

Abstract
Genotype imputation has been widely utilized for two reasons in the analysis of genome-wide association studies (GWAS). One reason is to increase the power for association studies when causal single nucleotide polymorphisms are not collected in the GWAS. The second reason is to aid the interpretation of a GWAS result by predicting the association statistics at untyped variants. In this article, we show that prediction of association statistics at untyped variants that have an influence on the trait produces is overly conservative. Current imputation methods assume that none of the variants in a region (locus consists of multiple variants) affect the trait, which is often inconsistent with the observed data. In this article, we propose a new method, CAUSAL-Imp, which can impute the association statistics at untyped variants while taking into account variants in the region that may affect the trait. Our method builds on recent methods that impute the marginal statistics for GWAS by utilizing the fact that marginal statistics follow a multivariate normal distribution. We utilize both simulated and real data sets to assess the performance of our method. We show that traditional imputation approaches underestimate the association statistics for variants involved in the trait, and our results demonstrate that our approach provides less biased estimates of these association statistics.
Keywords: causal variants, genome-wide association studies, imputation, summary statistics.
1. Introduction
Genome-wide association studies (GWAS) have been used to discover the genetic variants that affect the trait of interest (Hakonarson et al., 2007; Sladek et al., 2007; Zeggini et al., 2007; Yang et al., 2011; Köttgen et al., 2012; Lu et al., 2013; Ripke et al., 2013). GWAS collect information on genetic variants, typically single nucleotide polymorphisms (SNPs), from two populations. In this case, the two populations comprise a large number of individuals who carry a specific disease (cases) and those who do not (controls). GWAS estimate correlations between disease status and collected genetic variants. After estimating the correlations, we perform a statistical test to indicate whether each of the estimated correlations is statistically significant. The computed significant statistics are known as summary statistics or marginal statistics. In GWAS, due to cost considerations, only a subset of SNPs, called tag SNPs, are genotyped and SNPs that are not collected are referred to as untyped SNPs. Although genotypes of untyped SNPs are not collected, we can infer these variant genotypes using their correlations to the tag SNPs. The correlation between a pair of variants is referred to as linkage disequilibrium (LD) (Pritchard and Przeworski, 2001; Reich et al., 2001). Imputation is a process that uses LD to compute the genotypes of the missing variants (Marchini et al., 2007; Browning, 2008; Marchini and Howie, 2008, 2010; Howie et al., 2009, 2012; Li et al., 2009, 2010).
Genotype imputation requires two data sets. One data set is a set of individuals who are genotyped at all the SNPs, and this data set is referred to as the reference panel. The other data set, which is the data set of interest, consists of individuals who are only genotyped at the tag SNPs. We can impute the genotypes of untyped SNPs in the second data set by utilizing the correlations between SNPs that are learned from the reference panel. To use the imputed genotypes for GWAS, we compute the summary statistics of the imputed genotypes by applying the same statistical test as if the imputed SNPs are collected in the second data set. In this article, we use summary statistics and marginal statistics interchangeably. Summary statistics, such as z-scores, indicate the magnitude of the associations between genotypes and a phenotype of interest.
There are two methodologies for aiding GWAS analysis with imputation. The standard way of utilizing imputation in the GWAS analysis is to impute the genotypes and compute the summary statistics from the imputed genotypes (Marchini et al., 2007; Browning, 2008; Marchini and Howie, 2008, 2010; Howie et al., 2009, 2012; Li et al., 2009, 2010). More recently, a second class of methods has been developed that directly imputes the marginal statistics. These methods approximate the combined result of genotype imputation and association test results. It is shown that the statistics of tag SNPs and untyped SNPs follow a multivariate normal distribution (MVN) (Han et al., 2009; Kostem et al., 2011; Hormozdiari et al., 2014, 2015, 2016, 2017, 2018). Thus, given the LD between tag SNPs and untyped SNPs, we get a conditional distribution of statistics of untyped SNPs conditioning on the statistics of tag SNPs. Having the statistics of tag SNPs, we can impute the untyped SNPs with mean of the conditional distribution (Lee et al., 2013; Pasaniuc et al., 2014). These methods are shown to have similar accuracy of genotype imputation and are much faster to use for GWAS. Another benefit of the second class of methods is that these methods only require summary statistics to perform imputation while the first class of methods require individual's level genotype data that are not always available.
Genotype imputation has been widely utilized for two reasons in the analysis of GWAS. One reason is to increase the statistical power of association studies when the causal SNPs are not collected in the GWAS. The second reason is to aid the interpretation of GWAS results by predicting the association statistics at untyped variants. Unfortunately, all the existing methods assume a null-based model where all the variants are not causal. As a result, the computed summary statistics for untyped SNPs are lower than the true summary statistics when there exists a causal variant. Thus, the null-based imputation approach is conservative. These approaches are reasonable when the goal is to identify more genetic variants associated with the trait (Marchini et al., 2007; Browning, 2008; Marchini and Howie, 2008, 2010; Howie et al., 2009, 2012; Li et al., 2009, 2010). However, when the goal is to interpret the associated regions to identify the actual causal variants, this assumption will cause bias at variants that are actually causal.
In this article, we introduce a novel method for imputation of summary statistics under the assumption that some SNPs in a locus can be causal. Our approach uses the statistics at tag SNPs and LD patterns to infer which of the variants are causal, and performs imputation with this information taken into account. As shown in previous works (Han et al., 2009; Kostem et al., 2011; Hormozdiari et al., 2014, 2015), the joint distribution of marginal statistics follows MVN, and the mean of the distribution depends on which SNPs are causal. We compute the marginal statistics of the untyped SNPs conditional on the marginal statistics of tag SNP and the knowledge which SNPs are causal. Since we do not know which variants are causal within a region, we impute the marginal statistics of the untyped SNPs as a weighted average of all possible subsets of SNPs in the region to be causal. Unfortunately, considering all possible subsets of SNPs are intractable, so we assume that we have at most three causal SNPs in a locus. This assumption makes our approach applicable to larger loci in the genome without reducing the accuracy of our method. The idea of bounding the number of causal SNPs is widely used in fine-mapping literature (Hormozdiari et al., 2014, 2015, 2016).
We show that our method (CAUSAL-Imp) performs favorably in both simulated and real data. We apply our method to simulated data sets wherein we generated the marginal statistics. Then, we treat some of the SNPs as untyped and other SNPs as tagged. We apply CAUSAL-Imp and DIST*, which is our implementation of DIST (Lee et al., 2013). We use simulated data to illustrate that CAUSAL-Imp tends to impute summary statistics that are closer to the true generated summary statistics than DIST*. Next, we evaluate our performance utilizing the Northern Finland Birth Cohort (NFBC) data set (Sabatti et al., 2008). We treat the previously reported significant SNPs as untyped and try to impute their summary statistics using CAUSAL-Imp and DIST*. We show that CAUSAL-Imp imputes the associated statistics more accurately than previous approaches.
2. Results
2.1. Overview of CAUSAL-Imp
CAUSAL-Imp builds on methods that perform imputation on summary statistics. It is known that the statistics for a set of SNPs (SNPs in a locus) follow an MVN distribution with a variance–covariance matrix equal to the pairwise correlation between the genotypes (Han et al., 2009; Kostem et al., 2011; Hormozdiari et al., 2014, 2015). For simplicity, let us consider the case wherein one SNP is untyped and the rest are tag SNPs in a region; we have 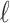 SNPs and the
SNPs and the 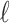 -th SNP is untyped. Let si be the marginal statistics of the i-th SNP. Let
-th SNP is untyped. Let si be the marginal statistics of the i-th SNP. Let 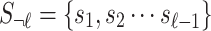 and
and 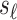 indicate the marginal statistics for the tag and untyped SNPs, respectively. In traditional methods that impute the summary statistics, the model of the joint distribution is as follows:
indicate the marginal statistics for the tag and untyped SNPs, respectively. In traditional methods that impute the summary statistics, the model of the joint distribution is as follows:
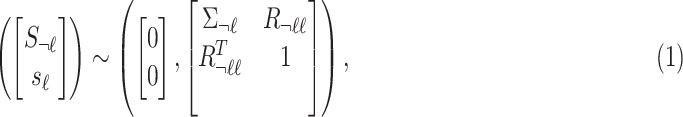 |
where 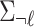 is a (
is a (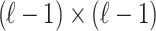 ) matrix of LD for all the SNPs excluding the
) matrix of LD for all the SNPs excluding the 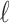 -th SNP and
-th SNP and 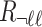 is a
is a 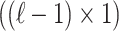 vector that represents the correlation of all the variants with the
vector that represents the correlation of all the variants with the 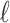 -th SNP, excluding the
-th SNP, excluding the  -th SNP. We can obtain the variance–covariance matrix of the model utilizing the correlation of genotypes from a reference panel, such as the 1000 Genomes data (Durbin et al., 2010; McVean et al., 2012). Then, given the association statistics at observed variants, we can use the conditional form of the multivariate normal to estimate the association statistics at the untyped variants. In traditional methods, marginal statistics of untyped SNPs conditioned on the marginal statistics of tag SNP is as follows:
-th SNP. We can obtain the variance–covariance matrix of the model utilizing the correlation of genotypes from a reference panel, such as the 1000 Genomes data (Durbin et al., 2010; McVean et al., 2012). Then, given the association statistics at observed variants, we can use the conditional form of the multivariate normal to estimate the association statistics at the untyped variants. In traditional methods, marginal statistics of untyped SNPs conditioned on the marginal statistics of tag SNP is as follows:
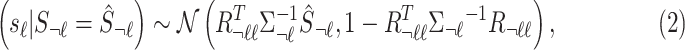 |
where 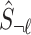 is the observed marginals statistics for all the tag SNPs. We impute the untyped SNP with the mean of the mentioned distribution
is the observed marginals statistics for all the tag SNPs. We impute the untyped SNP with the mean of the mentioned distribution 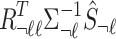 (Lee et al., 2013; Pasaniuc et al., 2014).
(Lee et al., 2013; Pasaniuc et al., 2014).
Our method, CAUSAL-Imp, takes into account the fact that some variants can be causal. Let us assume we only have one causal SNP and the i-th SNP is causal. Then, the marginal statistics for this SNP follows a normal distribution as follows: 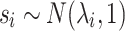 where
where  is the noncentrality parameter (NCP) for the i-th SNP that depends on the true effect size of the SNP toward the phenotype. We extend this to the case where the j-th SNP is not causal and is in LD with the causal SNP i. Then the marginal statistics for the j-th SNP is as follows:
is the noncentrality parameter (NCP) for the i-th SNP that depends on the true effect size of the SNP toward the phenotype. We extend this to the case where the j-th SNP is not causal and is in LD with the causal SNP i. Then the marginal statistics for the j-th SNP is as follows: 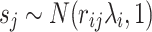 , where
, where 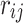 is the LD (genotype Pearson's correlation) between SNPs i and j. To provide a simplified description of this section, we assume that all causal variants have the same NCP. However, CAUSAL-Imp takes into account that causal variants can have different NCP values. We define any subset of SNPs that are causal as the causal status. Causal status indicates which SNPs are causal and which are not. We use 1 to indicate the variants that are causal and 0 to indicate the variants that are not causal. Let
is the LD (genotype Pearson's correlation) between SNPs i and j. To provide a simplified description of this section, we assume that all causal variants have the same NCP. However, CAUSAL-Imp takes into account that causal variants can have different NCP values. We define any subset of SNPs that are causal as the causal status. Causal status indicates which SNPs are causal and which are not. We use 1 to indicate the variants that are causal and 0 to indicate the variants that are not causal. Let 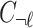 be a vector of size
be a vector of size 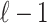 to represent the causal status of the first
to represent the causal status of the first 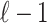 SNPs. Similarly, Let
SNPs. Similarly, Let 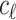 be a binary variable that indicates the causal status of the
be a binary variable that indicates the causal status of the 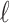 -th SNP. As shown in previous works (Han et al., 2009; Hormozdiari et al., 2014, 2015), the joint marginal statistics given the causal statistics is as follows:
-th SNP. As shown in previous works (Han et al., 2009; Hormozdiari et al., 2014, 2015), the joint marginal statistics given the causal statistics is as follows:
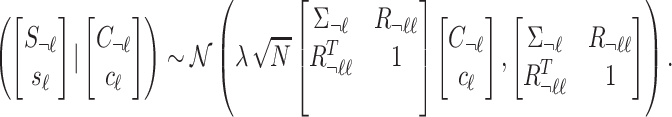 |
The summary statistics of untyped SNP (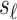 ) conditioning on the statistics of the tag SNPs (
) conditioning on the statistics of the tag SNPs (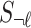 ) and the given causal status,
) and the given causal status, 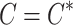 , are as follows:
, are as follows:
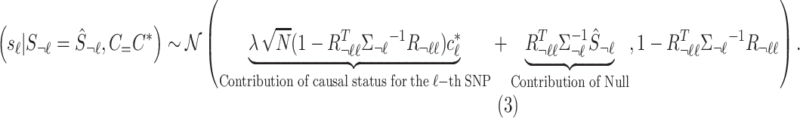 |
However, the true causal status is not known. Thus, CAUSAL-Imp considers all the possible causal statuses. We impute summary statistics as a weighted average of all the summary statistics computed for the unobserved variants for different causal status.
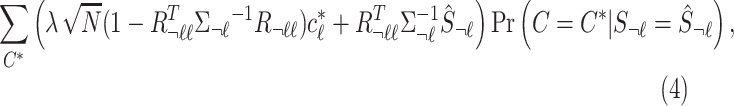 |
where 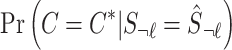 is the posterior probability of a causal status given the observed marginal statistics. Although we describe the method to consider all possible causal status, in practice, we allow up to three causal variants in a locus to reduce the computational complexity.
is the posterior probability of a causal status given the observed marginal statistics. Although we describe the method to consider all possible causal status, in practice, we allow up to three causal variants in a locus to reduce the computational complexity.
2.2. A motivating example
Figure 1 shows a simple region where we have 10 SNPs. In this example, we observe the statistics of three SNPs (SNP3, SNP7, and SNP10), which are indicated by the black arrows. The light triangles indicate the real marginal statistics for all the 10 SNPs. The rest of the SNPs are untyped. Given, the marginal statistics of these three SNPs, we want to impute the marginal statistics of other SNPs. In this example, as the marginal statistic of SNP10 is slightly inflated, we assume one of the SNPs in the region should be causal. In CAUSAL-Imp, we do not know the real causal SNPs, thus we consider all the possible causal statuses in this region. In this example, there are 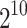 possible causal statuses. For a specific causal status, we impute the summary statistics of the seven unobserved SNPs utilizing the conditional MVN. The dark dots indicate the marginal statistics imputed by CAUSAL-Imp. The light dots indicate the marginal statistics imputed by DIST* [our implementation of DIST; Lee et al. (2013)], which assumes the null model wherein all variants are not causal. In this example, our imputed marginal statistics are closer to the true marginal statistics than those of DIST*.
possible causal statuses. For a specific causal status, we impute the summary statistics of the seven unobserved SNPs utilizing the conditional MVN. The dark dots indicate the marginal statistics imputed by CAUSAL-Imp. The light dots indicate the marginal statistics imputed by DIST* [our implementation of DIST; Lee et al. (2013)], which assumes the null model wherein all variants are not causal. In this example, our imputed marginal statistics are closer to the true marginal statistics than those of DIST*.
FIG. 1.

Motivating example for CAUSAL-Imp. Black arrows indicate the observed (tag) SNPs. Utilizing the fact that the observed marginal statistics of SNP10 is inflated, we can assume one of the SNPs in this region is causal. SNP, single nucleotide polymorphism.
Note that we perform our evaluations using our own implementation of the standard summary statistic method (DIST) (Lee et al., 2013), which we refer to as DIST*. The reason we used our own implementation is that these methods rely on many matrix operations that may result in numerical issues. The differences in linear algebra libraries dealing with numerical issues can cause differences in the results. By reimplementing DIST, our approach and DIST* share many parts of the implementation to eliminate this issue from the evaluation.
2.3. CAUSAL-Imp achieves better statistics than the existing methods in simulated data sets
To assess the performance of our method, we simulated marginal statistics utilizing the NFBC data set. The NFBC data set consists of 10 phenotypes and 331,476 genotypes measured in 5327 individuals. Since imputation is a regional analysis, we selected 20 regions from the NFBC and computed the LD between each pair of SNPs. In this setting, we use 100 SNPs for each locus. Then, we simulated the marginal statistics from the MVN distribution similar to the previous studies (Zaitlen et al., 2007; Hormozdiari et al., 2014, 2015), where we implant one causal SNP. We generated 1000 sets of summary statistics. We assume that 30% of the SNPs are tagged and that the rest of SNPs, including the causal SNP, are untyped. Then, we run CAUSAL-Imp and DIST* on the simulated data.
We compute the average distance between the imputed marginal statistics and the true simulated marginal statistics as a measure of accuracy. We use the 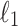 distance as a measure of accuracy, which is computed as follows:
distance as a measure of accuracy, which is computed as follows: 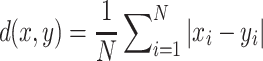 . We compute this distance for the causal SNP, shown in Figure 2A, and the other SNPs, shown in Figure 2B. We vary the power from 20% to 80%. We observe that the statistics imputed by our method are closer to the true statistics. We perform a similar experiment wherein we implant two causal variants in a locus. In this experiment, the imputed statistics from CAUSAL-Imp are closer to true statistics than those of DIST*. The results for this experiment are not shown due to space limitation.
. We compute this distance for the causal SNP, shown in Figure 2A, and the other SNPs, shown in Figure 2B. We vary the power from 20% to 80%. We observe that the statistics imputed by our method are closer to the true statistics. We perform a similar experiment wherein we implant two causal variants in a locus. In this experiment, the imputed statistics from CAUSAL-Imp are closer to true statistics than those of DIST*. The results for this experiment are not shown due to space limitation.
FIG. 2.

CAUSAL-Imp achieves better statistics than the existing methods in simulated data sets. We simulated marginal statistics for regions that are obtained from the NFBC data. We compared the imputed marginal statistics of our method and DIST*. Our method tends to impute statistics that are closer to the true estimated marginal statistics both for causal and noncausal SNPs. We use 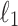 norm to compute the distance. We range the power on the causal SNPs from 20% to 80%. (A) Illustrates the results of the causal variants. (B) Illustrates the results of noncausal variants. NFBC, Northern Finland Birth Cohort.
norm to compute the distance. We range the power on the causal SNPs from 20% to 80%. (A) Illustrates the results of the causal variants. (B) Illustrates the results of noncausal variants. NFBC, Northern Finland Birth Cohort.
2.4. CAUSAL-Imp controls Type I error
We illustrate that CAUSAL-Imp performs better than existing methods. In addition, we need to show these methods control the Type I error. Imputed summary statistics that are controlled for Type I error under the null (no variant is causal) are not inflated or deflated. Genomic inflation is a metric used to check whether the Type I error is controlled (Devlin and Roeder, 1999). We expect the genomic inflation to be close to 1 when there exists no inflation or deflation of statistics. We simulated data under the null where no variant is causal. We consider 30% of the variants to be missing, and then we impute their summary statistics. The genomic inflation for the true summary statistics is 0.98, and the genomic inflation for CAUSAL-Imp is 0.93. However, the genomic inflation of DIST* and IMPUTE2 (Howie et al., 2009) is 0.80 and 1.02, respectively. Thus, CAUSAL-Imp controls the Type I error.
2.5. CAUSAL-Imp achieves better statistics than the existing methods in NFBC
The actual utility of our approach is in examining regions that contain associations where the actual causal variants are not collected. We simulate this scenario by taking actual associated regions in the NFBC data set and removing the peak-associated SNPs from each associated regions [which were reported in a previous study; Sabatti et al. (2008)]. We then apply CAUSAL-Imp, DIST*, and IMPUTE2 (Howie et al., 2009) to evaluate the accuracies of these methods on the peak SNPs. The results are given in Table 1. We observe that the imputed summary statistics from CAUSAL-Imp are closer to the estimated summary statistics than those of DIST*.
Table 1.
CAUSAL-Imp Achieves Better Statistics in Northern Finland Birth Cohort Data Set
| Phenotype | chr | rsID | True statistics | DIST* | CAUSAL-Imp | IMPUTE2 |
|---|---|---|---|---|---|---|
| TG | 2 | rs673548 | −5.444 | −5.37 | −5.38 | −4.46 |
| 8 | rs10096633 | −5.679 | −5.63 | −5.64 | −5.17 | |
| 15 | rs2624265 | 4.22 | 3.55 | 4.15 | 3.60 | |
| HDL | 15 | rs1532085 | 7.13 | 5.59 | 7.17 | 6.47 |
| 16 | rs3764261 | 12.01 | 8.23 | 8.28 | 6.47 | |
| 16 | rs255049 | 6.06 | 5.11 | 5.61 | 5.70 | |
| 17 | rs9891572 | 4.25 | 3.99 | 4.02 | 4.40 | |
| LDL | 1 | rs646776 | −7.70 | −7.92 | −7.92 | −6.96 |
| 2 | rs693 | 6.81 | 6.27 | 6.63 | 5.91 | |
| 11 | rs102275 | −4.51 | −4.43 | −4.44 | −4.54 | |
| 11 | rs174546 | −4.52 | −4.43 | −4.45 | −4.58 | |
| 11 | rs174556 | −4.69 | −4.73 | −4.75 | −4.62 | |
| 11 | rs1535 | −4.43 | −4.46 | −4.46 | −4.45 | |
| 19 | rs11668477 | −5.96 | −3.78 | −3.78 | −5.33 | |
| 19 | rs157580 | −5.161 | −2.6 | −5.24 | −4.20 | |
| CRP | 12 | rs2650000 | −7.08 | −5.25 | −7.36 | −6.05 |
| GLU | 2 | rs560887 | −6.97 | −6.21 | −6.80 | −5.69 |
| 7 | rs10244051 | 5.31 | 4.34 | 4.67 | 4.97 | |
| 7 | rs2191348 | 5.30 | 4.33 | 4.66 | 4.97 | |
| 11 | rs1447352 | −6.35 | −5.08 | −5.39 | −4.75 | |
| 11 | rs7121092 | −5.50 | −4.93 | −5.78 | −4.60 |
We run association on the NFBC data set. We consider the SNPs that are reported significant in a previous study (Sabatti et al., 2008). Then, we treat these SNPs as untyped and impute the marginal statistics using CAUSAL-Imp, DIST*, and IMPUTE2. Our method tends to produce summary statistics closer to the estimated marginal statistics than the two other methods.
TG, triglycerides; HDL, high-density lipoprotein; LDL, low-density lipoprotein; CRP, C-reactive protein; GLU, glucose.
Bold values indicate the best results.
3. Methods
3.1. A standard association statistics
In this study, we have a quantitative phenotype collected for n individuals at m SNPs. Let Y be a 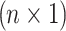 vector of phenotypic values where yj is the phenotypic values for j-th individual. Let G be an
vector of phenotypic values where yj is the phenotypic values for j-th individual. Let G be an 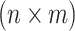 matrix of minor allele counts, where
matrix of minor allele counts, where 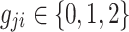 is the minor allele count for j-th individual at i-th SNP, and X be the normalized allele counts matrix G. Define β to be an
is the minor allele count for j-th individual at i-th SNP, and X be the normalized allele counts matrix G. Define β to be an 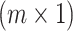 effect size vector, and
effect size vector, and 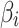 is the effect size of i-th SNP. For simplicity, we assume that both the phenotypic values and the allele counts at each SNP are normalized to have mean 0 and variance 1. Let
is the effect size of i-th SNP. For simplicity, we assume that both the phenotypic values and the allele counts at each SNP are normalized to have mean 0 and variance 1. Let 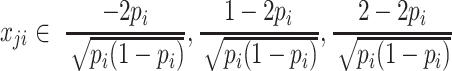 that is the normalized value for
that is the normalized value for 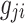 , where pi is the frequency of i-th SNP in the population. Assuming Fisher's polygenic model holds, we use the generative model,
, where pi is the frequency of i-th SNP in the population. Assuming Fisher's polygenic model holds, we use the generative model, 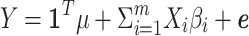 , where
, where 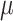 is the phenotypic mean of population,
is the phenotypic mean of population, 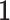 is an
is an 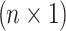 vector of 1, Xi is normalized minor allele counts at i-th SNP,
vector of 1, Xi is normalized minor allele counts at i-th SNP, 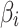 is effect size of i-th SNP, and
is effect size of i-th SNP, and 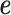 is a vector of measurement noise and environment contributions. We assume
is a vector of measurement noise and environment contributions. We assume 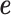 has a normal distribution with mean 0 and variance,
has a normal distribution with mean 0 and variance, 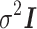 (
(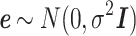 ).
).
In standard GWAS, effect size for each SNP is estimated one SNP at a time. Thus, to compute the marginal statistics for each SNP, we use the following model, 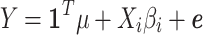 . We note there is a discrepancy between the generative model and testing model; as long as there is no population structure in the data, the estimated effect size is unbiased and follows a normal distribution with mean equal to the true value of effect size. Thus, we have
. We note there is a discrepancy between the generative model and testing model; as long as there is no population structure in the data, the estimated effect size is unbiased and follows a normal distribution with mean equal to the true value of effect size. Thus, we have 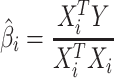 and
and 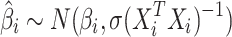 . We use “hat” for each variable to indicate the estimated value for that variable.
. We use “hat” for each variable to indicate the estimated value for that variable.
It is known that the marginal statistics for each SNP is computed as the ratio between the estimated effect size and the estimated variance. Let si indicate the marginal statistics estimated for the i-th SNP. As the marginal statistics follow a normal distribution, we can define the statistics as follows:
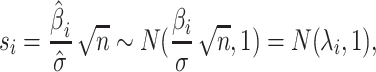 |
where 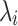 is the NCP for the i-th SNP and
is the NCP for the i-th SNP and 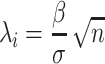 .
.
3.2. Indirect association statistics
To show the indirect association statistics, we assume that i-th variant is associated with the phenotype and j-th variant is correlated with the i-th variant. Thus, the estimated effect size and the marginal statistics for the j-th variant are computed as 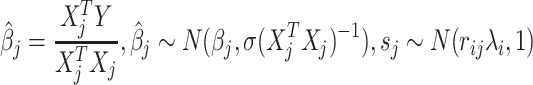 , where
, where 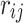 is the correlation between genotypes of i-th and j-th SNPs. Moreover, we estimate the correlation between the genotypes as
is the correlation between genotypes of i-th and j-th SNPs. Moreover, we estimate the correlation between the genotypes as 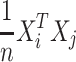 . We compute the covariance between the estimated marginal statistics for the i-th and j-th SNPs as
. We compute the covariance between the estimated marginal statistics for the i-th and j-th SNPs as 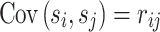 . Thus, the joint distribution of the marginal association statistics for the two SNPs given their NCPs follows an MVN:
. Thus, the joint distribution of the marginal association statistics for the two SNPs given their NCPs follows an MVN:
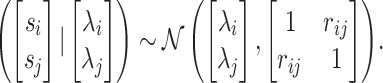 |
3.3. Traditional summary statistics imputation when one SNP is untyped
In this section, we show how traditional summary statistics imputation approaches (Lee et al., 2013; Pasaniuc et al., 2014) work under the scenario when only one SNP is untyped in a locus. Let us say we have 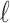 SNPs in a region where
SNPs in a region where 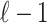 of the SNPs are tagged and only the last SNPs is untyped. We select the
of the SNPs are tagged and only the last SNPs is untyped. We select the 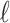 -th SNP to be untyped just for simplicity. Let si indicate the marginal statistics of i-th SNP. Let
-th SNP to be untyped just for simplicity. Let si indicate the marginal statistics of i-th SNP. Let 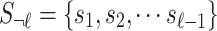 be an
be an 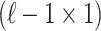 vector of association statistics,
vector of association statistics, 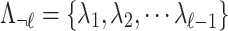 be an
be an 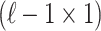 vector of NCPs, and
vector of NCPs, and 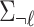 be an
be an 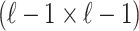 matrix of the pairwise correlation coefficients for the tag SNPs. For the untyped SNP, we use
matrix of the pairwise correlation coefficients for the tag SNPs. For the untyped SNP, we use 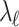 to indicate the unknown NCP. We want to impute the association statistic
to indicate the unknown NCP. We want to impute the association statistic 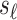 , and let
, and let 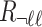 denote the
denote the 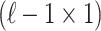 vector of the correlation coefficients between
vector of the correlation coefficients between 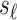 and the
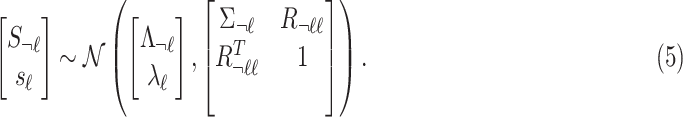
and the 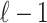 tag SNPs. Thus the joint distribution of the association statistics of the untyped SNP,
tag SNPs. Thus the joint distribution of the association statistics of the untyped SNP, 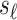 , and the
, and the 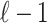 tag SNPs,
tag SNPs, 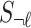 , follows a MVN, which can be expressed as follows:
, follows a MVN, which can be expressed as follows:
 |
Under the null assumption where 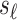 and
and 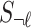 are not associated,
are not associated, 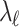 and
and 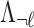 are 0's. Using this equation, we can generate a distribution of the statistics of untyped SNP,
are 0's. Using this equation, we can generate a distribution of the statistics of untyped SNP, 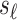 condition on the observed summary statistics,
condition on the observed summary statistics, 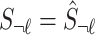 . The conditional distribution follows a MVN, which is computed as follows:
. The conditional distribution follows a MVN, which is computed as follows: 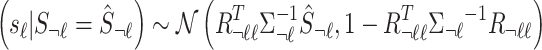 . Thus, utilizing this equation, the traditional summary statistics imputation approaches impute the statistics of the untyped SNP as
. Thus, utilizing this equation, the traditional summary statistics imputation approaches impute the statistics of the untyped SNP as 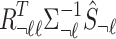 .
.
3.4. Traditional summary statistics imputation when more than one SNP is untyped
In this section, we show how traditional summary statistics imputation approaches (Lee et al., 2013; Pasaniuc et al., 2014) work under the scenario where more than one SNP is untyped in a locus. We use 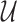 and
and 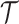 to indicate the set of untyped and tag SNPs, respectively. Let
to indicate the set of untyped and tag SNPs, respectively. Let 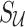 and
and 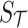 indicate the unobserved summary statistics of untyped SNPs and observe summary statistics of tag SNPs, respectively. We use
indicate the unobserved summary statistics of untyped SNPs and observe summary statistics of tag SNPs, respectively. We use 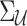 and
and 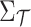 to denote
to denote 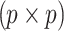 and
and 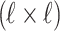 matrices of pairwise correlation coefficients obtained from the untyped SNPs and tag SNPs, respectively. We want to impute unobserved summary statistics
matrices of pairwise correlation coefficients obtained from the untyped SNPs and tag SNPs, respectively. We want to impute unobserved summary statistics 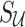 using both observed
using both observed 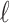 SNPs and p unobserved SNPs. In this case,
SNPs and p unobserved SNPs. In this case, 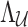 is a
is a 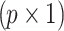 vector of NCPs of untyped SNPs and
vector of NCPs of untyped SNPs and 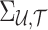 denotes the
denotes the 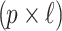 matrix of the correlation coefficients between the p untyped SNPs and the
matrix of the correlation coefficients between the p untyped SNPs and the 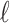 tag SNPs. The joint distribution of the association statistics of the untyped SNP
tag SNPs. The joint distribution of the association statistics of the untyped SNP 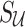 and the tag SNPs
and the tag SNPs 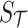 follows an MVN, which can be expressed as follows:
follows an MVN, which can be expressed as follows:
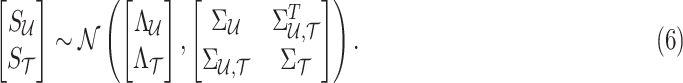 |
Under the null assumption that the untyped SNPs and tag SNPs are not associated, the NCPs of both 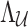 and
and 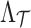 are 0's. Using Equation (6), we can generate a distribution of the statistics of the untyped SNPs,
are 0's. Using Equation (6), we can generate a distribution of the statistics of the untyped SNPs, 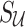 , conditioned on the observed statistics,
, conditioned on the observed statistics, 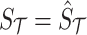 . The conditional distribution follows an MVN, which is computed as follows:
. The conditional distribution follows an MVN, which is computed as follows:
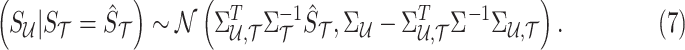 |
Thus, utilizing the mentioned equation, the traditional summary statistics imputation approaches impute the statistic of the untyped SNPs as 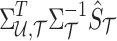 .
.
3.5. CAUSAL-Imp summary statistics imputation with fixed NCP
Recall that having 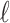 SNPs whose summary statistics are observed and p SNPs whose summary statistics are unobserved, we have a MVN expressed as Equation (6). Instead of assuming that all
SNPs whose summary statistics are observed and p SNPs whose summary statistics are unobserved, we have a MVN expressed as Equation (6). Instead of assuming that all 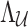 and
and 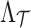 are 0's, our method considers that any subset of SNPs are causal. We introduce C to denote the causal status of the SNPs. Causal status is an
are 0's, our method considers that any subset of SNPs are causal. We introduce C to denote the causal status of the SNPs. Causal status is an 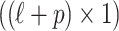 vector of 0's and 1's where ci indicates the causal status of the i-th SNP. Each SNP can have two possible causal statuses 0 or 1, where 0 indicates the SNP is not causal and 1 indicates the SNP is causal. For simplicity, we assume that the NCPs for all the causal variants are the same and equal to
vector of 0's and 1's where ci indicates the causal status of the i-th SNP. Each SNP can have two possible causal statuses 0 or 1, where 0 indicates the SNP is not causal and 1 indicates the SNP is causal. For simplicity, we assume that the NCPs for all the causal variants are the same and equal to 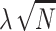 . Later, we will relax this assumption. There are
. Later, we will relax this assumption. There are 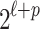 possible causal statuses for C, which is denoted by the set
possible causal statuses for C, which is denoted by the set 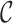 (in practice we only consider up to three causal variants in locus, thus CAUSAL-Imp needs to consider at most
(in practice we only consider up to three causal variants in locus, thus CAUSAL-Imp needs to consider at most 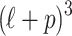 causal statuses). The causal status is consisted of two parts, the causal status of tag SNPs, which we denote by
causal statuses). The causal status is consisted of two parts, the causal status of tag SNPs, which we denote by 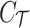 , and the causal status of untyped SNPs, which we denote by
, and the causal status of untyped SNPs, which we denote by 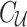 . The joint distribution of observed and unobserved summary statistics in Equation (7) can be expressed as follows:
. The joint distribution of observed and unobserved summary statistics in Equation (7) can be expressed as follows: 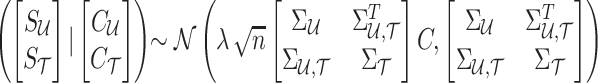 . Using this equation, we can compute the distribution of the untyped statistics,
. Using this equation, we can compute the distribution of the untyped statistics, 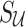 , conditional on the observed statistics,
, conditional on the observed statistics, 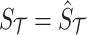 , and the known causal status,
, and the known causal status, 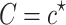 . This conditional distribution follows a multivariate normal that is expressed as follows:
. This conditional distribution follows a multivariate normal that is expressed as follows:
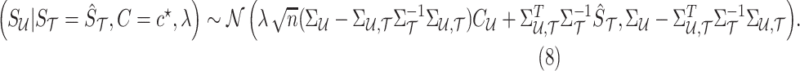 |
We want to compute the probability of summary statistics of untyped SNPs given the summary statistics of the tag SNPs, 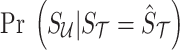 . Utilizing the total probability and Baye's rule, we have
. Utilizing the total probability and Baye's rule, we have
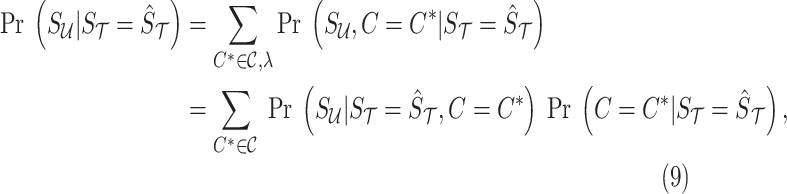 |
where 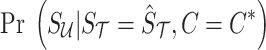 is computed from Equation (8), and
is computed from Equation (8), and 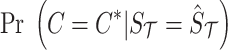 is computed as follows:
is computed as follows:
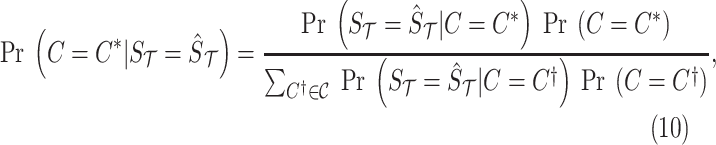 |
where 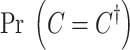 is the prior of the causal status. Similar to most of the fine-mapping methods, for the prior, we assume that SNPs are independent and the probability of an SNP to be causal is equal to 0.01 (Hormozdiari et al., 2014, 2015). This prior implies a sparsity prior on the causal status. Moreover,
is the prior of the causal status. Similar to most of the fine-mapping methods, for the prior, we assume that SNPs are independent and the probability of an SNP to be causal is equal to 0.01 (Hormozdiari et al., 2014, 2015). This prior implies a sparsity prior on the causal status. Moreover, 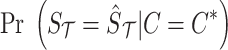 is the likelihood of observed summary statistics given the causal status
is the likelihood of observed summary statistics given the causal status 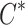 . The observed summary statistics, given the causal status, follows a normal distribution and is computed as follows:
. The observed summary statistics, given the causal status, follows a normal distribution and is computed as follows:
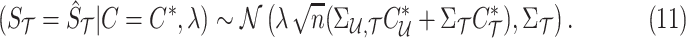 |
Utilizing Equations (8), (10), and (11), we compute the value of 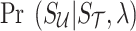 from Equation (9). Thus, we impute
from Equation (9). Thus, we impute 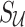 as the mean of
as the mean of 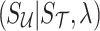 as follows:
as follows:
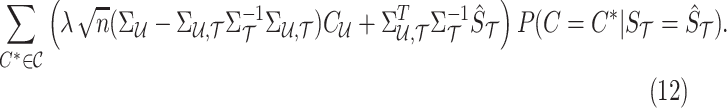 |
3.6. CAUSAL-Imp summary statistics imputation
In previous sections, we assume that the NCPs of the causal variants are fixed and their values are known. In this section, we relax this assumption. We utilize CAVIAR-model (Hormozdiari et al., 2014, 2015, 2016) that is used in fine-mapping frameworks. In CAVIAR-model, the joint distribution of marginal statistics (S) given the vector of NCPs (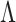 ) follows an MVN distribution that is expressed as
) follows an MVN distribution that is expressed as 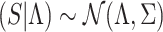 . In addition, the vector of NCPs given the causal status (C) follows an MVN distribution that is expressed as
. In addition, the vector of NCPs given the causal status (C) follows an MVN distribution that is expressed as 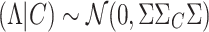 , where
, where 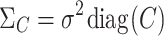 and
and 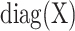 creates a diagonal matrix where the i-th diagonal element is assigned to xi. Using the conjugate prior, we have the following:
creates a diagonal matrix where the i-th diagonal element is assigned to xi. Using the conjugate prior, we have the following:
 |
Thus, utilizing the same statistical framework in CAUSAL-Imp, we have the following:
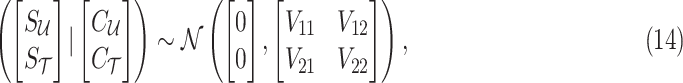 |
where
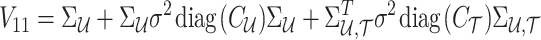 |
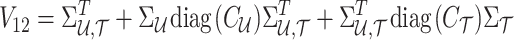 |
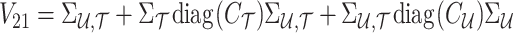 |
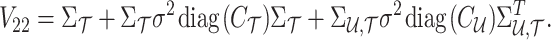 |
Using the MVN conditional distribution, we have
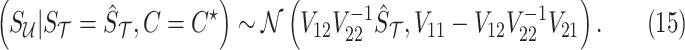 |
Thus, for a given causal status, the optimal value for the imputed marginal statistics is the mean of the mentioned distribution, which is 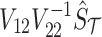 . It is worth mentioning that both
. It is worth mentioning that both 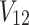 and
and 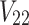 depend on the vector of causal status
depend on the vector of causal status 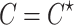 . CAUSAL-Imp utilizes Equation (15) instead of Equation (8).
. CAUSAL-Imp utilizes Equation (15) instead of Equation (8).
4. Discussion
Genotype imputation is widely used to predict the genotypes of untyped SNPs that are not collected in a data set by utilizing the correlation (LD) between the untyped SNPs and the tag SNPs whose genotypes are collected. We propose a new method, CAUSAL-Imp, which combines the principle of fine mapping and summary statistics imputation. CAUSAL-Imp computes the summary statistics for unobserved SNPs by conditioning on the statistics of the observed SNPs and given causal status. CAUSAL-Imp considers all the possible causal statuses where any subset of SNPs can be causal. Thus, the imputed summary statistic is the weighted average of all the summary statistics computed for the unobserved variants for different causal statuses.
Our approach builds upon the recently developed summary statistics framework for imputation (Lee et al., 2013; Pasaniuc et al., 2014). Imputation methods utilizing hidden Markov models (HMMs) to impute individual level data were developed almost 10 years ago (Marchini et al., 2007; Browning, 2008; Marchini and Howie, 2008, 2010; Howie et al., 2009, 2012; Li et al., 2010) and have been improved ever since. In our approach, we incorporate idea of a causal variant and implicitly are then taking the phenotype into account when performing the imputation. It is theoretically possible to extend the HMM-based imputation approaches to take into account causal variants and phenotypes. However, the implementation of such an approach would be incredibly complicated.
Acknowledgments
Y.W., F.H., J.W.J.J., and E.E. are supported by National Science Foundation grants 0513612, 0731455, 0729049, 0916676, 1065276, 1302448, 1320589, 1331176, and 1815624, and National Institutes of Health grants K25-HL080079, U01-DA024417, P01-HL30568, P01-HL28481, R01-GM083198, R01-ES021801, R01-MH101782, and R01-ES022282. E.E. is supported, in part, by the NIH BD2 K award, U54EB020403. We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- Browning S.R. 2008. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum. Genet. 124, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B., and Roeder K. 1999. Genomic control for association studies. Biometrics 55, 997–1004 [DOI] [PubMed] [Google Scholar]
- Durbin R.M., Altshuler D.L., Abecasis G.R., et al. . 2010. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H., Grant S.F.A., Bradfield J.P., et al. . 2007. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448, 591–594 [DOI] [PubMed] [Google Scholar]
- Han B., Kang H.M., and Eskin E. 2009. Rapid and accurate multiple testing correction and power estimation for millions of correlated markers. PLoS Genet. 5, e1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., Gazal S., van de Geijn B., et al. . 2018. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet. 50, 1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., Kichaev G., Yang W.-Y., et al. . 2015. Identification of causal genes for complex traits. Bioinformatics 31, i206–i213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., Kostem E., Kang E.Y., et al. . 2014. Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., van de Bunt M., Segrè A.V., et al. . 2016. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 99, 1245–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., Zhu A., Kichaev G., et al. . 2017. Widespread allelic heterogeneity in complex traits. Am. J. Hum. Genet. 100, 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B., Fuchsberger C., Stephens M., et al. . 2012. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B.N., Donnelly P., and Marchini J. 2009. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostem E., Lozano J.A., and Eskin E. 2011. Increasing power of genome-wide association studies by collecting additional single-nucleotide polymorphisms. Genetics 188, 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen A., Albrecht E., Teumer A., et al. . 2012. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 45, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D., Bigdeli T.B., Riley B.P., et al. . 2013. Dist: Direct imputation of summary statistics for unmeasured SNPs. Bioinformatics 29, 2925–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Vitart V., Burdon K.P., et al. . 2013. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat. Genet. 45, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Willer C., Sanna S., et al. . 2009. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 10, 387–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Willer C.J., Ding J., et al. . 2010. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34, 816–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J., and Howie B. 2008. Comparing algorithms for genotype imputation. Am. J. Hum. Genet. 83, 535–539; author reply 539–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J., and Howie B. 2010. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 [DOI] [PubMed] [Google Scholar]
- Marchini J., Howie B., Myers S., et al. . 2007. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 [DOI] [PubMed] [Google Scholar]
- McVean G.A., Altshuler D.M., Durbin R.M., et al. . 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasaniuc B., Zaitlen N., Shi H., et al. . 2014. Fast and accurate imputation of summary statistics enhances evidence of functional enrichment. Bioinformatics 30, 2906–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J.K., and Przeworski M. 2001. Linkage disequilibrium in humans: Models and data. Am. J. Hum. Genet. 69, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D.E., Cargill M., Bolk S., et al. . 2001. Linkage disequilibrium in the human genome. Nature 411, 199–204 [DOI] [PubMed] [Google Scholar]
- Ripke S., O'Dushlaine C., Chambert K., et al. . 2013. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45:1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatti C., Service S.K., Hartikainen A.-L., et al. . 2008. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 41, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R., Rocheleau G., Rung J., et al. . 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885 [DOI] [PubMed] [Google Scholar]
- Yang J., Manolio T.A., Pasquale L.R., et al. . 2011. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 43:519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitlen N., Kang H.M., Eskin E., et al. . 2007. Leveraging the hapmap correlation structure in association studies. Am. J. Hum. Genet. 80, 683–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E., Weedon M.N., Lindgren C.M., et al. . 2007. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316, 1336–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]