
Figure 5.
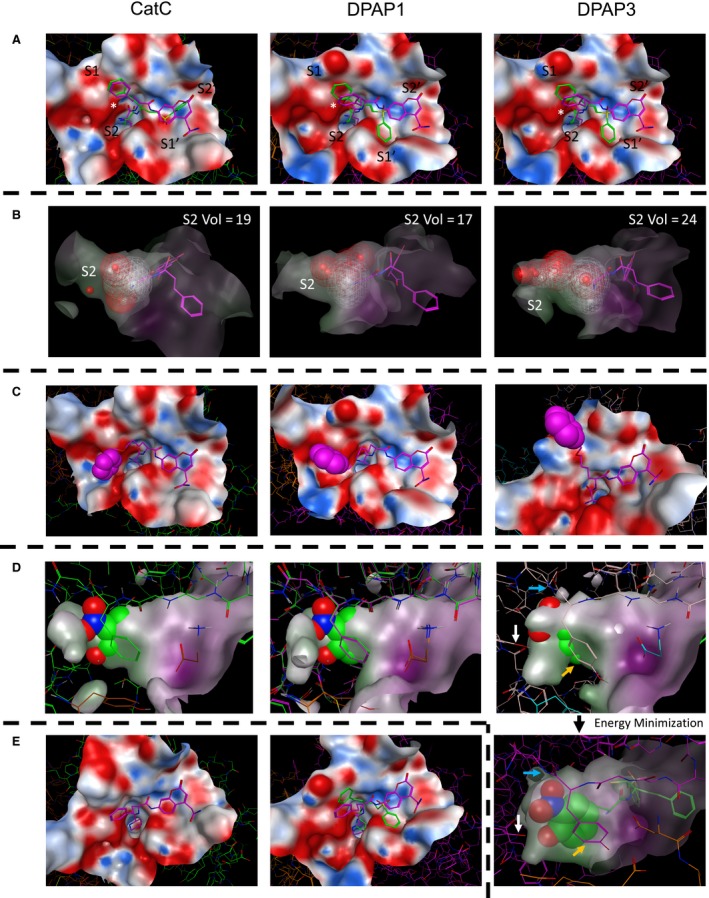
Docking studies on DPAPs. For all panels, the docked structures of compounds into the crystal structure of CatC or the homology models of DPAP1 and DPAP3 are shown on the left, middle and right images respectively. Inhibitors are shown in green, substrates in purple and the surface of the active sites within 6 Å of the ligand as an electrostatic surface. (A) Top views of nVal‐hPhe‐VS and nVal‐hPhe‐ACC docked into the active site of the different DPAPs. The position of the S2 to S2′ pockets is indicated, and that of the exclusion domain N‐terminal Asp is marked with an asterisk. (B) Side view of nVal‐hPhe‐VS docked into the active site of the different DPAPs illustrating the difference in size of the S2 pocket. The dashed volume area defines the volume of the S2 pocket for each DPAP and was calculated by using the ‘Site Finder’ function of MOE. The number on the upper right of each image indicates the calculated relative volume of the S2 pocket. (C) Images illustrating the flexibility of the nLeu(o-Bzl) side chain when docking the nVal‐Leu(o‐Bzl)‐ACC substrate into the different active sites. The benzyl group (spacefill atoms) can reach into different conserved groves distal from the S2 and S1 pockets. (D) Side view of the S2 pocket after modelling the nTyr(NO 2)‐hPhe‐VS inhibitor into the structures of each DPAP after covalent modification of the catalytic cysteine by the vinyl sulfone group. The surface of the S2 pocket is shown to illustrate steric clashes between the P2 side chain (spacefill atoms) and the S2 pocket. The structure of the inhibitor bound to DPAP3 was further refined by allowing energy minimization of residues within 4.5 Å of the Tyr(NO 2) side chain (lower right image). Note that in DPAP3, the NO 2 group might form hydrogen bonds with the amide bond of Ile552 (blue arrows) and with the hydroxylic group of Tyr716 (white arrows) at the bottom of the S2 pocket. We also observed potential stacking interactions between Tyr551 (orange arrows) and the free amine of the inhibitors, as well as hydrophobic interactions with the Tyr(NO 2) side chain. (E) Docking of hPro‐hPhe‐ACC into the structure of CatC and of hPro‐hPhe‐VS and hPro‐hPhe‐ACC into the model of DPAP1. Note that neither of these compounds could be docked into the DPAP3 model, nor hPro‐hPhe‐VS into the CatC structure.