

Abstract
Aims
To evaluate a novel tetrahydroisoquinoline derivative YR4‐42 as a selective peroxisome proliferator‐activated receptor γ (PPARγ) modulator (SPPARM) and explore its anti‐diabetic effects in vitro and in vivo.
Materials and methods
Using two standard full PPARγ agonists rosiglitazone and pioglitazone as controls, the PPARγ binding affinity and transactivation action of YR4‐42 were evaluated using biochemical and cell‐based reporter gene assays. The capacity of YR4‐42 to recruit coactivators of PPARγ was also assessed. The effects of YR4‐42 on adipogenesis and glucose consumption and PPARγ Ser273 phosphorylation were investigated in 3T3‐L1 adipocytes. The effects of YR4‐42 and pioglitazone, serving as positive control, on glucose and lipids metabolism were investigated in high‐fat diet‐induced obese (DIO) C57BL/6J mice. The expression of PPARγ target genes involved in glucose and lipid metabolism was also assessed in vitro and in vivo.
Results
In vitro biochemical and cell‐based functional assays showed that YR4‐42 has much weaker binding affinity, transactivation, and recruitment to PPARγ of the coactivators thyroid hormone receptor‐associated protein complex 220 kDa component (TRAP220) and PPARγ coactivator 1‐α (PGC1α) compared to full agonists. In 3 T3‐L1 adipocytes, YR4‐42 significantly improved glucose consumption without a lipogenesis effect, while blocking tumour necrosis factor α‐mediated phosphorylation of PPARγ at Ser273, thereby upregulating the expression of the PPARγ Ser273 phosphorylation‐dependent genes. Furthermore, in DIO mice, oral administration of YR4‐42 ameliorated the hyperglycaemia, with a similar insulin sensitization effect to that of pioglitazone. Importantly, YR4‐42 also improved hyperlipidaemia‐associated hepatic steatosis without weight gain, which avoids a major side effect of pioglitazone. Thus, YR4‐42 appeared to selectively modulate PPARγ responses. This finding was supported by the gene expression analysis, which showed that YR4‐42 selectively targets PPARγ‐regulated genes mapped to glucose and lipid metabolism in DIO mice.
Conclusions
We conclude that YR4‐42 is a novel anti‐diabetic drug candidate with significant advantages compared to standard PPARγ agonists. YR4‐42 should be further investigated in preclinical and clinical studies.
Keywords: adipogenesis, diabetes, insulin resistance, selective PPARγ modulators
1. INTRODUCTION
Insulin resistance is a common pathology for Type 2 diabetes mellitus, which results in systemic disorders of glucose and lipid metabolism.1, 2 Insulin sensitizers, such as the thiazolidinediones (TZDs), are effective oral medications against diabetes. The classic TZDs are agonists of the nuclear receptor peroxisome proliferator‐activated receptor γ (PPARγ), which is highly expressed in adipose tissue. PPARγ is a master regulator of adipogenesis and lipid metabolism, as well as a key modulator of insulin sensitivity3, 4, 5; however, the benefits of the well‐established TZDs as insulin‐sensitizing drugs are compromised by several undesirable side effects, such as weight gain, fluid retention, bone loss and risk of congestive heart failure. These limitations have led to serious safety concerns and tight restrictions in clinical therapy, and even drug withdrawal in some countries.4, 6
To overcome these disadvantages, a new generation of PPARγ agonists inducing fewer side effects, but harbouring insulin‐sensitizing abilities are being developed. For example, INT131 has been reported to be a selective PPARγ modulator (SPPARM) that provides strong anti‐diabetic efficacy with fewer side effects compared to the full PPARγ agonist rosiglitazone.7, 8 Although some SPPARMs exhibit high affinity to PPARγ, they have only a very limited transactivation effect on PPARγ.7 SR1664, another newly developed PPARγ agonist, can block the inhibitory effect of Cdk5‐mediated phosphorylation on PPARγ.9 In our previous research, we noticed that TZDs, such as pioglitazone, could induce hepatic triglyceride (TG) accumulation and then aggravate liver steatosis in diabetic mice by regulating their hepatic gene expression profile.10, 11 The aim of the present study was to identify novel SPPARMs with much fewer side effects than those caused by TZDs.
In the present paper, we report a newly synthesized tetrahydroisoquinoline derivative, YR4‐42, which has low PPARγ transactivation potency and low PPARγ coactivator selective recruitment capacity. We evaluated the anti‐diabetic efficacy of YR4‐42, both ex vivo and in vivo, and compared it with TZDs, thus exploring the potential of YR4‐42 as an anti‐diabetic drug candidate.
2. MATERIALS AND METHODS
2.1. Materials and reagents
Pioglitazone and rosiglitazone, two standard TZDs agents, were produced by Jiang Su Heng Rui Medicine Co. Ltd (Jiangsu, China), PPARα agonist GW7647 and PPARδ agonist GW501516 were purchased from Sigma‐Aldrich (St Louis, Missouri), PPARγ antagonist GW9662 was purchased from Cayman Chemicals (St Louis, Missouri). The synthesis of a series of tetrahydroisoquinoline derivatives, including YR4‐42, was reported by Prof. Liu previously.12 YR4‐42 was obtained through recrystallization. The purity was 98.36%, as determined by liquid chromatography high‐resolution mass spectrum and the calculated high‐resolution mass spectrum (electrospray ionization) was C29H32F2NO5 [M + H]+ 512.2243 (found 512.2243, 0.2243 ppm; Supplementary information 1). The LanthaScreen PPARα/γ/δ Competitive Binding Assay kit (PV4894) and PPARγ Coactivator Assay Kit (PV4548) were obtained from Life Technologies (Carlsbad, California). Plasma TG and total cholesterol kits were obtained from BioSino, Inc. (Beijing, China). Free fatty acid (FFA) assay kits were from Sekisui Medical (Tokyo, Japan). SYBR® Premix Ex Taq™ II, used for real‐time PCR, was purchased from TaKaRa Bio (Otsu, Japan). Antibodies used in Western blot assays were: anti‐PPARγ (Cell Signaling Technology, #2435, dilute 1:2000), anti‐pPPARγ at Ser273 (Bioss, bs‐4888R, dilute 1:1000), anti‐βactin (Santa Cruz Biotechnology, sc‐47 778, dilute1:5000), secondary antibody conjugated with horseradish peroxidase (ZSGB‐BIO, Inc., Beijing, China, dilute1:2000).
2.2. Animals
Male C57BL/6J mice (6 weeks' old, body weight 18‐19 g) were obtained from the Experimental Animal Centre, Chinese Academy of Medical Sciences (Beijing, China) and housed at a constant temperature (22 ± 3°C) and in a 12‐hour light:12‐hour dark cycle. The mice were fed with a high‐fat diet (60% of calories from fat; Research Diets, Inc., Beijing, China) for 10 weeks to initiate a diet‐induced‐obese (DIO) model, as previously reported.11 The high‐fat diet was continuously provided during the whole compound treatment period. All animal experiments were performed strictly in compliance with Chinese guidelines, including the Standards for Laboratory Animals (GB14925‐2001) and the Guideline on the Humane Treatment of Laboratory Animals (MOST 2006a), and all animal procedures were approved by Beijing Administration Office for Laboratory Animals (approval number: SCXK‐Beijing‐2014‐0004).
2.3. Plasmids constructions of PPAR subtypes and mutants
The full‐length human PPARγ (Gene bank: NM_015869) and RXRα (Gene bank: NM_002957.5) coding sequences were inserted into the vector pcDNA3.1 to construct the plasmids pcDNA3.1‐hPPARγ and pcDNA3.1‐hRXRα. Plasmids expressing the ligand‐binding domain (LBD) of human PPARα/γ/δ were constructed as follows: PPARαLBD (Ser167 to Tyr468, Gene bank: NM_001001928), PPARγLBD (Ser204 to Tyr505, Gene bank: NM_015869) and PPARδLBD (Ser139 to Tyr441, Gene bank: NM_006238.4) were fused with the DNA‐binding domain (DBD) fragment (Met1‐Ser147) of Gal4 obtained from plasmid pGBKT7 to connect into plasmid pcDNA3.1, resulting in the construction of pcDNA3.1‐hPPARα/γ/δLBD‐Gal4DBD. The upstream activator sequence (UAS) of Gal4, 5′CGGAAGACTCTCCTCCG3′, or the PPARγ responsive element (PPRE), 5′AAACTAGGTC3′, was inserted into the plasmid Peak12 Sx Syn luci to construct the luciferase reporter plasmid Peak12‐Gal4UAS‐luci or Peak12‐PPRE‐luci.
PPARγ mutation constructions were established by mutation on indicated amino acid sites in pcDNA3.1‐hPPARγLBD‐Gal4DBD, using TransStart FastPfu PCR SuperMix (Transgen Biotech, Beijing, China). Ser273, His449 and Tyr473 were mutated to Ala273, Ala449 and Ala473, respectively, which were then named as pcDNA3.1‐hPPARγLBDM273/449/473‐Gal4DBD. (Detailed information on these plasmids is presented in Supplementary information 2.)
2.4. Cell culture and induction of adipocytes differentiation
The mouse fibroblast cell line 3T3‐L1 was purchased from American Type Culture Collection. Human embryonic kidney 293E cell line, Chinese hamster ovary cells and mouse liver cell line NCTC‐1469 were obtained from the Cell Resource Centre, Peking Union Medical College (headquarters of National Infrastructure of Cell Line Resources). Their species origin was verified with PCR, and the identities of the cell lines were authenticated with short tandem repeat (STR) profiling (ATCC STR profile database). All the information can be viewed on the website (http://cellresource.cn). All cells were cultured with DMEM/high glucose medium (Gibco, Waltham, Massachusetts) containing 10% fetal bovine serum (Gibco) and 100 mg/L penicillin‐streptomycin, and maintained in an incubator at 37°C and humidified with 5% CO2‐containing atmosphere.
For induction of mature adipocytes, confluent 3T3‐L1 cells were cultured in differentiation‐inducing medium (DMEM high glucose medium added with 10% fetal bovine serum, 1.0 μM dexamethasone, 0.5 mM 3‐Isobutyl‐1‐methylxanthine, 10 μg/mL insulin) for 2 days. The cells were then cultured in maintaining medium (DMEM high glucose medium added with 10% fetal bovine serum, 10 μg/mL insulin) for another 10 days.
To determine the lipid accumulation capacity of adipocytes, Oil‐Red O staining for lipid droplets was performed and the intracellular TG levels were measured using a commercial kit (Applygen, Beijing, China). To detect the ability of adipocytes to consume glucose, glucose concentration in cell medium was determined before and after mature adipocytes were treated by compounds for 24 hours using the glucose oxidase method.
To determine PPARγ phosphorylation by CDK5, mature 3T3‐L1 adipocytes were pre‐treated with indicated compounds for 45 minutes, then 50 ng/mL tumour necrosis factor (TNF)α was added for another 30‐minute treatment. Cells were then collected and lysed for Western blotting for PPARγ and PPARγ Ser273 phosphorylation. When performing Western blotting analysis, the total protein in the cell lysate was determined using a BCA assay kit (Applygen) with the same amount always loaded for SDS‐PAGE electrophoresis. Band intensity was quantitatively analysed using a Gel‐PRO Analyser (Media Cybernetics, Rockville, Maryland).
Real‐time PCR was performed to analysis the gene expression in 3T3‐L1 adipocytes after 7‐day treatment of YR4‐42 and rosiglitazone, respectively, by use of synthesized primer oligos, listed in Supplementary information 3.
When we performed experiments with cultured cells, we always standardized the results to the total cell numbers.
2.5. Transactivation assay of PPARα/γ/δ and PPARγ mutants
The transactivation assay of PPAR has been described previously.13, 14 Briefly, to determine PPRE‐regulated transactivation, 293E cells were co‐transfected with the plasmids of pcDNA3.1‐hPPARγ, pcDNA3.1‐hRXRα, Peak12‐PPRE‐luci and the internal control pRL‐TK plasmid (Promega, Madison, Wisconsin); to determine transactivation assay on the LBD of PPAR subunits, 293E cells were co‐transfected with the plasmids of pcDNA3.1‐hPPARα/γ/δLBD‐Gal4DBD, Peak12‐Gal4UAS‐luci and the internal control pRL‐TK plasmid, using lipofectamine 2000 (Life Technologies). After 24 hours, transfected cells were harvested and seeded again into a 96‐well plate and treated with compounds or drugs, as described in figure 1 and supplementary figure S2. After 24‐hour treatments with chemicals, cells were lysed and dual‐luciferase activity were detected using firefly and renilla luciferase substrates (Vigorous, Beijing, China).
To determine transactivation of PPARγ mutants induced by compounds, Chinese hamster ovary cells were transfected with pcDNA3.1‐hPPARα/γ/δLBD‐Gal4DBD or pcDNA3.1‐hPPARγLBDM273/449/473‐Gal4DBD, and the following procedures were followed as described above for the transactivation assay of PPARγ subunits in 293E cells.
2.6. Molecular simulation studies between YR4‐42 and PPARγ
Molecular simulation studies were performed using the Discovery Studio 2017 software package (BIOVIA, San Diego, California). The complex structure of PPARγ was obtained from the Protein Data Bank (code: 5Y2O).15 The docking calculation was performed with CDocker protocol using default settings. All calculations were carried out on a DELL Precision T5500 workstation.
2.7. Animal treatments and in vivo pharmacological evaluation of YR4‐42
We chose pioglitazone as the positive control for in vivo activity evaluation as it was the most widely used insulin sensitizer in the clinic to date and showed comparable in vitro activity to YR4‐42 in our research.
We randomly divided 16‐week‐old DIO‐C57BL/6J mice into three groups (n = 12 per group) and then administered by oral gavage 0.5% CMC‐Na (vehicle group), pioglitazone (25 mg/kg) and YR4‐42 (50 mg/kg) for 38 days. Additionally, a control group of 12 age‐ and gender‐matched C57BL/6J mice were fed standard chow (Research Diets, Inc.) and were then administered 0.5% CMC‐Na by oral gavage for the same time.
Blood biochemistry markers were measured using individual commercial kits as described above and established methods as previously reported.10, 14 Briefly, the body weight and daily food intake of each mouse was monitored every 2 days. Fasting blood glucose and lipids levels were determined once every week. An insulin tolerance test was performed on day 14. At the end of the experiment (day 38), the mice were killed, and plasma samples were stored for downstream biochemical analysis. Tissue samples were immediately removed, weighed and stored at −80°C for subsequent analysis. When we analysed tissue samples, we always normalized the result to the total tissue weight.
Real‐time PCR was performed to analyse the gene expression in muscle, adipose tissue and liver in mice by use of the synthesized primer oligos listed in Supplementary information 3.
2.8. Data analysis
All values are presented as mean ± SEM. Data were analysed using one‐way ANOVA, with Bonferroni's correction or Student's t‐test. P values <0.05 were taken to indicate statistical significance.
3. RESULTS
3.1. YR4‐42 selectively activates PPARγ with a unique agonism different from that of conventional TZDs
Details of the procedures used to synthesize a series of tetrahydroisoquinoline derivatives targeting PPARγ were as previously reported12 and synthesis of YR4‐42 is shown in Figure S1. Like rosiglitazone and pioglitazone, YR4‐42 contains an aromatic tail and a polar head by combining a tetrahydroisoquinoline motif and a phenylpropionic acid motif, and its PPARγ activation was confirmed using PPRE‐regulated transactivation assay, which suggested a structure‐based agonism (Figure 1A).
Figure 1.
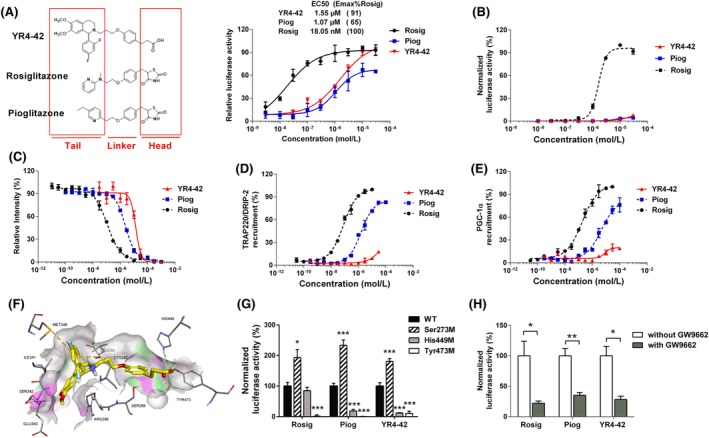
YR4‐42 selectively activates peroxisome proliferator‐activated receptor γ (PPARγ) with a unique agonism different from conventional thiazolidinediones (TZDs). A, Structure‐based PPARγ responsive element (PPRE) transactivation of PPARγ ligands. Structure as well as PPARγ transactivation activity of YR4‐42 is compared to rosiglitazone and pioglitazone. B, Transactivation assay of YR4‐42 on the PPARγ ligand‐binding domain (LBD). The concentration of compounds ranged from 10 nmol/L to 50 μmol/L. Two classic TZDs, rosiglitazone (Rosig) and pioglitazone (Piog), were used as positive controls. Relative activity of YR4‐42 were normalized to the percentage of the activity of rosiglitazone at the indicated concentrations. C, Affinity evaluation of YR4‐42 and PPARγ by Lanthascreen PPAR competitive binding assay. Concentrations of compounds ranged from 10 pmol/L to 1 mmol/L. Relative intensities of YR4‐42, rosiglitazone and pioglitazone at the indicated concentrations were normalized to the intensity of the negative control (solvent DMSO only). D, and E, Determination of the interaction between YR4‐42 and PPARγ coactivators, thyroid hormone receptor‐associated protein complex 220 kDa component (TRAP220) D, and PPARγ coactivator 1‐α (PGC1α) PGC1α E, using LanthaScreen TR‐FRET PPARγ Coactivator Assay. F, Molecular simulation between YR4‐42 and PPARγ (Protein Data Bank code 5Y2O), ‐CDocker interaction energy = 29.82 kcal/Mol. G, Transactivation assay of YR4‐42 to PPARγ wild‐type (WT), and PPARγ mutated at Ser273, His449 as well as at Tyr473. Concentration of YR4‐42, rosiglitazone and pioglitazone was 10 μmol/L. H, Transactivation of PPARγ induced by YR4‐42 with or without 20 μmol/L of PPARγ antagonist GW9662. The concentration of YR4‐42, rosiglitazone and pioglitazone was 10 μmol/L. All results were plotted from duplicate independent experiments and data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, vs WT or the indicated group
To investigate the transactivation ability of PPARα/γ/δ LBDs, we used a PPARα/γ/δLBD‐Gal4‐luciferase reporter assay to evaluate the selective agonist capabilities of YR4‐42 on the various PPAR subunits. As expected, YR4‐42 activated PPARγ in a concentration‐dependent manner. Importantly, PPARγ transactivation induced by YR4‐42 was comparable to pioglitazone, but much weaker than rosiglitazone, a well‐known full agonist of PPARγ (Figure 1B). Notably, YR4‐42 showed 26% activation potency of PPARα at a concentration of 10 μM (Figure S2A) and had no effect on PPARδ transactivation (Figure S2B).
We next used LanthaScreen TR‐FRET PPAR competitive binding assay to evaluate the binding affinity between YR4‐42 and the different PPAR subunits, which demonstrated that YR4‐42 bound to PPARγ with a Ki value of 5744 nmol/L, while the Ki values of rosiglitazone and pioglitazone binding to PPARγ were 38 nmol/L and 860 nmol/L, respectively, suggesting that YR4‐42 cannot bind PPARγ as strongly as well‐established TZDs (Figure 1C). Nevertheless, YR4‐42 did not bind to PPARα (Figure S2C) nor to PPARδ (Figure S2D). The expression of targeted genes of PPARα16 and PPARδ17 were determined by real‐time PCR in mouse hepatic NCTC‐1469 cells after 24‐hour treatment with YR4‐42; the result suggested that YR4‐42 had no significant effect on PPARα‐ and PPARδ‐targeted genes, but YR4‐42 could induce expression of PPARα itself (Figure S2E,F).
Specifically and efficiently recruiting coactivators is recognized as a special characteristic of selective PPARγ agonists.7 We then performed LanthaScreen TR‐FRET PPARγ coactivator assay to determine whether YR4‐42 could activate PPARγ by recruiting its coactivators. Indeed, our data suggested YR4‐42 could recruit thyroid hormone receptor‐associated protein complex 220 kDa component (TRAP220) and PPARγ coactivator 1‐α (PGC1α), the two standard PPARγ coactivators; however, the capacity of YR4‐42 to induce TRAP220 or PGC1α to interact with PPARγ was not as strong as rosiglitazone and pioglitazone (Figure 1D,E). Furthermore, we carried out a simulation assay to investigate the binding machinery between YR4‐42 and PPARγ, suggesting that YR4‐42 bound to PPARγ via hydrogen bonding with His449 as well as Pi‐sulfur interactions with Met348 and Cys285 (Figure 1F).
To investigate the key binding sites involved in the interaction between YR4‐42 and PPARγ, we constructed several PPARγ mutants and assessed the transactivation of PPARγ induced by YR4‐42 (Figure 1G). The transactivation capacity of PPARγ was significantly inhibited when His449 was mutated, which was consistent with the previous molecular simulation study. We also observed that the Ser273 mutation increased while Tyr473 mutation decreased the transactivation of PPARγ, effects similar to those observed for rosiglitazone and pioglitazone, indicating that YR4‐42 shares the same binding sites as these TZDs. Moreover, GW9662, an established PPARγ antagonist, inhibited the transactivation induced by YR4‐42 to the same extent as rosiglitazone and pioglitazone, suggesting that YR4‐42, like those TZDs, had a similar competitive binding of GW9662 (Figure 1H).
3.2. YR4‐42 selectively regulates adipogenesis and blocks Ser273 phosphorylation of PPARγ in adipocytes
We then investigated whether YR4‐42 could regulate adipocyte differentiation similarly to other TZDs. Mouse 3T3‐L1 cells were induced to adipocytes and at the same time treated with YR4‐42, rosiglitazone and pioglitazone, respectively, for 7 days (Figure 2A,B), or mature 3T3‐L1 adipocytes were treated with YR4‐42, rosiglitazone and pioglitazone, respectively, for 2 days (Figure 2C,D). Lipid droplet staining with Oil‐red O showed that YR4‐42 treatment induced much fewer lipid droplets than rosiglitazone and pioglitazone at different concentrations (Figure 2A,C). Next, we determined intracellular TG levels in 3T3‐L1 adipocytes treated with these chemicals. As expected, YR4‐42 caused much less intracellular TG accumulation than TZDs (Figure 2B,D).
Figure 2.
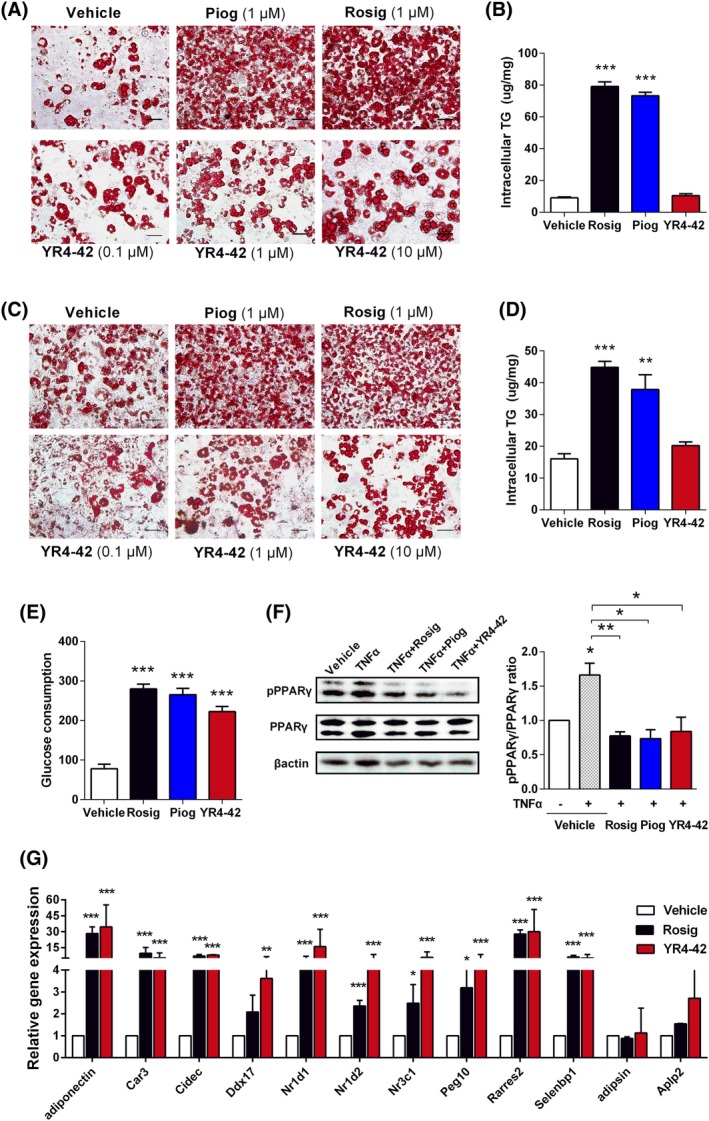
YR4‐42 selectively regulates adipogenesis and blocks Ser273 phosphorylation of peroxisome proliferator‐activated receptor γ (PPARγ) in adipocytes. A, 3T3‐L1 adipocytes were treated with compounds at the indicated concentration for 7 days during the period of differentiation and then stained with Oil‐red O. Depicted are representative images for lipid droplets induced by different PPARγ agonists. Scale bar 50 μm. B, To quantify lipid accumulation in adipocytes differentiation and adipogenesis, intracellular triglyceride (TG) levels were measured after 3T3‐L1 adipocytes were incubated with 10 μmol/L of each indicated compound for 7 days. C, Mature 3T3‐L1 adipocytes were treated with compounds at the indicated concentration for 2 days and then stained with Oil‐red O. Depicted are representative images for lipid droplets induced by different PPARγ agonists. Scale bar 50 μm. D, Intracellular TG levels were measured after mature 3T3‐L1 adipocytes were incubated with 10 μmol/L of each indicated compound for 2 days. E, Glucose consumption of 3T3‐L1 adipocytes after incubation of 10 μmol/L of each indicated compound for 24 hours. F, Regulation of PPARγ Ser273 phosphorylation by YR4‐42. Mature 3T3‐L1 adipocytes were pre‐incubated with 10 μmol/L of compounds and then induced by 50 ng/mL TNFα, after which, intracellular protein levels of PPARγ and phosphorylated PPARγ were determined by Western blot analysis. G, Gene expression analysis by real‐time PCR in 3T3‐L1 adipocytes treated by 10 μmol/L of YR4‐42 and rosiglitazone, respectively, for 7 days. All the experiments were performed independently at least three times. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, vs vehicle or the indicated group
Although YR4‐42 showed weaker PPARγ agonism and less adipogenesis effect compared to TZDs, determination of glucose consumption by adipocytes indicated that YR4‐42 promoted the glucose disposal capacity of adipocytes, which was similar to TZDs (Figure 2E).
It has been demonstrated that TNFα can induce phosphorylation of PPARγ on Ser273 and thereby inhibit the activation of PPARγ in a CDK5‐dependent manner.18 Our results showed that YR4‐42 efficiently blocked TNFα‐induced Ser273 phosphorylation of PPARγ, thus leading to PPARγ activation. This finding also supported the notion that the mutation of Ser273 on PPARγ could increase the transactivation induced by YR4‐42 (Figure 2F).
In 3T3‐L1 adipocytes after treatment of compounds, we investigated gene expression profiling which had been previously reported as PPARγ Ser273 phosphorylation‐dependent,9, 18 the majority of these genes were significantly upregulated on YR4‐42 treatment (Figure 2G), suggesting that regulation of Ser273 phosphorylation is a key process for YR4‐42 to activate PPARγ.
3.3. YR4‐42 ameliorates hyperglycaemia, hyperlipidaemia and hepatic steatosis in DIO mice without weight gain
As previous findings suggested that YR4‐42 is a PPARγ agonist with equivalent transactivation and weaker affinity compared to pioglitazone, we administrated the DIO mice with either pioglitazone (25 mg/kg, according to previous reports10, 11, 19) or YR4‐42 (50 mg/kg) for 38 days to compare the potential in vivo effects of YR4‐42 and pioglitazone. The body weights of DIO mice increased significantly compared to control mice fed with normal chow diet, and pioglitazone administration further enhanced weight gain, interestingly, YR4‐42 treatment significantly decreased the body weight of DIO mice (Figure 3A,B) by attenuating intraperitoneal fat accumulation (Figure 3C). YR4‐42 had no obvious influence on liver weight, which was different from pioglitazone (Figure 3D), although the weight ratio of liver to body increased (Table S1), which might be attributable to the massive loss of body weight. We also noted that YR4‐42 treatment had no significant effect on the food intake of the mice (Table S2). As the half lethal dose of YR4‐42 is >2 g/kg (Table S3), we believe that loss of body weight was neither a toxic effect nor appetite suppression induced by YR4‐42.
Figure 3.
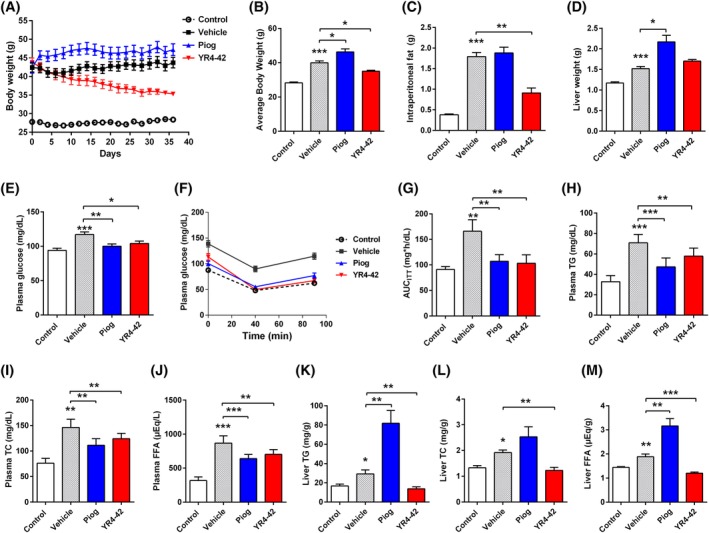
YR4‐42 ameliorates hyperglycaemia, hyperlipidaemia and hepatic steatosis of diet‐induced obese (DIO) mice without weight gain. DIO‐C57BL/6J mice were divided into three groups: Vehicle group (oral gavage of 0.5%CMC‐Na); Piog group (oral gavage of pioglitazone, 25 mg/kg) and YR4‐42 group (oral gavage of YR4‐42, 50 mg/kg). C57BL/6J mice fed with standard chow and oral gavage of 0.5%CMC‐Na represented the control group. A, Body weights of mice determined continuously during 38 days of oral administration. B, Average body weight of mice after 38 days of oral administration. C, D, Intraperitoneal fat C, and liver D of mice were weighted at the end of oral administration. E, Fasting blood glucose levels, determined on the 7th day. F, Insulin tolerance test, performed on the 14th day. G, Area under the curve of insulin tolerance test (AUCITT)measurements on the 14th day. H, Plasma triglyceride (TG), I, total cholesterol (TC), J, free fatty acid (FFA) levels in the mice, determined at the end of oral administration (on the 38th day). K, Liver TG, L, TC and M, FFA levels in the mice, determined at the end of oral administration (on the 38th day). All experiments were performed independently. Data are shown as mean ± SEM of every group, n = 12. *P < 0.05, **P < 0.01, ***P < 0.001, vs control group or the indicated group
Fasting blood glucose determination on the 7th day of compound administration showed that YR4‐42 rescued the hyperglycaemia of DIO mice as effectively as pioglitazone (Figure 3E), which was in line with the result of insulin tolerance test performed on the 14th day after the compound administration (Figure 3F,G), suggesting YR4‐42 displayed similar efficacy in controlling blood glucose and improving insulin resistance compared to pioglitazone.
Long‐term administration of high‐fat diet‐induced hyperlipidaemia in DIO mice, which could be characterized by higher lipid levels in plasma compared to normal chow‐fed mice. As with pioglitazone, YR4‐42 administration significantly attenuated the hyperlipidaemia by decreasing plasma TG, total cholesterol and FFA levels (Figure 3H‐J).
The DIO mice had the pathological features of hepatic steatosis and, importantly, pioglitazone administration resulted in exacerbated hepatic steatosis and increased hepatic TG, total cholesterol and FFA levels, which was previously confirmed by the transcriptional upregulation of genes associated with lipid metabolism.11 In contrast, YR4‐42 administration significantly decreased the concentration of hepatic TG, total cholesterol and FFA (Figure 3K‐M). Meanwhile, the representative histological sections of the mouse liver (Figure S3) illustrated that hepatic steatosis of DIO mice was significantly improved after 38 days of YR4‐42 treatment, suggesting that YR4‐42, compared to pioglitazone, had a great advantage to attenuate hepatic steatosis in DIO mice.
3.4. YR4‐42 selectively regulates gene expression involved in glucose and lipids metabolism in vivo
To further understand the mechanism whereby YR4‐42 regulates glucose and lipid metabolism, we collected muscle tissues of DIO mice after treatment with YR4‐42 (50 mg/kg) for 38 days. Genes annotated to glucose metabolism, such as Glut2, Glut4 and Pgc1α, were significantly upregulated (Figure 4A), which suggests that YR4‐42 might improve glucose homeostasis and mitochondrial biosynthesis by selectively inducing PPARγ's target genes.20, 21 Compared to pioglitazone, YR4‐42 has a different spectrum of targeted genes.
Figure 4.
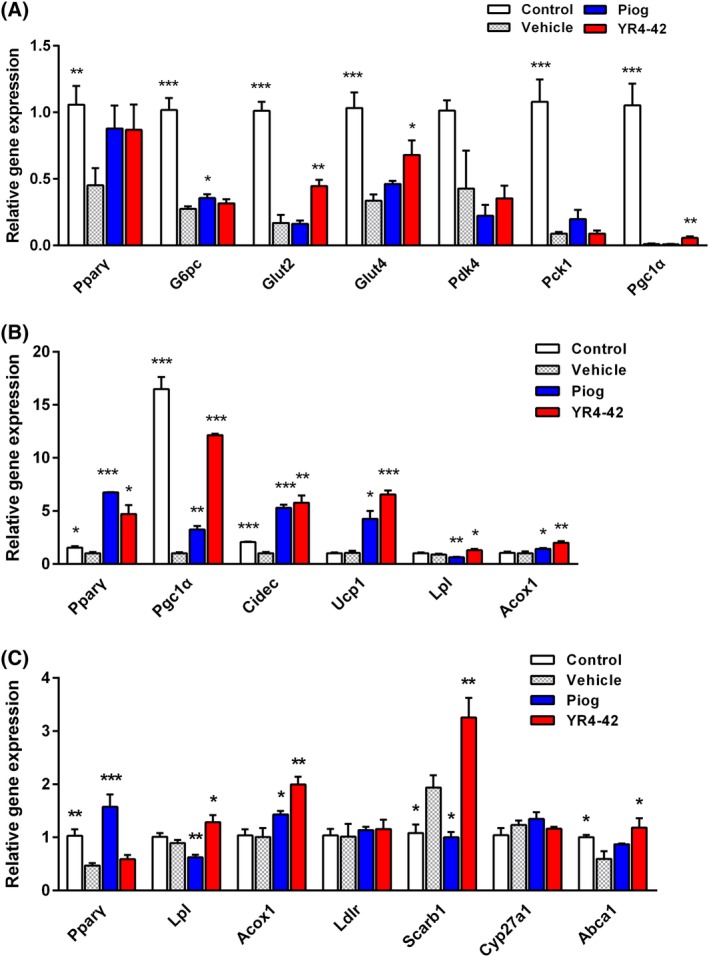
YR4‐42 selectively regulates expression of genes involved in glucose and lipids metabolism in diet‐induced obese (DIO) mice. Gene expression in muscles, brown adipose tissue and liver in DIO mice was determined by real‐time PCR after treatment with YR4‐42 or pioglitazone. A, Expression level of genes related to glucose metabolism in the gastrocnemius of DIO mice after 38 days of oral administration of YR4‐42 (50 mg/kg) and pioglitazone (25 mg/kg). B, Expression changes of genes associated with thermogenesis and lipolysis as well as fatty acid oxidation in the brown adipose tissue at the back neck of DIO mice. C, Genes annotated to fatty acids and cholesterol metabolism in the liver of DIO mice. Data are shown as mean ± SEM of every group, n = 4‐6. *P < 0.05, **P < 0.01, ***P < 0.001, vs vehicle group
Brown adipose tissue is the thermogenic subcutaneous fat depot mainly accumulated at the back neck (the paravertebral area) of mice. Genes involved in thermogenesis, such as Pgc1α,Cidec and Ucp1, which were recognized as PPARγ‐targeted genes,22, 23 were significantly upregulated by YR4‐42 treatment (Figure 4B). Improvement of thermogenesis in fat depots might contribute into elevated energy metabolism and loss of body weight in DIO mice. Moreover, lipolysis and fatty acid oxidation associated genes, such as Lpl and Acox1, were induced in response to YR4‐42 treatment, suggesting YR4‐42 could induce fat turnover in adipose tissues.
We noted that, in DIO mice, YR4‐42 attenuated hepatic steatosis by decreasing TG, total cholesterol and FFA in the liver, which was quite distinct from the action of pioglitazone. We therefore compared, in particular, the expression level of Pparγ and genes responsible for fatty acid and cholesterol metabolism in the liver. In DIO mice, YR4‐42 induced Lpl, Acox1 (genes engaged in lipid hydrolysis and fatty acid oxidation), Scarb1 (genes engaged in cholesterol transporting into liver) and Abca1 (genes engaged in cholesterol efflux), which might partially explain why YR4‐42 decreased TG, total cholesterol and FFA levels in the liver. Notably, YR4‐42 had no obvious influence on Pparγ in the liver of DIO mice, while pioglitazone significantly upregulated Pparγ expression. In addition, the regulatory effects on two genes, Lpl and Scarb1, resulting from the treatment of YR4‐42 and pioglitazone, were in the opposite direction (Figure 4C), which indicated differential transcription regulation on Pparγ and its targeted genes in the liver after treatment with YR4‐42 and pioglitazone.
4. DISCUSSION
The use of SPPARMs is considered a new approach for PPARγ‐targeted drugs. Several studies have evaluated SPPARMs in comparison to the established stronger agonists such as rosiglitazone and pioglitazone24, 25, 26, 27, 28, 29; however, to date, results have not been able to determine, especially at the molecular level, the characteristics of SPPARM or how to identify an ideal SPPARM. In the present study, we concluded that YR4‐42 is a novel SPPARM based on its PPARγ transactivation activity, PPARγ binding and affinity, and its ability to recruit PPARγ coactivators. We showed that YR4‐42 has similar PPARγ binding sites to those of the well‐established TZDs, and is competitively inhibited by the PPARγ antagonist GW9662, but introduces mild transactivation on PPARγ and selectively recruits the coactivators, TRAP220 and PGC1α. The coactivators, especially TRAP220, are essential for adipogenesis induced by PPARγ,30 which may provide an explanation for the selective regulation of adipogenesis by YR4‐42. In addition, YR4‐42 significantly induced PGC1α expression in the muscle and adipose tissue, indicating YR4‐42 possibly regulates mitochondrial biosynthesis and thermogenesis in a PGC1α‐mediated pathway and this needs further exploration.
Another strategy in the development of an effective SPPARM is to inhibit PPARγ phosphorylation.9 We found that YR4‐42 can block TNFα‐induced, CDK5‐dependent PPARγ Ser273 phosphorylation and displays weak adipogenesis which is line with the previous report.18 Furthermore, we identified Ser273 on PPARγ as one of the key interacting sites for YR4‐42, which is the same site for other TZDs; however, we could not distinguish YR4‐42 from pioglitazone in an interaction model with PPARγ, and thus need to investigate further how YR4‐42 blocks phosphorylation of PPARγ by CDK5 as well as the underlying mechanism of TRAP220 and PGC1α recruitment.
The desired pharmacological and clinical effect of PPARγ agonists is to eliminate insulin resistance in vivo, including amelioration of hyperglycaemia and hyperlipidaemia.4, 6 Clinical studies showed that SPPARMs have equivalent efficacy in terms of glycaemic control compared to the strong PPARγ agonist, but with much fewer side effects, such as fluid accumulation and body weight gain.29 Our data showed that YR4‐42 has a similar insulin‐sensitizing activity to that of pioglitazone, as well as comparable potency to ameliorate hyperglycaemia and hyperlipidemia in DIO mice. Interestingly, YR4‐42 treatment also improved hepatic steatosis and induced weight loss which was mainly attributed to fat mass loss. We further explored the underlying mechanism of YR4‐42 attenuating hyperglycaemia and lipid disorder and found that YR4‐42 can selectively induce the expression of genes involved in glucose and lipid metabolism in muscle, adipose tissue and the liver. Interestingly, YR4‐42's direct effect on PPARγ transactivation and its targeted genes differs from pioglitazone; the specific mechanism of YR4‐42 regulating PPARγ‐targeted genes requires further exploration.
INT131, a representative SPPARM which has progressed through to phase 2 clinical trials, was reported to show similar capacity to reduce HbA1c but with less fluid accumulation and weight gain as compared to TZDs. Consistent with INT131 and the SPPARM design, YR4‐42 also reduced hyperglycaemia and hyperlipidaemia without weight gain in a mouse model of type 2 diabetes. Notably, YR4‐42 uniquely improved hepatic steatosis in DIO mice, which indicates that YR4‐42 has potential for use in the treatment of the non‐alcoholic fatty liver disease coexisting with type 2 diabetes. Although it is recognized that pioglitazone has beneficial effects on lipid metabolism and hepatic steatosis in humans,31, 32 YR4‐42 is still worth further study for its less typical TZD' side effects. The anti‐diabetic and lipid‐lowering effects of YR4‐42 in the present study were, however, based on a rodent model, therefore, in the future we still need to evaluate whether YR4‐42 has beneficial effects in humans.
Taken together, the results of the present study show that our newly developed SPPARM, YR4‐42, can selectively regulate adipogenesis and block Ser273 phosphorylation of PPARγ in adipocytes. In a DIO mouse model, oral administration of YR4‐42 can ameliorate hyperglycaemia and hyperlipidaemia and associated hepatic steatosis, with significantly fewer side effects than pioglitazone. Therefore, we propose that YR4‐42 is a promising anti‐diabetic drug candidate and should be further investigated in preclinical and clinical studies.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Yi Huan and Xuan Pan designed the study, performed experiments, analyzed the data, prepared figures and write the manuscript. Xuan Pan synthesized the compound YR4‐42. Jun Peng, Chunming Jia, Sujuan Sun, Guoliang Bai, Xing Wang, Tian Zhou, Rongcui Li, Shuainan Liu, Caina Li, Quan Liu participated in experiments and helped the data analysis. Zhufang Shen and Zhanzhu Liu coordinated the study and are responsible for this study.
Supporting information
Figure S1. Reagents and conditions in synthesis: (a) SOCl2, toluene, reflux, 2 h; (b) NaOH, CH2Cl2, r.t., 5 h; (c) POCl3, toluene, reflux, 2 h; (d) NaBH4, CH3OH, r.t., 1 h; (e) 1‐bromo‐3‐chloropropane, K2CO3, 2‐butanone, reflux, 2 h; (f) K2CO3, DMF, 80°C, 2 h; (g) KOH, CH3OH, reflux, 1 h, then HCl.
Figure S2. YR4‐42 has no effect on transactivation and affinity of PPARα and PPARδ. A. and B. Transactivation of YR4‐42 on PPARα (A) and PPARδ (B). The concentration range of YR4‐42 was set from 10 nmol/L to 100 μmol/L (A) or from 1 nmol/L to 10 μmol/L (B), the activity of YR4‐42 at the indicated concentrations was normalized to the percentage of activity of the identified PPARα agonist GW7647 or PPARδ agonist GW501516. C and D. Evaluation of the affinity of compounds to PPARα (C) and PPARδ (D) by Lanthascreen PPAR competitive binding assay. The concentration range of compounds was ranged from 500 pmol/L to 100 μmol/L, and intensity of compound at the indicated concentrations was normalized to the percentage of intensity of negative control (Solvent DMSO only). E and F. Expression of genes targeted to PPARα (E) and PPARδ (F) was determined by Real‐time PCR after mouse liver cell NCTC‐1469 was treated by YR4‐42 or indicated PPARα/δ agonists. Data are expressed as mean ± SEM of three parallel experiments. *P < 0.05, **P < 0.01, ***P < 0.001, vs vehicle group.
Figure S3. YR4‐42 ameliorates hepatic steatosis in diet‐induced obese (DIO) mice. The visual histological sections of the mouse liver after 38 days of treatment. Control: C57BL/6J mice fed with standard chow and oral gavage of 0.5% CMC‐Na; Vehicle: C57BL/6J mice fed with high‐fat diet (DIO mice) and treated with oral gavage of 0.5% CMC‐Na; Piog: DIO mice treated with oral gavage of pioglitazone, 25 mg/kg; YR4‐42: DIO mice treated with oral gavage of YR4‐42, 50 mg/kg. Scale bar: 50 μm.
Table S1. Weight index of intraperitoneal fat and liver.
Table S2. Average daily food intake.
Table S3. Acute toxicity test on YR4‐42.
Supplementary Information
ACKNOWLEDGMENTS
This work was financially supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016‐I2M‐4‐001,2017‐I2M‐1‐010) and the Drug Innovation Major Project in China (2018ZX09711001‐003‐005, 2018ZX09711‐001‐005‐014). We thank Prof. Chengyu Jiang in Peking Union Medical College for providing the Peak12‐luciferase vector.
Huan Y, Pan X, Peng J, et al. A novel specific peroxisome proliferator‐activated receptor γ (PPARγ) modulator YR4‐42 ameliorates hyperglycaemia and dyslipidaemia and hepatic steatosis in diet‐induced obese mice. Diabetes Obes Metab. 2019;21:2553–2563. 10.1111/dom.13843
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/dom.13843.
Funding information This work was financially supported by the CAMS Innovation Fund for Medical Sciences (CIFMS; 2016‐I2M‐4‐001, 2017‐I2M‐1‐010) and the Drug Innovation Major Project in China (2018ZX09711001‐003‐005, 2018ZX09711‐001‐005‐014).
Contributor Information
Zhanzhu Liu, Email: liuzhanzhu@imm.ac.cn.
Zhufang Shen, Email: shenzhf@imm.ac.cn.
REFERENCES
- 1. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53:1270‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840‐846. [DOI] [PubMed] [Google Scholar]
- 3. Kahn BB, McGraw TE. Rosiglitazone, PPARgamma, and type 2 diabetes. N Engl J Med. 2010;363:2667‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Savage DB. PPAR gamma as a metabolic regulator: insights from genomics and pharmacology. Expert Rev Mol Med. 2005;7:1‐16. [DOI] [PubMed] [Google Scholar]
- 6. Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Higgins LS, Mantzoros CS. The development of INT131 as a selective PPARgamma modulator: approach to a safer insulin sensitizer. PPAR Res. 2008;2008:936906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kintscher U, Goebel M. INT‐131, a PPARgamma agonist for the treatment of type 2 diabetes. Curr Opin Investig Drugs. 2009;10:381‐387. [PubMed] [Google Scholar]
- 9. Choi JH, Banks AS, Kamenecka TM, et al. Antidiabetic actions of a non‐agonist PPARgamma ligand blocking Cdk5‐mediated phosphorylation. Nature. 2011;477:477‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng J, Huan Y, Jiang Q, Sun SJ, Jia CM, Shen ZF. Effects and potential mechanisms of pioglitazone on lipid metabolism in obese diabetic KKAy mice. PPAR Res. 2014;2014:538183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jia C, Huan Y, Liu S, et al. Effect of chronic pioglitazone treatment on hepatic gene expression profile in obese C57BL/6J mice. Int J Mol Sci. 2015;16:12213‐12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu R, Zhou YL, Huan Y, Liu Q, Shen ZF, Liu ZZ. Design, synthesis, and PPARalpha/gamma agonistic activity of novel tetrahydroisoquinoline derivatives. Yao Xue Xue Bao. 2011;46:311‐316. [Article in Chinese]. [PubMed] [Google Scholar]
- 13. Huan Y, Peng J, Wang Y, et al. Establishment and application of screening methods for non‐agonist PPARgamma ligand. Yao Xue Xue Bao. 2014;49:1658‐1664. [Article in Chinese]. [PubMed] [Google Scholar]
- 14. Lei L, Liu Q, Liu S, et al. Antidiabetic potential of a novel dual‐target activator of glucokinase and peroxisome proliferator activated receptor‐gamma. Metabolism. 2015;64:1250‐1261. [DOI] [PubMed] [Google Scholar]
- 15. Lee MA, Tan L, Yang H, Im YG, Im YJ. Structures of PPARgamma complexed with lobeglitazone and pioglitazone reveal key determinants for the recognition of antidiabetic drugs. Sci Rep. 2017;7:16837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol. 2015;62:720‐733. [DOI] [PubMed] [Google Scholar]
- 17. Lee CH, Olson P, Hevener A, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006;103:3444‐3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi JH, Banks AS, Estall JL, et al. Anti‐diabetic drugs inhibit obesity‐linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aoyama T, Ikejima K, Kon K, Okumura K, Arai K, Watanabe S. Pioglitazone promotes survival and prevents hepatic regeneration failure after partial hepatectomy in obese and diabetic KK‐A(y) mice. Hepatology. 2009;49:1636‐1644. [DOI] [PubMed] [Google Scholar]
- 20. Im SS, Kim JW, Kim TH, et al. Identification and characterization of peroxisome proliferator response element in the mouse GLUT2 promoter. Exp Mol Med. 2005;37:101‐110. [DOI] [PubMed] [Google Scholar]
- 21. Koves TR, Li P, An J, et al. Peroxisome proliferator‐activated receptor‐gamma co‐activator 1alpha‐mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid‐induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588‐33598. [DOI] [PubMed] [Google Scholar]
- 22. Boström P, Wu J, Jedrychowski MP, et al. A PGC1‐alpha‐dependent myokine that drives brown‐fat‐like development of white fat and thermogenesis. Nature. 2012;481:463‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barreau C, Labit E, Guissard C, et al. Regionalization of browning revealed by whole subcutaneous adipose tissue imaging. Obesity (Silver Spring). 2016;24:1081‐1089. [DOI] [PubMed] [Google Scholar]
- 24. Doshi LS, Brahma MK, Bahirat UA, Dixit AV, Nemmani KV. Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin Investig Drugs. 2010;19:489‐512. [DOI] [PubMed] [Google Scholar]
- 25. Furukawa A, Arita T, Fukuzaki T, et al. Synthesis and biological evaluation of novel (−)‐Cercosporamide derivatives as potent selective PPARgamma modulators. Eur J Med Chem. 2012;54:522‐533. [DOI] [PubMed] [Google Scholar]
- 26. Liu W, Lau F, Liu K, et al. Benzimidazolones: a new class of selective peroxisome proliferator‐activated receptor gamma (PPARgamma) modulators. J Med Chem. 2011;54:8541‐8554. [DOI] [PubMed] [Google Scholar]
- 27. Taygerly JP, McGee LR, Rubenstein SM, et al. Discovery of INT131: a selective PPARgamma modulator that enhances insulin sensitivity. Bioorg Med Chem. 2013;21:979‐992. [DOI] [PubMed] [Google Scholar]
- 28. Tan Y, Muise ES, Dai H, et al. Novel transcriptome profiling analyses demonstrate that selective peroxisome proliferator‐activated receptor gamma (PPARgamma) modulators display attenuated and selective gene regulatory activity in comparison with PPARgamma full agonists. Mol Pharmacol. 2012;82:68‐79. [DOI] [PubMed] [Google Scholar]
- 29. DePaoli AM, Higgins LS, Henry RR, Mantzoros C, Dunn FL. Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care. 2014;37:1918‐1923. [DOI] [PubMed] [Google Scholar]
- 30. Ge K, Guermah M, Yuan CX, et al. Transcription coactivator TRAP220 is required for PPAR gamma 2‐stimulated adipogenesis. Nature. 2002;417:563‐567. [DOI] [PubMed] [Google Scholar]
- 31. Belfort R, Harrison SA, Brown K, et al. A placebo‐controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297‐2307. [DOI] [PubMed] [Google Scholar]
- 32. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reagents and conditions in synthesis: (a) SOCl2, toluene, reflux, 2 h; (b) NaOH, CH2Cl2, r.t., 5 h; (c) POCl3, toluene, reflux, 2 h; (d) NaBH4, CH3OH, r.t., 1 h; (e) 1‐bromo‐3‐chloropropane, K2CO3, 2‐butanone, reflux, 2 h; (f) K2CO3, DMF, 80°C, 2 h; (g) KOH, CH3OH, reflux, 1 h, then HCl.
Figure S2. YR4‐42 has no effect on transactivation and affinity of PPARα and PPARδ. A. and B. Transactivation of YR4‐42 on PPARα (A) and PPARδ (B). The concentration range of YR4‐42 was set from 10 nmol/L to 100 μmol/L (A) or from 1 nmol/L to 10 μmol/L (B), the activity of YR4‐42 at the indicated concentrations was normalized to the percentage of activity of the identified PPARα agonist GW7647 or PPARδ agonist GW501516. C and D. Evaluation of the affinity of compounds to PPARα (C) and PPARδ (D) by Lanthascreen PPAR competitive binding assay. The concentration range of compounds was ranged from 500 pmol/L to 100 μmol/L, and intensity of compound at the indicated concentrations was normalized to the percentage of intensity of negative control (Solvent DMSO only). E and F. Expression of genes targeted to PPARα (E) and PPARδ (F) was determined by Real‐time PCR after mouse liver cell NCTC‐1469 was treated by YR4‐42 or indicated PPARα/δ agonists. Data are expressed as mean ± SEM of three parallel experiments. *P < 0.05, **P < 0.01, ***P < 0.001, vs vehicle group.
Figure S3. YR4‐42 ameliorates hepatic steatosis in diet‐induced obese (DIO) mice. The visual histological sections of the mouse liver after 38 days of treatment. Control: C57BL/6J mice fed with standard chow and oral gavage of 0.5% CMC‐Na; Vehicle: C57BL/6J mice fed with high‐fat diet (DIO mice) and treated with oral gavage of 0.5% CMC‐Na; Piog: DIO mice treated with oral gavage of pioglitazone, 25 mg/kg; YR4‐42: DIO mice treated with oral gavage of YR4‐42, 50 mg/kg. Scale bar: 50 μm.
Table S1. Weight index of intraperitoneal fat and liver.
Table S2. Average daily food intake.
Table S3. Acute toxicity test on YR4‐42.
Supplementary Information