A were purified from E. coli (Fig. S1) and then injected onto a Superose 6 size-exclusion chromatography column. Although WT DNMT3A eluted as two major peaks as described above, the mutant protein eluted predominantly near the void volume (Fig. 3A). This could be explained by two possible mechanisms, either the mutant protein preparation was entirely aggregated or R882H shifts the equilibrium of ordered DNMT3A oligomers to higher-order multimers. To distinguish between these two possibilities, we added a stoichiometric amount of purified DNMT3L (Fig. S1) to the R882H protein in attempt to disrupt any higher-order mutant oligomer, and we ran the DNMT3A–DNMT3L complex over a size-exclusion column. The R882H–DNMT3L complex mostly runs as a lower molecular weight species, with the major peak eluting at the expected volume of a tetramer, whereas DNMT3L injected alone eluted as a monomer as expected (Fig. 3A). This demonstrates that the observed high-molecular-weight species of the mutant is not mis-folded/aggregated protein, but rather is a very large, ordered oligomer that can be broken down into smaller complexes in the presence of DNMT3L. To verify this was not a biochemical artifact, we expressed Myc-tagged WT DNMT3A and FLAG-tagged R882H DNMT3A in HEK293 cells, lysed the cells by mechanical disruption, and ran the lysate over a Superose 6 size-exclusion column. The amount of WT or mutant DNMT3A in each fraction was determined by Western blotting. We found that Myc–WT DNMT3A elutes in five fractions, with the majority of protein eluting around the expected mass of a tetramer (Fig. S2, A and B). Consistent with the observed oligomeric state of the mutant recombinant protein, FLAG–R882H DNMT3A from mammalian cell lysate elutes in only four fractions, with most of the protein eluting near the void volume of the size-exclusion column (Fig. S2, A and B).
Figure 3.

Examining the oligomeric state of R882H DNMT3A. A, size-exclusion chromatograms of DNMT3A and DNMT3L proteins. Mixed complexes were pre-incubated for 2 h at room temperature prior to injection onto a Superose 6 column. Peak components were confirmed by SDS-PAGE analysis. 500 μg of each protein was injected. B, quantification of nuclear puncta in NIH-3T3 cells after transfection with YFP–DNMT3A with or without co-transfection of CFP–DNMT3L. Error bars represent S.D. F732A and R885A have a statistically significant lower number of nuclear puncta compared with WT, p < 0.05 (*). There is no difference in the number of mutant DNMT3A nuclear puncta compared with WT upon DNMT3L co-expression. C, representative images are from B. Scale bars, 5 μm.
To demonstrate that WT and R882H DNMT3A can form oligomers in live cells, we expressed full-length YFP-tagged DNMT3A in NIH-3T3 cells to look for any obvious changes in sub-nuclear localization. It has been shown that WT DNMT3A localizes to heterochromatin regions (visualized as a punctate pattern in the nuclei of NIH-3T3 cells), which is highly dependent on the ability of DNMT3A to form oligomers (30, 43–45). After transient transfection, we clearly see that both WT and R882H (as well as R882C) form a similar strong punctate pattern indicative of oligomer formation, which is reduced upon co-expression of DNMT3L (Fig. 3, B and C). As a control, we see that expression of known oligomer-disrupting mutants F732A and R885A alone has a reduced number of nuclear puncta relative to WT (Fig. 3, B and C), as reported previously (30). Together, these data demonstrate that R882H stabilizes full-length DNMT3A oligomer formation in a purified system using recombinant protein, in mammalian cell lysates, and in live cells expressing tagged DNMT3A protein.
Consistent with prior studies and the observed loss of DNMT3A function in leukemia patients carrying the R882H mutation (18, 31, 46), we find that R882H displays reduced enzymatic activity relative to WT, with a roughly 20-fold reduction in apparent kcat (Fig. S3). Previous reports have proposed that the R882H mutation has dominant-negative behavior in both mouse models and human cell lines overexpressing the mutant (18, 26, 27); however, this was recently challenged in another study using mostly the purified catalytic domain in a reconstituted biochemical assay (33). In our hands, the addition of varying amounts of full-length R882H to a fixed amount (100 nm) of full-length WT DNMT3A caused a marked and dose-dependent reduction in methyltransferase activity, consistent with dominant-negative behavior (Fig. 4A). In contrast, adding additional WT DNMT3A to 100 nm WT protein caused an increase in activity (Fig. 4A). Notably, the decrease in specific activity occurred more drastically with the addition of mutant than WT DNMT3A (Fig. 4B). Given that R882H causes a shift in the DNMT3A oligomer equilibrium toward larger complexes and we have shown that large full-length oligomers have lower enzymatic activity relative to smaller complexes, this raised the possibility that the R882H mutation exerts its dominant-negative function by shifting the distribution of R882H–WT DNMT3A complexes to higher-order oligomers. Indeed, injection of a mixture of R882H–WT recombinant DNMT3A protein at a 1:1 ratio on a size-exclusion column reveals the complex exists mostly as a large oligomer (Fig. 3A), similar to mutant alone. A comparable shift in the oligomeric distribution of DNMT3A was observed in cell lysates after co-expression of Myc–WT and FLAG–R882H DNMT3A in HEK293 cells (Fig. S2B), where the dominant-negative behavior of the mutant was also confirmed (Fig. S2C). The addition of oligomer-disrupting DNMT3L was able to increase the activity of the WT–R882H complex in a biochemical assay to levels greater than WT alone at a one–to–one 3A–to–3L ratio (Fig. 4C). Interestingly, although the addition of DNMT3L to WT DNMT3A alone was capable of activating the WT protein 4.5-fold, DNMT3L activated R882H >10-fold under the same conditions (Fig. 4D). This would be expected if the primary mechanism of DNMT3L activation is through oligomer disruption to form a more active tetrameric species. Relative to WT, R882H DNMT3A has a larger proportion of protein existing as a large oligomer with low activity; therefore, it should be stimulated to a greater extent by oligomer disruption than WT.
Figure 4.

Dominant-negative behavior of R882H DNMT3A and rescue by DNMT3L. A, total DNA methyltransferase activity of 100 nm WT DNMT3A measured in the presence of additional amounts of R882H or WT DNMT3A. Mixtures of WT and mutant protein were pre-incubated at room temperature for 2 h. Error bars represent S.D., n = 4. B, DNA methyltransferase activity from A normalized to total DNMT3A present. Error bars represent S.D., n = 4. C, 100 nm WT DNMT3A activity in the presence of an equal concentration of R882H after a 2-h pre-incubation. Activity of the WT (100 nm), R882H (100 nm), and DNMT3L (200 nm) complex (a one–to–one DNMT3A–to–DNMT3L) ratio after a 2-h pre-incubation is shown. Error bars represent S.D., n = 4. D, activity of 100 nm WT DNMT3A alone and in the presence of 100 nm DNMT3L. Activity of 100 nm R882H DNMT3A alone and the presence of 100 nm DNMT3L is shown. WT and R882H DNMT3A activity alone is normalized to 1. Error bars represent S.D., n = 3.
Next, we determined whether DNMT3A could be activated through oligomer disruption in the absence of DNMT3L by generating point mutations along the oligomer-forming RD interface of the catalytic domain. Arg-885 and Asp-876 form electrostatic interactions with the opposite monomer to stabilize the RD interface. Mutation of these amino acids to alanine has been shown to prevent oligomer formation in NIH-3T3 cells as demonstrated and discussed above. Full-length R885A and D876A DNMT3A were purified from E. coli (Fig. S1) and injected onto a size-exclusion column to examine their oligomeric state. In contrast to WT and R882H, both interface-disrupting mutants demonstrated a significant shift from large oligomers to a smaller species, with the predominant peaks eluting around the expected volume of dimers and monomers (Fig. 5A). This shift in equilibrium was not complete, as some fraction of R885A and D876A DNMT3A still eluted as a high-molecular-weight species. Both R885A and D876A had reduced enzymatic activity relative to WT, displaying comparable activity to R882H (Fig. 5B), confirming that DNMT3A dimers and monomers have low enzymatic activity (29, 31). Given that these interface mutants shift the equilibrium of oligomer to smaller species, we would expect that DNMT3L would have reduced ability to activate these proteins relative to WT or R882H DNMT3A. Indeed, we find that DNMT3L is incapable of activating R885A and is only able to activate D876A a modest 2-fold, compared with DNMT3L's ability to activate WT 4.5-fold and R882H >10-fold (Fig. S4).
Figure 5.

Characterization of R885A and D876A oligomer-disrupting mutants and their effect on WT and R882H DNMT3A activity. A, chromatograms of R885A and D876A injected onto a Superose 6 size-exclusion column. B, total activity of 100 nm WT, R882H, R885A, and D876A alone compared with the activity of 100 nm WT protein mixed with an equal amount of the indicated DNMT3A protein after a 2-h pre-incubation. Error bars represent S.D., n = 6. C, activity of 100 nm WT DNMT3A titrated with R885A or D876A after a 2-h pre-incubation. Activity of R885A and D876A alone at the indicated concentrations is shown for comparison. The reaction was stopped after 90 min. Error bars represent S.D., n = 2. D, activity of 100 nm WT DNMT3A in the presence of varying concentrations of DNMT3L after a 2-h pre-incubation. Error bars represent S.D., n = 3. E, activity of 200 nm R882H DNMT3A in the presence of an equal amount of R885A or D876A after a 2-h pre-incubation. The reaction was stopped after 4 h. Error bars represent S.D., n = 3.
We would expect that after mixing these interface disrupting mutants with WT or R882H DNMT3A, the resulting complex would have higher activity than WT, R882H, or interface mutant DNMT3A alone. This is because the oligomer-disrupting mutations would break down higher-order WT or R882H oligomers into smaller, more active complexes. Upon mixing RD interface mutations R885A or D876A with WT protein at a one–to–one ratio, we see that the total activity of the resulting complex is greater than the sum of WT plus R885A or D876A alone (Fig. 5B). Upon titrating R885A or D876A into WT DNMT3A, we find that ratios of mutant to WT DNMT3A greater than one–to–one continue to activate the WT protein (Fig. 5C). This is in contrast to DNMT3L, which achieves maximal activation at a one–to–one DNMT3A–to–DNMT3L ratio (Fig. 5D), indicative of a very-tight binding interaction between DNMT3A and DNMT3L (8). As expected, R885A and D876A also activate R882H in a synergistic manner (Fig. 5E). The weaker total activation of WT and R882H by the interface-disrupting mutations compared with DNMT3L can be explained by two observations. One, the interface-disrupting mutations do not fully break down DNMT3A oligomers into smaller species, as some fractions of these two RD interface mutants run as large multimers by size-exclusion chromatography (Fig. 5A). Two, the DNMT3A–DNMT3L interaction is of higher affinity than the DNMT3A–DNMT3A interaction. Therefore, DNMT3L is fully capable of breaking up DNMT3A oligomers at stoichiometric concentrations, whereas the interface mutations are not.
It is important to note that relative to WT, full rescue of R882H DNMT3A activity with DNMT3L could not be achieved (Fig. 6A). Although the addition of DNMT3L could activate R882H >10-fold, the absolute activity of the R882H DNMT3A–DNMT3L complex remained suppressed relative to the WT DNMT3A–DNMT3L complex, remaining about 5-fold lower (Fig. 6A). This observation suggests that the R882H point mutation is interfering with DNMT3A methyltransferase activity by some mechanism in addition to shifting the equilibrium of DNMT3A oligomers to higher order, less active multimers. Given that the mutation is close to the DNA-binding surface of DNMT3A (Fig. 6B), we next investigated whether Arg-882 is involved in DNA binding. In an equilibrium DNA-binding assay, measuring the change in fluorescence polarization of a 30-bp FAM-labeled dsDNA oligonucleotide upon DNMT3A binding, we find that R882H has a roughly 2-fold elevated Kd value relative to WT (Fig. 6C). The off-rate of R882H for DNA is also apparently faster than WT DNMT3A. WT or R882H was pre-incubated with FAM-labeled DNA, and equilibrium was achieved prior to the addition of a 1000-fold excess of unlabeled DNA. Although the off-rate of both proteins was too fast to accurately measure with available laboratory instrumentation, the off-rate of WT appears to be significantly slower than R882H (Fig. 6D). Although all R882H DNMT3A had dissociated from labeled DNA by the time measurements could be made, at least 40% of WT protein remained bound at this early time point prior to fully dissociating within 5 min (Fig. 6D). In these same experiments, DNMT3L has no effect on equilibrium binding or apparent residence time for both WT and R882H (Fig. 6, C and D). This is consistent with an earlier study, showing no difference in DNMT3A DNA-binding affinity in the presence and absence of DNMT3L (38). During this work, a WT DNMT3A crystal structure was published with DNA bound (36). We and others note that Arg-882 appears to be making a contact with the phosphate backbone of the bound DNA (Fig. 6B). In the same structure, DNMT3L does not make any contacts with DNA. This provides the explanation for the observed DNA-binding defect of the R882H mutation.
Figure 6.

Arg-882 involvement in DNA binding. A, methyltransferase activity of WT and R882H DNMT3A at saturating concentrations of DNA and SAM substrates after a 2-h pre-incubation with an equal amount of DNMT3L. Error bars represent S.D., n = 3. B, crystal structure of WT DNMT3A bound to DNA (PDB code 5YX2), where Arg-882 appears to be forming an interaction with the DNA phosphate backbone. C, binding affinity of WT and R882H DNMT3A for FAM-labeled dsDNA in the absence and presence of DNMT3L measured by fluorescence polarization. D, relative residence time of WT compared with R882H DNMT3A for DNA in the absence and presence of DNMT3L. 100 nm DNMT3A pre-bound to 10 nm FAM-DNA was chased with 10 μm unlabeled DNA and fluorescence polarization measured over time.
Discussion
In this study, we establish the mechanism by which the R882H DNMT3A hot spot mutation exerts its dominant-negative behavior, dramatically reducing the DNA methyltransferase activity of both mutant and WT DNMT3A. The R882H mutation impairs DNMT3A function by two mechanisms. One, R882H stabilizes the formation of large DNMT3A oligomers, which are intrinsically less active than smaller species. Two, the R882H mutations remove a key binding interaction with substrate DNA. We observe that the R882H mutation shifts the equilibrium of both mutant alone and a one–to–one mixture of R882H–WT DNMT3A oligomer from smaller tetramers to very large multimers by size-exclusion chromatography. These large multimers of the full-length DNMT3A protein are less active than smaller complexes, as determined by simply measuring the total DNA methyltransferase activity at varying concentrations of enzyme. Upon the addition of DNMT3L, the large DNMT3A oligomers are broken down into tetramers, which is accompanied by an increase in methyltransferase activity (Fig. 7). This new model of DNMT3A regulation by oligomer formation and R882H dysfunction is supported by the use of point mutations along the oligomerization interfaces of the catalytic domain. We show that the RD mutants, R885A and D876A, both form mostly dimers and monomers with very low catalytic activity in solution. When forming a complex with WT or R882H DNMT3A, these mutations synergistically enhance the DNA methyltransferase activity of the resulting complex. This reveals that even in the absence of DNMT3L, oligomer disruption alone enhances the activity of DNMT3A. Although DNMT3L clearly prevents oligomer formation to stimulate DNMT3A activity, it is possible that DNMT3L also stimulates DNMT3A activity through additional mechanisms. In prior studies, there has been some speculation that DNMT3L itself binds DNA and that this may enhance DNMT3A activity; however, it is now clear from the DNMT3A–DNMT3L–DNA crystal structure that DNMT3L does not make any interactions with substrate DNA during catalysis (36). Our data are in agreement with this observation. In our hands, DNMT3L does not significantly influence full-length WT or R882H DNMT3A DNA binding.
Figure 7.
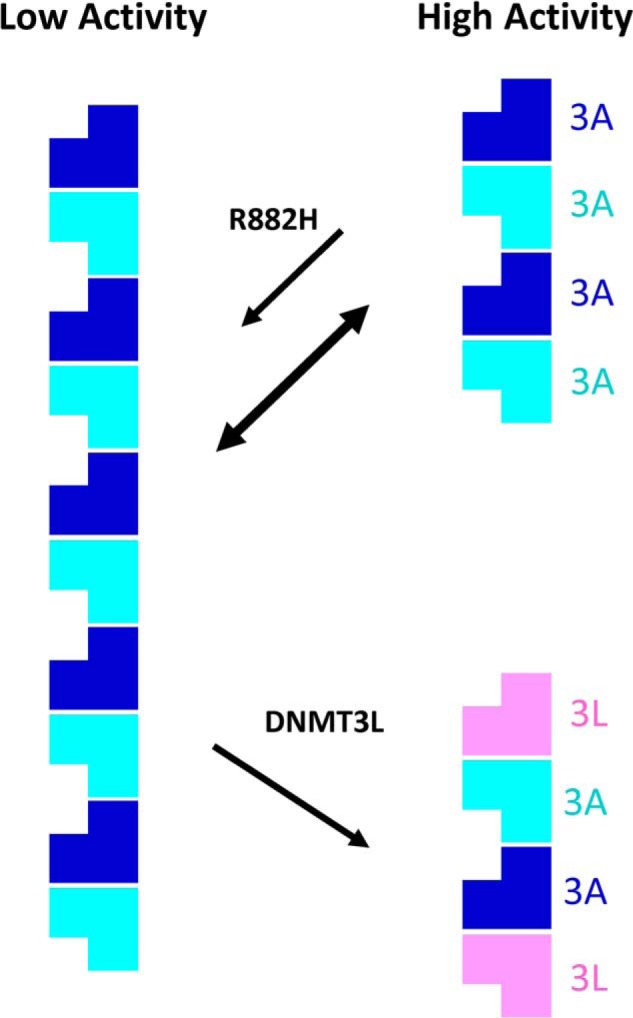
Model of DNMT3A regulation by oligomer formation and the influence of R882H and DNMT3L. DNMT3A exists in an equilibrium between very large oligomeric multimers and smaller complexes about the size of a tetramer. Here, large oligomeric DNMT3A species are significantly less active than smaller complexes. The R882H hot spot mutation found in leukemia exerts its dominant-negative behavior in part by binding WT DNMT3A and shifting the distribution of DNMT3A complexes from smaller, more active tetramers to larger oligomers with low activity. DNMT3L activates DNMT3A by breaking up large oligomers into smaller complexes.
It is clear from the recent WT DNMT3A–DNA co-crystal structure that DNMT3A makes many contacts with DNA (36). There are multiple key interactions between DNMT3A and the DNA phosphate backbone, major and minor grooves and with the flipped base in the enzyme-active site. Mutation of many of these amino acids causes a sharp reduction in enzymatic activity, with similar total activity to the R882H hot spot mutation (36). Mutation of several of these amino acids even leads to complete ablation of enzymatic activity (36). Yet, only Arg-882 mutations are selected for at a high frequency in hematopoietic cancers. This suggests that disruption of DNMT3A DNA binding alone is insufficient for driving leukemogenesis. It is therefore tempting to speculate that the R882H mutation provides an advantage to cancer cells primarily through higher-order oligomer stabilization rather than disruption of DNA binding.
Previous studies of the R882H mutation in the context of the isolated catalytic domain suggested that this mutation disrupts the RD interface, preventing the formation of active DNMT3A tetramers (29, 31). We note that in the DNMT3A crystal structures there are many amino acids along the RD interface that are clearly stabilizing DNMT3A–DNMT3A binding (34–36). Mutation of a number of these amino acids has been shown to disrupt this binding interface, both in this study and in others (29–31). In cancer, mutations of these RD interface-stabilizing amino acids are observed occasionally, but are not selected for anywhere near the same frequency as Arg-882 mutations. This is likely because heterozygous mutation of these amino acids would be activating to the WT allele rather than inhibitory, as we show here for the first time. Although the R885A and D876A mutations at the RD interface have very low enzymatic activity on their own, we clearly show that upon mixing with WT or R882H, the total DNA methyltransferase activity of the resulting DNMT3A complex is synergistically increased. The previously proposed model of R882H-mediated disruption of DNMT3A complexes is therefore not compatible with the observed loss of function in cancer patients or the numerous cellular and in vivo models developed to study this mutation.
Prior biochemical studies investigating DNMT3A regulation by oligomer formation and the biochemical dysfunctional mechanism of the R882H mutation have primarily utilized the N-terminally truncated catalytic domain (29–33). Here, we clearly demonstrate that the full-length protein behaves very differently than the isolated catalytic domain. Although the DNA methyltransferase activity of the catalytic domain appears to be unaffected by protein concentration and oligomer distribution, the full-length protein exhibits substantially-reduced enzymatic activity as the oligomer equilibrium shifts from smaller to larger multimers. Given that the R882H mutation influences oligomer distribution, previous studies utilizing only the isolated catalytic domain may have missed key aspects of the mutant behavior. This may be why the dominant-negative function the R882H mutation observed in vivo and confirmed here in reconstituted biochemical assays for the full-length DNMT3A protein was recently challenged (33). Clearly, the DNMT3A N-terminal unstructured regions, PWWP and/or ADD domains, are playing an important role here. The mechanism by which this occurs is unclear and will require further investigation. Structural and functional studies have clearly demonstrated that the ADD domain plays a key role in regulating DNMT3A methyltransferase activity and substrate binding (35, 41). The PWWP domain binds to DNA, causing substrate inhibition of DNMT3A enzymatic activity under some conditions (47). Both the PWWP and ADD domains bind histone ligands, which has been shown to be important for both DNMT3A recruitment to heterochromatin regions and activation of enzymatic activity (35, 39, 40). It is also possible that histone tail binding to the ADD or PWWP domains influences the oligomeric state and activity of DNMT3A. The N-terminal unstructured region has unknown function, but it likely plays an important regulatory role given that the DNMT3A2 isoform missing this region is selectively expressed during embryonic development but not in differentiated cell types (45).
In summary, our work provides insight into the molecular mechanisms by which the R882H hot spot mutation suppresses the methyltransferase activity of the WT–R882H DNMT3A complex. This dominant-negative behavior of the R882H mutant clearly depends on the ability of the mutant protein to form multimeric complexes with the WT allele. This raises the possibility that small molecules targeting the WT–R882H-binding interface could disrupt this interaction and break down large oligomers into smaller complexes to restore the DNA methyltransferase activity of DNMT3A. This may have a therapeutic benefit for a large population of leukemia patients harboring the R882H hot spot mutation, who currently have few options for treatment.
Experimental procedures
Protein purification
Both full-length and catalytic domain(612–912) human DNMT3As were cloned into the pET28b vector and expressed in E. coli with an N-terminal His tag. E. coli cultures were grown in LB medium to an absorbance of 0.8 at 37 °C prior to cooling the temperature to 16 °C for overnight induction with 1 mm IPTG. Cells were pelleted by centrifugation and resuspended in lysis buffer (50 mm Tris, pH 8.0, 300 mm NaCl, 1 mm TCEP, 0.1% Triton X-100, and 10 mm imidazole) prior to being lysed on ice by sonication (60% amplitude, three times 30 s). Cell debris was cleared by centrifugation at 30,000 rpm for 30 min. The supernatant from 2 liters of culture was then diluted to a final volume of 350 ml in lysis buffer prior to purification on an AKTA FPLC with a 5-ml His trap. Dilution of the lysate was found to be important for recovering DNMT3A protein from the lysate. When concentrated, the DNMT3A protein in the lysate does not readily bind the Ni column, likely due to the formation of large DNMT3A oligomers. This is especially true for the full-length R882H protein. The His trap column was washed, and DNMT3A protein was eluted in a step gradient of imidazole (50, 100, 200, and 500 mm) in column buffer (50 mm Tris, pH 8.0, 300 mm NaCl, and 1 mm TCEP). Pure elution fractions were combined and dialyzed overnight at 4 °C in dialysis buffer (50 mm Tris, pH 8.0, 300 mm NaCl, and 1 mm TCEP). After dialysis, DNMT3A protein was concentrated to between 0.5 and 1 mg/ml using a 100 kDa MWCO centrifugation filter, then frozen and stored at −80 °C. DNMT3A and DNMT3L proteins were subject to size-exclusion chromatography analysis on a Superose 6 10/300 SEC column. The column was pre-equilibrated with column buffer (50 mm Tris, pH 8.0, 150 mm NaCl, and 1 mm TCEP). 500 μg of protein was injected in column buffer with a flow rate of 0.5 ml/min. Components of each elution peak were confirmed by SDS-PAGE and Coomassie staining.
Full-length human DNMT3L was expressed in E. coli in the pET28b vector. E. coli cultures were grown in LB medium to an absorbance of 0.8 at 37 °C prior to cooling the temperature to 16 °C for overnight induction with 1 mm IPTG. Cells from 12 liters of E. coli culture were pelleted by centrifugation and resuspended in 100 ml of lysis buffer (50 mm Tris, pH 8.0, 300 mm NaCl, 1 mm TCEP, 0.1% Triton X-100, and 10 mm imidazole) prior to being lysed on ice by sonication (60% amplitude, three times 30 s). Cell debris was cleared by centrifugation at 30,000 rpm for 30 min. The supernatant was loaded directly onto a His trap column using an AKTA FPLC. The column was washed, and DNMT3L protein was eluted in a step gradient of imidazole (50, 100, and 250 mm) in column buffer (50 mm Tris, pH 8.0, 300 mm NaCl, and 1 mm TCEP). Pure fractions were dialyzed overnight at 4 °C in 50 mm Tris, pH 8.0, 300 mm NaCl, and 1 mm TCEP, then concentrated to 3 mg/ml, and stored at −80 °C.
Methyltransferase assay
DNMT3A protein was diluted into assay buffer (50 mm Tris, pH 8.0, 20 mm NaCl, and 1 mm TCEP) at varying concentrations prior to the addition of DNA and [3H]SAM (specific activity 18 Ci/mmol, PerkinElmer Life Sciences) to initiate the reaction. For experiments with mixed WT and mutant DNMT3A or DNMT3L, the protein was preincubated in assay buffer for 2 h (unless otherwise indicated) prior to initiating the reaction. The final reaction volume was 50 μl. Product was produced linearly with time up to 5 h. Reaction times for each experiment were 2 h unless otherwise indicated. The reactions were stopped with 1 mm cold SAM. The 50-μl reaction was then diluted into 300 μl of assay buffer containing 40 μl of DEAE resin. The solution was mixed by rotation for 30 min in a spin cup filter (PierceTM Spin Cups, Paper Filter no. 69700, Life Technologies, Inc.) for the DNA to bind to the DEAE resin. The resin was collected and washed in wash buffer (50 mm Tris, pH 8.0, 20 mm NaCl, and 1 mm TCEP) three times by centrifugation in the spin cup. DEAE resin was then resuspended in 200 μl of water, transferred to 4 ml of scintillation fluid, and counted on a Micro Beta 2 scintillation counter.
Mammalian cell expression and cell lysate analysis
N-terminally Myc-tagged full-length WT DNMT3A and N-terminally FLAG-tagged full-length R882H DNMT3A were subcloned into the pcDNA3.1 vector and transiently expressed in adherent HEK293 cells. Cells were plated in a 10-cm dish at 50% confluency 1 day prior to transfection. Cells were transfected with FuGENE transfection reagent (Promega), then scraped, washed in PBS, and pelleted 24 h later. Cell pellets were resuspended in 800 μl of lysis buffer (25 mm Tris, pH 7.8, 150 mm NaCl, and 1 mm TCEP) prior to lysis by Dounce homogenization on ice. The lysate was centrifuged for 10 min at 14,000 rpm. 400 μl of the soluble fraction of the cell lysate was then run on a Superose 6 size-exclusion column in 25 mm Tris, pH 7.8, 150 mm NaCl, and 1 mm TCEP. Two-ml fractions were collected, and DNMT3A protein was detected by Western blotting (LICOR) using anti-Myc (9E10) or anti-FLAG (M2) antibodies. GAPDH was detected using an anti-GAPDH antibody (Cell Signaling, catalog no. 5174S).
DNMT3A DNA methyltransferase activity in each fraction was determined using a slightly modified protocol described above. 25 μl of each fraction containing DNMT3A protein was mixed with [3H]SAM (3 μm) and DNA (dIdC, 10 μm bp) substrates. The reaction was allowed to proceed for 4 h at room temperature prior to stopping the reaction with 1 mm unlabeled SAM. The reaction mixture was then incubated with DEAE resin, and the amount of radiolabeled DNA was measured as described above.
DNA-binding assays
In an equilibrium binding experiment, FAM-labeled 30-bp dsDNA (5′-FAM-TCGCTGTCGTACGTCGCGTCTGTGAGTCGA-3′) was diluted in assay buffer (50 mm Tris, pH 8.0, 20 mm NaCl, and 1 mm TCEP) to a final concentration of 10 nm prior to the addition of varying concentrations of DNMT3A or DNMT3A–DNMT3L complex. The mixture was allowed to reach equilibrium over 30 min at room temperature. Fluorescence polarization was measured on an M1000 Tecan plate reader in a 384-well plate (Corning catalog no. 3820) in a final volume of 15 μl. To measure the relative residence time of WT and R882H DNMT3A for DNA, 10 nm FAM-labeled 30-bp dsDNA was incubated with 100 nm DNMT3A for 30 min at room temperature to reach equilibrium. 10 μm unlabeled dsDNA was then added, and dissociation of the DNMT3A–FAM–DNA complex was measured continuously on an M1000 Tecan plate reader as described above.
Subnuclear localization
NIH-3T3 cells were plated at a density of 2E4 cells/well in a 96-well plate in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. The following day, cells were washed with PBS and then transfected with 200 ng of plasmid YFP–DNMT3A with or without 200 ng of CFP–DNMT3L using Lipofectamine 3000. 24 h after transfection, cells were washed and fixed with paraformaldehyde prior to being permeabilized with 0.1% Triton X-100 in PBS and 4′,6-diamidino-2-phenylindole staining. Cells were imaged using GE IN Cell 2000 high-content screening system with a ×40 objective lens. The number of nuclear puncta in YFP- or YFP/CFP-positive cells were counted and normalized to the nuclear area.
Author contributions
T.-V. N., S. Y., Y. W., R. D., J. K., and D. M. B. data curation; T.-V. N., S. Y., Y. W., A. R., A. S., R. D., J. K., M. W., P. G. S., N. A. L., P. Z., P. F., F. H. V., and D. M. B. formal analysis; T.-V. N., A. R., A. S., R. D., M. W., P. G. S., N. A. L., P. Z., P. F., F. H. V., and D. M. B. investigation; T.-V. N., N. A. L., F. H. V., and D. M. B. writing-original draft; T.-V. N., N. A. L., F. H. V., and D. M. B. writing-review and editing; A. R., M. W., P. G. S., P. Z., P. F., F. H. V., and D. M. B. project administration; M. W., P. G. S., L. Y., P. Z., P. F., F. H. V., and D. M. B. resources; M. W., P. G. S., N. A. L., L. Y., P. Z., P. F., F. H. V., and D. M. B. supervision; M. W., P. G. S., L. Y., P. F., and D. M. B. funding acquisition; P. F., F. H. V., and D. M. B. conceptualization; F. H. V. and D. M. B. methodology; D. M. B. validation; D. M. B. visualization.
Supplementary Material
The authors have declared a conflict of interest. Competing financial interests are as follows: T.-V. N., S. Y., A. R., A. S., R. D., M. W., P. G. S., N. L., L. Y., P. Z., P. F., F. H. V., and D. M. B. are all current or former employees of H3 Biomedicine, Inc. Y. W. declares no competing financial interests.
This article contains Figs. S1–S4.
- DNMT
- DNA methyltransferase
- FAM
- 6-carboxyfluorescein
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- AML
- acute myeloid leukemia
- DMR
- differentially-methylated region
- HSC
- hematopoietic stem cell
- SAM
- S-adenosylmethionine
- TCEP
- tris(2-carboxyethyl)phosphine
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- CFP
- cyan fluorescent protein.
References
- 1. Lyko F. (2018) The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92 10.1038/nrg.2017.80 [DOI] [PubMed] [Google Scholar]
- 2. Okano M., Bell D. W., Haber D. A., and Li E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- 3. Chen T., Ueda Y., Dodge J. E., Wang Z., and Li E. (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605 10.1128/MCB.23.16.5594-5605.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Challen G. A., Sun D., Mayle A., Jeong M., Luo M., Rodriguez B., Mallaney C., Celik H., Yang L., Xia Z., Cullen S., Berg J., Zheng Y., Darlington G. J., Li W., and Goodell M. A. (2014) Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15, 350–364 10.1016/j.stem.2014.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aapola U., Lyle R., Krohn K., Antonarakis S. E., and Peterson P. (2001) Isolation and initial characterization of the mouse Dnmt3l gene. Cytogenet. Genome Res. 92, 122–126 10.1159/000056881 [DOI] [PubMed] [Google Scholar]
- 6. Hata K., Okano M., Lei H., and Li E. (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 7. Aapola U., Kawasaki K., Scott H. S., Ollila J., Vihinen M., Heino M., Shintani A., Kawasaki K., Minoshima S., Krohn K., Antonarakis S. E., Shimizu N., Kudoh J., and Peterson P. (2000) Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 65, 293–298 10.1006/geno.2000.6168 [DOI] [PubMed] [Google Scholar]
- 8. Chedin F., Lieber M. R., and Hsieh C.-L. (2002) The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. U.S.A. 99, 16916–16921 10.1073/pnas.262443999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z.-X., Mann J. R., Hsieh C.-L., Riggs A. D., and Chédin F. (2005) Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell. Biochem. 95, 902–917 10.1002/jcb.20447 [DOI] [PubMed] [Google Scholar]
- 10. Gowher H., Liebert K., Hermann A., Xu G., and Jeltsch A. (2005) Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 280, 13341–13348 10.1074/jbc.M413412200 [DOI] [PubMed] [Google Scholar]
- 11. Van Emburgh B. O., and Robertson K. D. (2011) Modulation of Dnmt3b function in vitro by interactions with Dnmt3L, Dnmt3a and Dnmt3b splice variants. Nucleic Acids Res. 39, 4984–5002 10.1093/nar/gkr116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suetake I., Shinozaki F., Miyagawa J., Takeshima H., and Tajima S. (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 279, 27816–27823 10.1074/jbc.M400181200 [DOI] [PubMed] [Google Scholar]
- 13. Bourc'his D., Xu G. L., Lin C. S., Bollman B., and Bestor T. H. (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539 10.1126/science.1065848 [DOI] [PubMed] [Google Scholar]
- 14. Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 10.1056/NEJMoa0903840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papaemmanuil E., Gerstung M., Bullinger L., Gaidzik V. I., Paschka P., Roberts N. D., Potter N. E., Heuser M., Thol F., Bolli N., Gundem G., Van Loo P., Martincorena I., Ganly P., Mudie L., et al. (2016) Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 10.1056/NEJMoa1516192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan X.-Q., Peng L., Zeng W.-J., Jiang B.-Y., Li G.-C., and Chen X.-P. (2016) DNMT3A Arg-882 mutations predict a poor prognosis in AML: a meta-analysis from 4474 patients. Medicine 95, e3519 10.1097/MD.0000000000003519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spencer D. H., Russler-Germain D. A., Ketkar S., Helton N. M., Lamprecht T. L., Fulton R. S., Fronick C. C., O'Laughlin M., Heath S. E., Shinawi M., Westervelt P., Payton J. E., Wartman L. D., Welch J. S., Wilson R. K., et al. (2017) CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 168, 801–816.e13 10.1016/j.cell.2017.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russler-Germain D. A., Spencer D. H., Young M. A., Lamprecht T. L., Miller C. A., Fulton R., Meyer M. R., Erdmann-Gilmore P., Townsend R. R., Wilson R. K., and Ley T. J. (2014) The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454 10.1016/j.ccr.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Challen G. A., Sun D., Jeong M., Luo M., Jelinek J., Berg J. S., Bock C., Vasanthakumar A., Gu H., Xi Y., Liang S., Lu Y., Darlington G. J., Meissner A., Issa J.-P. J., et al. (2011) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 44, 23–31 10.1038/ng.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tadokoro Y., Ema H., Okano M., Li E., and Nakauchi H. (2007) De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 204, 715–722 10.1084/jem.20060750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dai Y.-J., Wang Y.-Y., Huang J.-Y., Xia L., Shi X.-D., Xu J., Lu J., Su X.-B., Yang Y., Zhang W.-N., Wang P.-P., Wu S.-F., Huang T., Mi J.-Q., Han Z.-G., et al. (2017) Conditional knockin of Dnmt3a R878H initiates acute myeloid leukemia with mTOR pathway involvement. Proc. Natl. Acad. Sci. U.S.A. 114, 5237–5242 10.1073/pnas.1703476114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu R., Wang P., Parton T., Zhou Y., Chrysovergis K., Rockowitz S., Chen W.-Y., Abdel-Wahab O., Wade P. A., Zheng D., and Wang G. G. (2016) Epigenetic perturbations by Arg-882–mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 30, 92–107 10.1016/j.ccell.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cole C. B., Russler-Germain D. A., Ketkar S., Verdoni A. M., Smith A. M., Bangert C. V., Helton N. M., Guo M., Klco J. M., O'Laughlin S., Fronick C., Fulton R., Chang G. S., Petti A. A., Miller C. A., and Ley T. J. (2017) Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J. Clin. Invest. 127, 3657–3674 10.1172/JCI93041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scourzic L., Couronné L., Pedersen M. T., Della Valle V., Diop M., Mylonas E., Calvo J., Mouly E., Lopez C. K., Martin N., Fontenay M., Bender A., Guibert S., Dubreuil P., Dessen P., et al. (2016) DNMT3AR882H mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 30, 1388–1398 10.1038/leu.2016.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu J., Wang Y.-Y., Dai Y.-J., Zhang W., Zhang W.-N., Xiong S.-M., Gu Z.-H., Wang K.-K., Zeng R., Chen Z., and Chen S.-J. (2014) DNMT3A Arg-882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 111, 2620–2625 10.1073/pnas.1400150111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu R., Wang J., Ren Z., Yin J., Wang Y., Cai L., and Wang G. G. (2019) A model system for studying the DNMT3A hot spot mutation (DNMT3AR882) demonstrates a causal relationship between its dominant-negative effect and leukemogenesis. Cancer Res. 79, 3583–3594 10.1158/0008-5472.CAN-18-3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S. J., Zhao H., Hardikar S., Singh A. K., Goodell M. A., and Chen T. (2013) A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 122, 4086–4089 10.1182/blood-2013-02-483487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C., Payton J. E., Baty J., Welch J., Harris C. C., Lichti C. F., Townsend R. R., Fulton R. S., Dooling D. J., et al. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 10.1056/NEJMoa1005143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holz-Schietinger C., Matje D. M., Harrison M. F., and Reich N. O. (2011) Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J. Biol. Chem. 286, 41479–41488 10.1074/jbc.M111.284687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jurkowska R. Z., Rajavelu A., Anspach N., Urbanke C., Jankevicius G., Ragozin S., Nellen W., and Jeltsch A. (2011) Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J. Biol. Chem. 286, 24200–24207 10.1074/jbc.M111.254987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holz-Schietinger C., Matje D. M., and Reich N. O. (2012) Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 287, 30941–30951 10.1074/jbc.M112.366625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holz-Schietinger C., and Reich N. O. (2010) The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J. Biol. Chem. 285, 29091–29100 10.1074/jbc.M110.142513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emperle M., Dukatz M., Kunert S., Holzer K., Rajavelu A., Jurkowska R. Z., and Jeltsch A. (2018) The DNMT3A R882H mutation does not cause dominant negative effects in purified mixed DNMT3A/R882H complexes. Sci. Rep. 8, 13242 10.1038/s41598-018-31635-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jia D., Jurkowska R. Z., Zhang X., Jeltsch A., and Cheng X. (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251 10.1038/nature06146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo X., Wang L., Li J., Ding Z., Xiao J., Yin X., He S., Shi P., Dong L., Li G., Tian C., Wang J., Cong Y., and Xu Y. (2015) Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644 10.1038/nature13899 [DOI] [PubMed] [Google Scholar]
- 36. Zhang Z.-M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S. B., Wang Y., Wang G. G., and Song J. (2018) Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391 10.1038/nature25477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veland N., Lu Y., Hardikar S., Gaddis S., Zeng Y., Liu B., Estecio M. R., Takata Y., Lin K., Tomida M. W., Shen J., Saha D., Gowher H., Zhao H., and Chen T. (2019) DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 47, 152–167 10.1093/nar/gky947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kareta M. S., Botello Z. M., Ennis J. J., Chou C., and Chédin F. (2006) Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 281, 25893–25902 10.1074/jbc.M603140200 [DOI] [PubMed] [Google Scholar]
- 39. Chen T., Tsujimoto N., and Li E. (2004) The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 24, 9048–9058 10.1128/MCB.24.20.9048-9058.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R. Z., Ragozin S., and Jeltsch A. (2010) The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285, 26114–26120 10.1074/jbc.M109.089433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li B.-Z., Huang Z., Cui Q.-Y., Song X.-H., Du L., Jeltsch A., Chen P., Li G., Li E., and Xu G.-L. (2011) Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 21, 1172–1181 10.1038/cr.2011.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emperle M., Rajavelu A., Reinhardt R., Jurkowska R. Z., and Jeltsch A. (2014) Cooperative DNA binding and protein/DNA fiber formation increases the activity of the Dnmt3a DNA methyltransferase. J. Biol. Chem. 289, 29602–29613 10.1074/jbc.M114.572032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bachman K. E., Rountree M. R., and Baylin S. B. (2001) Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 276, 32282–32287 10.1074/jbc.M104661200 [DOI] [PubMed] [Google Scholar]
- 44. Ge Y.-Z., Pu M.-T., Gowher H., Wu H.-P., Ding J.-P., Jeltsch A., and Xu G.-L. (2004) Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Biol. Chem. 279, 25447–25454 10.1074/jbc.M312296200 [DOI] [PubMed] [Google Scholar]
- 45. Chen T., Ueda Y., Xie S., and Li E. (2002) A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 277, 38746–38754 10.1074/jbc.M205312200 [DOI] [PubMed] [Google Scholar]
- 46. Yamashita Y., Yuan J., Suetake I., Suzuki H., Ishikawa Y., Choi Y. L., Ueno T., Soda M., Hamada T., Haruta H., Takada S., Miyazaki Y., Kiyoi H., Ito E., Naoe T., et al. (2010) Array-based genomic resequencing of human leukemia. Oncogene 29, 3723–3731 10.1038/onc.2010.117 [DOI] [PubMed] [Google Scholar]
- 47. Purdy M. M., Holz-Schietinger C., and Reich N. O. (2010) Identification of a second DNA-binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch. Biochem. Biophys. 498, 13–22 10.1016/j.abb.2010.03.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.