
Figure 1. Generation and characterization of full BUB1 gene deletion cell lines.
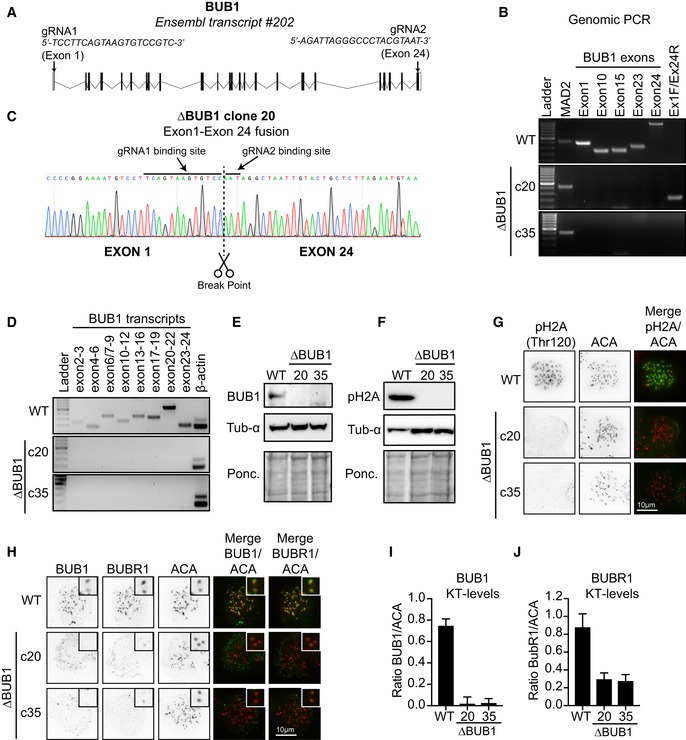
(A) Schematic overview of the genomic BUB1 locus and targeting gRNAs used to generate the BUB1 knockouts. (B) Whole‐genomic DNA was extracted from WT HAP1 cells and from each indicated clone and analyzed by PCR for the presence of the indicated BUB1 exons. A product amplifying the first exon of MAD2 was used as a positive control. Primers spanning exon 1 and exon 24 amplified a product in clone 20. (C) The product amplified form clone 20 using primers for exon 1 and exon 24 was analyzed by Sanger sequencing. A fusion between exons 1 and 24 could be confirmed and was induced at the guide binding sites. (D) BUB1 transcripts were amplified from cDNA generated from WT HAP1 cells or the two ∆BUB1 clones. Amplification of β‐actin was used as a positive control. Primer pairs were designed as such that they at least spanned 1 intron to ensure specific amplification. (E, F) WT and ∆BUB1 clones were analyzed by Western blot. α‐Tubulin and Ponceau S were used as loading controls. (G, H) Immunofluorescence images of WT and ∆BUB1 cells treated with nocodazole for 3 h before fixation. Cells were stained for either H2A‐pThr210 or BUB1 (green) combined with BUBR1 (green) and centromeres/ACA (red). Scale bar: 10 μm. (I, J) in (H). All visible kinetochore pairs in at least 10 cells were quantified, corrected for cytoplasmic levels, and normalized for ACA.
Data information: See Appendix Supplementary Methods for experimental details.