

Abstract
One of the long-standing mysteries of evolutionary genomics is the source of the wide phylogenetic diversity in genome nucleotide composition (G+C vs. A+T), which must be a consequence of interspecific differences in mutation bias, the efficiency of selection for different nucleotides, or a combination of the two. We demonstrate that although genomic G+C composition is strongly driven by mutation bias, it is also substantially modified by direct selection and/or as a by-product of biased gene conversion. Moreover, G+C composition at four-fold redundant sites is consistently elevated above the neutral expectation, more so than for any other classes of sites.
For some classes of genomic sites, G+C nucleotide composition covers nearly the full range of possible variation (frequencies of ∼0.0 to ∼1.0) across species1–5. It is commonly thought that the contribution of mutation to such variation can be determined from the nucleotide content of four-fold redundant (synonymous) sites within codons or from the composition of rare variants, and analyses of this type have led to the idea that mutation is universally biased in the direction of A+T6–8. However, selection on such sites can bias such interpretations. To eliminate such issues, we use direct estimates of the mutation spectra derived from mutation-accumulation (MA) experiments and/or parent-offspring trios for 37 diverse species.
Of the data sets analyzed herein, 25 involve published data (summarized in Ref. 9 with respect to mutation rates), and 12 involve long-term MA experiments in diverse microbial species reported here for the first time (Supplementary Dataset 1: Tables 1–3). Each new MA experiment involves the complete genome sequencing of ∼50 lines serially transferred through single-cell bottlenecks for thousands of cell divisions, which effectively eliminates the ability of natural selection to significantly modify the accumulation of all but the small fraction of extremely deleterious mutations (which in any case are irrelevant to the following analyses, as they do not accumulate evolutionarily; Ref. 9). From the resultant spectra for base-substitution mutations (typically based on dozens to hundreds of de novo mutations), letting m be the ratio of the per-nucleotide mutation rate in the G+C → A+T direction to the reciprocal rate, the expected equilibrium G+C composition under neutrality (where mutation is the only directional evolutionary force) is
| (1) |
Comparison of the observed genome-wide nucleotide compositions of the study species to these neutral expectations reveals several general patterns (Figure 1). First, mutation biases in unicellular species may be in either the A+T or G+C directions (leading to less than or greater than 0.5, respectively), although the former is most common, and no characterized multicellular eukaryote has mutation bias in the G+C direction. Second, regardless of the class of DNA or the phylogenetic grouping, with few exceptions, genome-wide G+C composition is close to or substantially above the neutral expectation, implying that the existence of a near universal direction force(s) favoring G+C content. Third, the primary exception to this pattern involves 0-fold redundant sites (where all nucleotide substitutions lead to amino-acid changes) in bacteria with endogenous mutation pressure towards G+C ( > 0.5), where selection for amino acids containing A+T in such codon positions apparently takes precedence over other G+C enhancing forces. This tendency is reflected in the diminished slope in the regression involving such sites (Supplementary Dataset 1: Table 4). Fourth, for 2- and 4-fold redundant sites (where 2 and 4 nucleotides encode for the same amino acid), G+C composition is particularly strongly elevated, by an average amount that is essentially independent of the neutral expectation, but with considerable variation. The strong elevation for 4-fold redundant sites implies the existence of general forces favoring G+C independent of the implications for the proteome.
Figure 1. Relationship between genome-wide nucleotide composition and the neutral expectation.
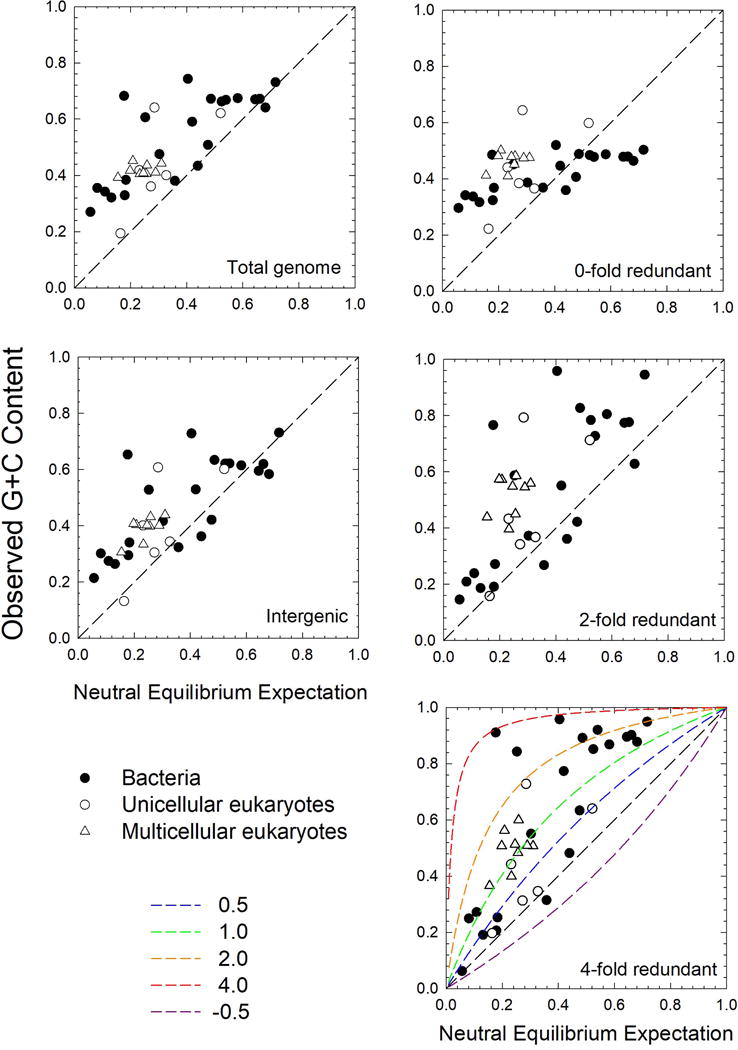
The data are subdivided into three major groups of organisms. The diagonal dashed lines denote agreement with the neutral expectation, with points above the diagonal reflecting conditions in which there is selection for elevated G+C content. For reference, the lower panel provides isoclines of expected genome compositions under selection, with values of the composite parameter S = ϕNes being equivalent to the ratio of the power of selection in favor of G+C content relative to the power of genetic drift. The neutral equilibrium expectation is calculated from Equation (1), and the observed G+C content is based on direct observation of genome contents. All data can be found in Supplementary Dataset 1: Tables 1–5.
The magnitude of the strength of selection required to account for the deviation of G+C composition at 4-fold redundant sites relative to the neutral expectation can be estimated by noting that in the presence of selection, Equation (1) generalizes to
| (2) |
where S = ϕNes, with being the effective population size, ϕ = 2 or 4 for haploids and diploids respectively, and s being the average selective advantage of G/C nucleotides over A/T10–11. S for each genomic category is shown in Supplementary Dataset 1: Table 5. In Figure 1, lines of expectation for for various values of S (equivalent to the ratio of the power of selection s to the power of drift 1/(ϕNe)) show that S (in favor of G+C) is generally in the range of 0.5 to 4. Thus, some selective force in favor of G+C composition is pervasive and relatively strong, although not strong enough to entirely overcome the mutational expectations.
The results for four-fold redundant sites are of relevance to the common usage of measures of standing variation at such positions to estimate Ne under the assumption of neutrality (drift-mutation equilibrium), which leads to an expected average heterozygosity of ϕNeu, where u is the mean mutation rate per nucleotide site. From a rearrangement of Equation (15) in Ref. 12, the ratio of heterozygosity under drift-mutation-selection equilibrium and the neutral expectation is
| (3) |
solution of which shows that when mutation is strongly biased towards A+T but selection strongly favors G+C, the expected nucleotide diversity can be several-fold greater than the neutral expectation ( ), which would lead to the same proportional overestimation of Ne when the mutation rate is factored out (Figure 2). When mutation bias and selection operate in the same direction, π can be downwardly biased up to a few-fold with respect to the neutral condition. Thus, relative estimates of Ne derived from silent-site variation can be off by several fold (when compared with each other) if selection is moderately strong and there are strong differences in mutation bias among contrasted species, which based on the wide range of estimated is clearly the case.
Figure 2. Expected equilibrium levels of within-population nucleotide diversity scaled by the neutral expectation.
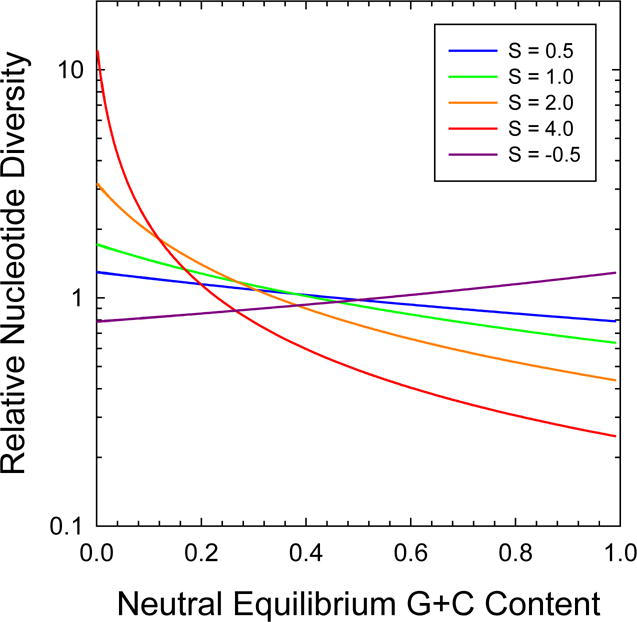
Derived from Equation (3) in the text, with various strengths of selection (S) color coded as in Figure 1.
Our results imply a near universal pervasive mechanism operating to increase G+C content, as previously inferred indirectly from polymorphism data for G+C-rich genomes7. However, the sources of such selection remain unclear. Given that the substantial number of species in this study inhabit a wide range of environments and are derived from a diversity of bacterial and eukaryotic lineages, consistent directional selection in favor of G+C is not readily reconciled by ecological and/or genetic-background arguments. Moreover, given that such selection is experienced by both silent and replacement codon sites, arguments based on protein-sequence constraints and transcription fidelity are not compelling. Likewise, because the pattern extends to intergenic (largely noncoding DNA), arguments based on gene expression and translation speed/accuracy13–14 do not seem to apply. Although gene expression levels within species are correlated with local gene G+C composition, all but one r2 values involving these variables are ≤ 0.02, and the signs of the relationships are inconsistent (Supplementary Dataset 1: Table 6). One general force that may be of relevance is DNA stability, in that G:C pairs involve three hydrogen bonds, whereas A:T pairs involve only two.
An alternative explanation for near universal pressure towards G+C content involves gene conversion, which results from the repair of heteroduplex DNA arising from recombination between two nonidentical sequences and if biased can operate like selection at the population-genetic level. In every organism that has been closely scrutinized, eukaryotes and bacteria, gene conversion has been found to be biased in the direction of G+C (Refs. 15-21), although the molecular mechanisms encouraging such universal behavior are unknown. Most attempts to estimate S associated with codon bias (which may be driven by biased gene conversion) have yielded estimates on the order of 0.1 to 4.0 in diverse phylogenetic groups4 (although not always in the G+C direction), and our results (Figure 1) are fully compatible with this magnitude of selection.
Because effective population sizes vary among organisms by several orders of magnitude, this small range in S suggests that there must be a roughly inverse relationship between Ne and s, whatever the force encouraging G+C content. Under a scenario of natural selection, such a condition is expected under any concave fitness function for increasing G+C content, as the selective advantage of incremental changes would then diminish with increased G+C (further out on the fitness plateau), and larger population sizes would enhance the efficiency of selection for higher G+C content. However, a scenario of biased gene conversion requires a rather different set of conditions – the magnitude of the biasing force (towards G+C) would have to increase with decreasing Ne. In principle, this might occur if a large fraction of GC conversions were deleterious, as natural selection opposing conversion-driven GC would be reduced in the face of increased random genetic drift9. This would, however, also require a very strong increase in the biasing force in small populations because biased gene conversion depends on both the asymmetric force and the recombination rate per nucleotide site, with the latter actually scaling negatively with Ne (Ref. 4).
In summary, our results conclusively support the idea that genome-wide nucleotide composition is strongly influenced by mutation bias at all classes of sites, but that phylogenetically general directional forces beyond mutation (natural selection and/or biased gene conversion) play a role as well. The positive association between neutral G+C-composition expectations and actual utilization at 0-fold redundant sites demonstrates that even amino-acid usage is dictated at least in part by mutation pressure, with the G+C content of such sites differing more than two-fold between genomes with strong mutation bias towards A+T vs. those with bias towards G+C (Figure 1; Supplementary Dataset 1: Table 1). However, despite this gradient, G+C utilization at 0-fold redundant sites is generally substantially greater than the neutral expectation when the latter is < 0.5, so the possibility that such content is influenced by the same selection pressures favoring G+C content at silent sites cannot be ruled out.
Finally, although the ultimate sources of variation in the mutation spectrum (which drives the wide range of variation in nucleotide composition among species) are unknown, they may involve effectively neutral processes. Owing to the predominance of deleterious mutations, selection is expected to generally drive the genome-wide mutation rate down to some level beyond which further advantages are offset by the power of random genetic drift9. However, any particular mutation rate can be compatible with a wide-range of mutational spectra, which may be free to wander over evolutionary time, conditional on the maintenance of a constant genome-wide deleterious rate22. Notably, when the prevailing mutation pressure towards A+T is in conflict with the forces favoring G+C content (which is true for most taxa), the average genome-wide mutation rate per nucleotide site will be indirectly inflated, owing to the elevated abundance of more mutable (G and C) nucleotides.
Methods
G/C composition calculation
Mutation spectra, strain culturing, and reference-genome information for the 37 species in this study are presented in Supplementary Dataset 1: Table 1. We enumerated all sites of the genomes to calculate genome-wide G+C nucleotide compositions. For the G/C nucleotide composition at different functional sites of the genomes, we parsed out: 1) the second nucleotides of all codons except stop codons to delineate 0-fold redundant sites; 2) the third nucleotides of codons for Asn, Asp, Cys, Gln, Glu, His, Lys, and Tyr amino acids for 2-fold redundant sites; 3) the third nucleotides of codons for Ala, Arg, Gly, Leu, Pro, Ser, Thr, and Val amino acids for 4-fold redundant sites; and 4) the nucleotides between the start and/or stop codons (or between UTRs when annotated) of two adjacent genes for intergenic DNA. Expression data of each gene were downloaded from the NCBI GEO database and the gene-specific G+C contents for 4-fold redundant sites were parsed as above. The statistical details of the relationship between gene expression and 4-fold redundant site G/C composition are in Supplementary Dataset 1: Table 6.
MA line transfers
For the 12 new microbial mutation-accumulation (MA) projects reported in this study, all lines were cultured under ideal conditions on solid agar plates, using procedures relied on in numerous previous studies summarized in Lynch et al.9 Within each study, all MA lines initiated from a single-cell progenitor, and were then single-cell transferred daily to weekly (depending on the growth rate, necessary for visual localization of colonies). Each month, numbers of cell divisions during each culturing cycle were estimated using colony-forming units from serial-dilution procedures.
Genome sequencing and raw data
Genomic DNA of the MA lines was extracted using the Wizard Genomic Purification Kit (Promega, Inc.). Illumina libraries for genome sequencing were then constructed using an optimized Nextera DNA library prep kit (Illumina Inc.), and 150 or 250 bp paired-end Illumina sequencing was done on a Hiseq2500 platform (Hubbard Center for Genome Studies, University of New Hampshire). Read trimming, mapping, and mutation rate calculations followed Long et al.23 Duplicate reads were removed using picard-tools-2.5.0 in GATK 3.6. Unique SNP and indel variants were analyzed with HaplotypeCaller, and standard hard-filtering parameters described by GATK Best Practices recommendations24–26. Candidate variants were identified visually with the Integrated Genome Viewer (IGV v. 2.3.5)27. All base-substitution and insertions/deletions identified are in Supplementary Dataset 1: Tables 2, 3.
Data availability
Raw reads of genome sequencing generated in this study are available in the National Center for Biotechnology Information Sequence Read Archive with BioProject no. PRJNA376572.
Supplementary Material
Online Supplementary Dataset 1: Tables 1–6, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Acknowledgments
Support was provided by the Multidisciplinary University Research Initiative awards W911NF-09-1-0444 and W911NF-14-1-0411 from the US Army Research Office to ML; National Institutes of Health awards R01-GM036827 and R35-GM122566 to ML; “Zhufeng Talent Program” startup grant 861701013155 from Ocean University of China to H.L.; R01-GM51986 and R35-GM122556 to YVB, and F32-GM083581 to DTK; and National Science Foundation grant DOB 1442246 to JTL. We thank T. G. Doak, P. Keightley, K. Morris, R. Ness, I. Ruiz-Trillo, S. Simpson, and W. K. Thomas, A. Uchimura, and Z. Ye for providing strains and/or technical help in data acquisition. We thank L. Duret for helpful comments.
Footnotes
Author Contributions H.L., W.S., and M.L. conceived and designed the study, performed data analyses, and wrote the manuscript. All authors contributed in data collection and provided input to the manuscript.
Competing interests
The authors declare no competing interests.
Literature Cited
- 1.Sueoka N. Proc Natl Acad Sci USA. 1962;48:582–592. doi: 10.1073/pnas.48.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu X, Hewett-Emmett D, Li WH. Genetica. 1998;102–3:383–391. [PubMed] [Google Scholar]
- 3.Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH. Proc Natl Acad Sci USA. 2004;101:3480–3485. doi: 10.1073/pnas.0307827100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynch M. The origin of genome architecture. Sinauer Associates, Inc; 2007. [Google Scholar]
- 5.Rocha EPC, Feil EJ. PLoS Genet. 2010;6:e1001104. doi: 10.1371/journal.pgen.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershberg R, Petrov DA. PLoS Genet. 2010;6:e1001115. doi: 10.1371/journal.pgen.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hildebrand F, Meyer A, Eyre-Walker A. PLoS Genet. 2010;6:e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch M. Proc Natl Acad Sci USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch M, et al. Nat Rev Genet. 2016;17:704–714. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- 10.Li WH. J Mol Evol. 1987;24:337–345. doi: 10.1007/BF02134132. [DOI] [PubMed] [Google Scholar]
- 11.Bulmer M. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McVean GAT, Charlesworth B. Genet Res. 1999;74:145–158. [Google Scholar]
- 13.Raghavan R, Kelkar YD, Ochman H. Proc Natl Acad Sci USA. 2012;109:14504–14507. doi: 10.1073/pnas.1205683109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelkar YD, Phillips DS, Ochman H. G3. 2015;5:1247–1252. doi: 10.1534/g3.115.016824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marais G, Mouchiroud D, Duret L. Genet Res. 2003;81:79–87. doi: 10.1017/s0016672302006079. [DOI] [PubMed] [Google Scholar]
- 16.Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. Nature. 2008;454:479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galtier N, Duret L, Glemin S, Ranwez V. Trends Genet. 2009;25:1–5. doi: 10.1016/j.tig.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Pessia E, et al. Genome Biol Evol. 2012;4:675–682. doi: 10.1093/gbe/evs052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams AL, et al. Elife. 2015;4:e04637. [Google Scholar]
- 20.Mugal CF, Weber CC, Ellegren H. Bioessays. 2015;37:1317–1326. doi: 10.1002/bies.201500058. [DOI] [PubMed] [Google Scholar]
- 21.Lassalle F, et al. PLoS Genet. 2015;11:e1004941. doi: 10.1371/journal.pgen.1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch M. Proc Natl Acad Sci USA. 2012;109:18851–18856. doi: 10.1073/pnas.1216130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long H, et al. Genome Biol Evol. 2016;8:3815–3821. doi: 10.1093/gbe/evw286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, et al. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DePristo MA, et al. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Auwera GA, et al. Curr Protoc Bioinformatics. 2013;43(11):10, 1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorvaldsdottir H, Robinson JT, Mesirov JP. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplementary Dataset 1: Tables 1–6, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Data Availability Statement
Raw reads of genome sequencing generated in this study are available in the National Center for Biotechnology Information Sequence Read Archive with BioProject no. PRJNA376572.